

Abstract
Mutations of GJB2, which encodes connexin 26, are the most common cause of hereditary hearing loss in many human populations. This study was initiated to determine the prevalence of GJB2 mutations in individuals with hearing loss from the Hazara Division in Pakistan. We recruited 70 participants with nonsyndromic deafness segregating as an apparently recessive trait and directly sequenced the GJB2 coding region from their DNA. The homozygous mutations c.71 G→A (p.W24X), c.104 T→G (p.I35S), and c.35delG (p.G12VfsX1) were identified as the cause of hearing loss in three participants (4.28%); in populations from other areas of Pakistan, frequencies of 6–7% have been observed. The mutations c.104 T→G and c.35delG were identified in Pakistan for the first time. These results confirm the low prevalence of GJB2 mutations in Hazara and suggest that mutations in other genes may play a significant role in the etiology of deafness in this population.
Keywords: Connexin 26, deafness, GJB2, hearing loss, Hazara, Pakistan
INTRODUCTION
Deafness is one of the most common sensory defects, and mutations in GJB2 at the DFNB1 locus are a significant cause of hearing loss in many human populations. GJB2 encodes connexin 26, a protein component of intercellular gap junctions, which play crucial physiological roles in the cochlea (Del Castillo and Del Castillo 2011). Most of the reported mutations of GJB2 are predicted to result in prematurely truncated proteins or in nonfunctional alleles. GJB2 mutations are responsible for up to 50% of all genetically inherited cases of hearing loss in many Caucasian populations (Denoyelle et al. 1998) and contribute to one-third of nonsyndromic hearing loss in the Greek population (Pampanos et al. 2002). Additionally, about 28% of autosomal nonsyndromic deafness in Turkey can be attributed to mutations in GJB2 (Bonyadi et al. 2009). In Pakistani families, however, only 6.1–6.9% of inherited hearing loss is caused by mutations in GJB2 (Santos et al. 2005; Yoong et al. 2011). More than 200 pathogenic mutations have been reported in GJB2 worldwide.
Hazara, in the province of Khyber Pakhtunkhwa of northern Pakistan, has been populated since ancient times. Alexander the Great, Ashoka the Great, and many other Indian kings ruled over Hazara. It was also part of the Turkish, Afghan, and British empires before Pakistan gained independence in 1947. Hazara's population of more than 4.5 million consists of two major ethnic groups, the Hindko and the Pashtun. A few other minor groups include the Kashmiri and Kohistani. The tribes include Karlal, Jadoon, Khawaja, Gujjar, Malik, Tanoli, Karlugh Turks, Abbasi, and Syed, among others. Some tribes trace their ancestry back to the invaders. No research on genetically inherited hearing loss has been previously conducted on the Hazara population.
MATERIALS AND METHODS
We studied a group of 70 deaf participants, residents of Hazara. Individuals with hearing loss were identified with the help of audiologists, centers for special education, and regional schools. Participants were visited multiple times to obtain family histories and to record phenotypes. The parents of all deaf individuals were first cousins, and none of the families had a previous history of hearing loss. A medical history was obtained from each participant to avoid including subjects with hearing loss due to other medical and environmental causes. Data were obtained about noise exposure, illness, accidents, antibiotic usage, abnormal hair and skin pigmentation, vestibular dysfunction, night blindness, infection, and tinnitus for all affected individuals. Goiters were excluded by palpation. Audiograms were obtained from the schools, but if the degree of hearing loss had not been previously documented, pure tone audiometry was performed in ambient noise conditions.
All participants were given Romberg and tandem gait tests to check vestibular function. Individuals were excluded from the study if additional clinical manifestations with hearing loss were observed. We also recruited 50 ethnically matched controls with no family history of hearing loss. A venous blood sample of 5–10 ml was collected from each participant. DNA was extracted from whole blood using a nonorganic method involving lysis, proteinase K digestion, salting out, and isopropanol precipitation. All procedures were approved by the Advanced Studies and Research Board of Hazara University, Mansehra, Pakistan, and the Institutional Review Board of the School of Biological Sciences, University of the Punjab, Lahore, Pakistan.
GJB2 consists of two exons, but the entire open reading frame of the gene resides in the second exon. We designed primers for PCR amplification of the coding exon of GJB2 with 80 bp of surrounding intronic and untranslated regions. GJB2 was amplified for all 70 deaf participants and 50 controls. The amplicons were precipitated with ethanol and sequenced with Big Dye Terminator version 3.1 (Applied Biosystems, Foster City, CA). Sequenced samples were electrophoresed on an ABI 3730 genetic analyzer, and the traces were inspected using SeqMan software (DNAstar Lasergene version 5.0.221.0).
RESULTS
Analysis of the sequencing data revealed the homozygous mutations c.71 G→A (p.W24X), c.104 T→G (p.I35S), and c.35delG (p.G12VfsX1) in three deaf individuals (Table 1; Fig. 1), for a prevalence of 4.28% among individuals with nonsyndromic deafness from the Hazara Division of Pakistan. There were 12 heterozygous individuals with the three known polymorphisms in GJB2: the p.R12H polymorphism was reported in 9 participants, p.V153 and p.M163V were each found in one participant, and one participant had p.R127H/V153I in a compound heterozygous state. The remaining 55 samples had a normal GJB2 sequence. None of the control samples was heterozygous for any of the mutations, suggesting that carriers of these variants are not common in this population.
Table 1.
GJB2 variations detected in deaf patients from Hazara, Pakistan
| Patient ID | cDNA Change | Amino Acid Change | Status | Hearing Loss |
|---|---|---|---|---|
| SIHT-13 | 71G→A | W24X | Homozygous | Profound |
| SIHT-28 | 104T→G | I35S | Homozygous | Severe to profound |
| SIHT-63 | 35delG | G12VfsX1 | Homozygous | Moderate to severe |
Fig. 1.
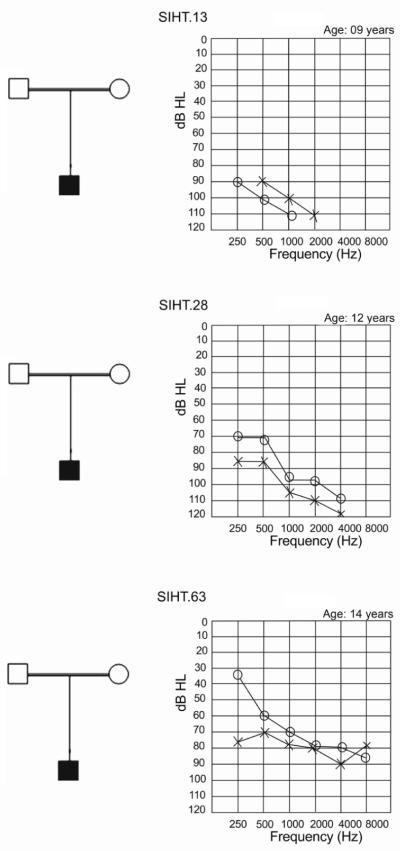
Simplified family pedigrees (left) and audiograms (right) for three deaf individuals with homozygous GJB2 mutations. The parents of each individual are first cousins. In the audiograms, hearing thresholds of the right ear are marked by circles, left ear by crosses. Study participant SIHT.13 (top), in whom the c.71 G→A mutation (p.W24X) was observed, had a profound hearing loss. Participant SIHT.28 (center), in whom the transversion mutation c.104 T→G (p.I35S) was observed, had a severe to profound hearing loss. Participant SIHT.63 (bottom), in whom a single nucleotide deletion (c.35delG) was observed, had a moderate to severe hearing loss.
DISCUSSION
The percentage of deafness in Hazara attributable to GJB2 mutations (4.28%) is slightly lower than that reported in previous studies of Pakistani families (6.1 and 6.9%) but is not significantly different. Since we studied sporadically affected individuals with no previous family history of hearing loss, there is a possibility that some individuals may have de novo mutations in genes responsible for dominantly inherited deafness or that some participants' deafness is due to other than genetic causes. As is true in other ethnic groups in Pakistan, many rare loci and genes may segregate in the Hazara population, with a cumulative effect on the etiology of deafness.
Previously, six variants in GJB2 were reported in Pakistani individuals with severe to profound hearing loss. The most common of these mutations are p.W77X and p.W24X (Santos et al. 2005). Pakistan and India have a shared history and culture, and p.W24X also has a high frequency in the Indian population (Mani et al. 2009). We found the p.W24X mutation in only one of our study participants (SIHT-13), however.
Similarly, the mutation c.35delG was identified in one deaf individual (SIHT-63) in our cohort. The c.35delG mutation has not been previously reported from Pakistan, although it is one of the most common mutations in Caucasian, Turkish, and many other populations. Because the c.35delG mutation is common to many populations, it is possible that it also persists in Hazara due to a founder effect. The individual SIHT-63 had a moderate to severe hearing loss. Affected individuals homozygous for the c.35delG mutation have been reported to manifest variable thresholds of hearing loss, with mild to profound degrees of deafness (Snoeckx et al. 2005). Some individuals with the c.35delG mutation in both alleles of GJB2 have significant residual hearing, suggesting the presence of genetic modifiers involved in ameliorating the phenotype of this mutation. To date, however, no such modifier has been localized or identified.
The mutation p.I35S was identified for the first time in Pakistan in a deaf individual (SIHT-28) participating in our study. This mutation was previously reported in Australian and Indian populations (Dahl et al. 2001; Mani et al. 2009) and was shown to impair trafficking of the protein to the plasma membrane. Given the shared ancestral history of Pakistan and India, it is tempting to speculate that p.I35S may be another ancestral mutation in the subcontinent.
In summary, our study established that GJB2 does not contribute to a high proportion of isolated hearing loss in the Hazara population, a finding in agreement with study results for other ethnic groups in Pakistan. Furthermore, two mutations, c.35delG and p.I35S, were identified for the first time in the Pakistani population.
ACKNOWLEDGMENTS
We are thankful to Dr. Mukhtiar Hassan for his support during MPhil thesis work at the Department of Biochemistry, Hazara University, Mansehra, Pakistan. We are also grateful to the participants in the study and to EDO social welfare Department Mansehra and Abbottabad. We also acknowledge the efforts of the Director of the Almunir Foundation for Disabled Children, Mansehra; the Director of the Al-Huda Speech Center, Abbottabad; and the principals of government schools for deaf children in Mansehra and Abbottabad for their help in identifying study participants. This research was supported by grant no. R01TW007608 from the Fogarty International Center and National Institute of Deafness and Other Communication Disorders, National Institutes of Health, USA.
REFERENCES
- Bonyadi M, Esmaeili M, Abhari M, Lotfi A. Mutation analysis of familial GJB2-related deafness in Iranian Azeri Turkish patients. Genet Test Mol Biomark. 2009;3:689–692. doi: 10.1089/gtmb.2009.0026. [DOI] [PubMed] [Google Scholar]
- Dahl HH, Saunders K, Kelly TM, Osborn AH, Wilcox S, Cone-Wesson B, Wunderlich JL, Dusart D, Kamarinos M, Gardner RJ, Dennehy S, Williamson R, Vallance N, Mutton P. Prevalence and nature of connexin 26 mutations in children with non-syndromic deafness. Med J Australia. 2001;175:191–194. doi: 10.5694/j.1326-5377.2001.tb143093.x. [DOI] [PubMed] [Google Scholar]
- Del Castillo FJ, Del Castillo I. The DFNB1 subtype of autosomal recessive nonsyndromic hearing impairment. Front Biosci. 2011;16:3252–3274. doi: 10.2741/3910. [DOI] [PubMed] [Google Scholar]
- Denoyelle F, Lina-Granade G, Plauchu H, Bruzzone R, Chaib H, Levi-Acobas, Weil FD, Petit C. Connexin 26 gene linked to a dominant deafness. Nature. 1998;393:319–320. doi: 10.1038/30639. [DOI] [PubMed] [Google Scholar]
- Mani RS, Ganapathy A, Jalvi R, Srikumari Srisailapathy CR, Malhotra V, Chadha S, Agarwal A, Ramesh A, Rangasayee RR, Anand A. Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss. Eur J Hum Genet. 2009;17:502–509. doi: 10.1038/ejhg.2008.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampanos A, Economides J, Iliadou V, Neou P, Leotsakos P, Voyiatzis N, Eleftheriades N, Tsakanikos M, Antoniadi T, Hatzaki A, Konstantopoulou I, Yannoukakos D, Gronskov K, Brondum-Nielsen K, Grigoriadou M, Gyftodimou J, Iliades T, Skevas A, Petersen MB. Prevalence of GJB2 mutations in prelingual deafness in the Greek population. Int J Pediatr Otorhinolaryngol. 2002;2:101–108. doi: 10.1016/s0165-5876(02)00177-5. [DOI] [PubMed] [Google Scholar]
- Santos RL, Wajid M, Pham TL, Hussan J, Ali G, Ahmad W, Leal SM. Low prevalence of Connexin 26 (GJB2) variants in Pakistani families with autosomal recessive non-syndromic hearing impairment. Clin Genet. 2005;67:61–68. doi: 10.1111/j.1399-0004.2005.00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoeckx RL, Huygen PL, Feldmann D, Marlin S, Denoyelle F, Waligora J, Mueller-Malesinska M, et al. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet. 2005;77:945–957. doi: 10.1086/497996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoong SY, Mavrogiannis LA, Wright J, Fairley L, Bennett CP, Charlton RS, Spencer N. Low prevalence of DFNB1 (connexin 26) mutations in British Pakistani children with nonsyndromic sensorineural hearing loss. Arch Dis Child. 2011;96:798–803. doi: 10.1136/adc.2010.209262. [DOI] [PubMed] [Google Scholar]