

Abstract
In addition to its important role in the regulation of somatic growth by acting as the major circulating transport protein for the insulin-like growth factors (IGFs), IGF binding protein-3 (IGFBP-3) has a variety of intracellular ligands that point to its function within major signaling pathways. The discovery of its interaction with the retinoid X receptor has led to the elucidation of roles in regulating the function of several nuclear hormone receptors including retinoic acid receptor-α, Nur77 and vitamin D receptor. Its interaction with the nuclear hormone receptor peroxisome proliferator-activated receptor-γ is believed to be involved in regulating adipocyte differentiation, which is also modulated by IGFBP-3 through an interaction with TGFβ/Smad signaling. IGFBP-3 can induce apoptosis alone or in conjunction with other agents, and in different systems can activate caspases −8 and −9. At least two unrelated proteins (LRP1 and TMEM219) have been designated as receptors for IGFBP-3, the latter with a demonstrated role in inducing caspase-8-dependent apoptosis. In contrast, IGFBP-3 also has demonstrated roles in survival-related functions, including the repair of DNA double-strand breaks through interaction with the epidermal growth factor receptor and DNA-dependent protein kinase, and the induction of autophagy through interaction with GRP78. The ability of IGFBP-3 to modulate the balance between pro-apoptotic and pro-survival sphingolipids by regulating sphingosine kinase 1 and sphingomyelinases may be integral to its role at the crossroads between cell death and survival in response to a variety of stimuli. The pleiotropic nature of IGFBP-3 activity supports the idea that IGFBP-3 itself, or pathways with which it interacts, should be investigated as targets of therapy for a variety of diseases.
Keywords: IGFBP-3, apoptosis, DNA damage repair, authophagy, sphingosine kinase, GRP78
Introduction
The insulin-like growth factors, IGF-I and IGF-II (encoded in humans by the genes IGF1 and IGF2) are ubiquitous growth factors that influence cell proliferation, differentiation, survival and migration. The IGFs, which are structurally and functionally related to insulin (Clemmons 2012), signal through the type 1 IGF receptor (IGF1R), a heterotetramer with tyrosine kinase activity, as well as the related insulin receptor (notably insulin receptor isoform A which mediates IGF-II signalling) and hybrids of the two receptor types (Martin and Baxter 2011; Siddle 2012). IGF-like signaling pathways have been strongly conserved through evolution and have been shown to modulate longevity in C. elegans (Lapierre and Hansen 2012).
In contrast to insulin, IGF-I and IGF-II are not known to be stored intracellularly, but are secreted by their cells of origin. In the extracellular environment and the circulation, they are predominantly bound by IGF binding proteins (IGFBPs), a family of six highly conserved proteins (Firth and Baxter 2002) encoded by the genes IGFBP1 to IGFBP6. These proteins are characterized by high affinity for both IGFs, with association constants above 109 L/mol (Baxter 2000; Forbes, et al. 2012). Although the IGFBP nomenclature has been applied to other proteins with weak structural homology to the IGFBPs, in particular mac25 (sometimes called IGFBP-7) and members of the CCN family, there is now wide consensus that only the six proteins with high affinity for the IGFs should be known as IGFBPs.
The conserved IGFBP structure can be divided into three domains of approximately equal size: cysteine-rich amino- and carboxy-terminal domains, which are highly conserved, joined by an unconserved central or linker domain. Both mutagenesis studies and structural determination by NMR and X-ray crystallography have revealed that high-affinity IGF binding involves residues in both the amino- and carboxy-terminal domains (Baxter 2000; Forbes et al. 2012). Two of the six IGFBPs (IGFBP-3 and IGFBP-5) form complexes in the circulation that contain either IGF-I or IGF-II, and a third protein, the acid-labile subunit or ALS (encoded by the IGFALS gene). The IGFBP binding site for ALS consists of a highly basic motif in the carboxy-terminal domain (Firth, et al. 2001; Firth, et al. 1998). Basic residues in this domain also form a bipartite nuclear localization signal (NLS) (Schedlich, et al. 2000). IGFBP-6, which also has a basic carboxy-terminal motif, is unable to bind ALS (Twigg, et al. 1998), but appears to have a functional NLS (Iosef, et al. 2008).
IGFBPs were first characterized for their IGF transport function in the circulation, where they are known to act as a reservoir of IGFs, mainly bound to IGFBP-3 in ternary complexes with ALS. Circulating IGFBPs regulate the bioavailability of the IGFs by controlling their egress from the circulation to the tissues (Baxter 1993; Payet, et al. 2004; Rajaram, et al. 1997). Over the past decade, it has become clear that IGFBPs have many ligands apart from the IGFs and ALS, and have significant functions in the pericellular and intracellular spaces in addition to the circulation. This review, concentrating on IGFBP-3, will discuss recent discoveries of other (i.e. non-IGF) ligands, and the biological functions in which they have been implicated. These interactions may explain, at least in part, the dichotomous growth-inhibitory and -stimulatory effects that have been attributed to IGFBP-3.
Nuclear hormone receptors
In 2000, Liu et al. reported the unexpected finding, by yeast two-hybrid screen, that IGFBP-3 interacts with the retinoid X receptor, RXRα (Liu, et al. 2000). RXRα is a member of the class II nuclear hormone receptors, and typically acts either as a homodimer or, more commonly, as a heterodimer with other members of this group including the retinoic acid receptors (RARs), the vitamin D receptor VDR, the thyroid hormone receptor TR, liver X receptors (LXRs) and peroxisome proliferator-activated receptors (PPARs) (Dawson and Xia 2012). This important study revealed IGFBP-3 as a potential transcriptional regulator, activating effects mediated through the RXR response element but inhibiting signaling through the RAR response element (RARE, activated by ligand binding to RXR-RAR heterodimers) (Liu et al. 2000).
Breast cancer cells with a basal-type molecular subtype have high IGFBP-3 expression, and growth inhibition of the basal cell lines Hs578T and MDA-MB-231 by the RARα ligand, all-trans-retinoic acid (atRA), was found to be modulated by endogenous IGFBP-3, such that immunoneutralizing IGFBP-3 sensitized both cell lines to atRA inhibition (Schedlich, et al. 2004). Further investigation showed that IGFBP-3 inhibited RARE transactivation by binding to RARα and blocking RXRα-RARα heterodimerization, with no effect on atRA binding. Thus, in this context, IGFBP-3 is potentially growth-stimulatory to these basal-type cell lines, by blocking their growth inhibition by RAR ligands. Similarly, IGFBP-3 has been found to block the RAR-dependent differentiation of myeloid leukemia cells, while it enhanced differentiation induced by an RXR-selective ligand (Ikezoe, et al. 2004).
Contrasting with its effect in breast cancer cells, in 22RV1 human prostate cancer cells, IGFBP-3 was reported to induce apoptosis by a mechanism involving another RXRα dimerization partner, Nur77 (Lee, et al. 2005). This IGFBP-3 effect involved the rapid mitochondrial translocation of RXRα-Nur77 dimers, resulting in cytochtome c release, and required direct cytoplasmic interaction between IGFBP-3 and Nur77 (Lee, et al. 2007), but the precise details of the mechanism are still unclear. IGFBP-3 also binds directly to VDR, as does the related binding protein IGFBP-5 (Schedlich, et al. 2007b), and IGFBP-3 is inhibitory to VDR transcriptional activity (Ikezoe et al. 2004).
The metabolic regulator PPARγ has an important role during adipogenic differentiation. Since PPARγ signals as a heterodimer with RXRα, it was proposed that IGFBP-3 might interfere with this process (Chan, et al. 2009), particularly since IGFBP-3 knockout mice are described as having increased adiposity (Yakar, et al. 2009). However, the adipogenic differentiation of bone marrow derived mesenchymal stem cells was not enhanced by IGFBP-3 knockout (Fritton, et al. 2010). Using the 3T3-L1 preadipocyte model of adipogenesis, both exogenous and overexpressed IGFBP-3 were found to be inhibitory to adipogenic differentiation (Chan et al. 2009). This inhibition was associated with the direct interaction of IGFBP-3 and PPARγ, demonstrated by coimmunoprecipitation from 3T3-L1 cell lysates, and the inhibition of PPARγ-RXRα dimerization and ligand-induced transcriptional activity. Recently we have observed the same effect of IGFBP-3 in human breast cancer cell lines although, paradoxically, IGFBP-3 did not reverse the growth-inhibitory effect of PPARγ ligands in these cells, but acted to enhance the growth inhibition (Pon et al., unpublished data).
A few studies have investigated structural determinants involved in nuclear receptor-IGFBP-3 interactions, using mutants of both RXRα and IGFBP-3, including the basic domain IGFBP-3 mutant originally characterized for its deficient ALS binding and nuclear translocation (Firth et al. 1998; Schedlich, et al. 1998). Using GST-pulldown assays, IGFBP-3 was found to interact with the DNA-binding domain (C domain) of RXRα. No binding was observed to the aminoterminal (A/B) domain, the hinge (D) domain, or the carboxyterminal ligand-binding (E/F) domain (Schedlich, et al. 2007a). Examining binding determinants on IGFBP-3, both aminoterminal residues (T58, R60) and basic carboxyterminal residues (220–222, 228–232) were found to be important for RXRα binding (Schedlich et al. 2007a). Similar residues are involved in IGFBP-3 interaction with PPARγ (Chan et al. 2009). Non-RXRα binding mutants were unable to block atRA signaling, demonstrating the essential role of the IGFBP-3-RXRα interaction in the inhibition of retinoic acid action. The 228–232 mutant also failed to inhibit PPARγ-dependent adipogenesis (Chan et al. 2009) and interestingly, appeared to exert a dominant negative effect in preventing breast cancer cell growth inhibition by the PPARγ ligand rosiglitazone, in contrast to wild-type IGFBP-3 which enhanced the rosiglitazone effect (Pon et al., unpublished data).
Thus it appears that, similar to the interaction between IGFBP-3 and the IGFs, its interaction with RXRα involves both aminoterminal and carboxyterminal residues. However, in contrast to IGF binding (Yan, et al. 2004), RXRα binding was not inhibited by mutating the key IGF-binding determinants, L77, L80 and L81 (Schedlich et al. 2007a). These residues are part of an LXXLL motif, sometimes termed the “NR box” and known to be involved in coactivator binding of nuclear receptors (Savkur and Burris 2004). The lack of involvement of these residues of IGFBP-3 in nuclear hormone receptor binding suggests that, despite its regulatory role, IGFBP-3 should not be categorized as a nuclear receptor coactivator, a conclusion also supported by its demonstrated interaction with the DNA-binding domain of RXRα.
LRP1 and TGFβ signaling
The ways in which extracellular IGFBP-3 regulates intracellular events – apart from simply preventing IGFs from binding and activating IGF1R – have remained elusive over many years. IGFBP-3 association with the surface of cells has been recognized for over two decades (Martin, et al. 1992) but the original characterization of its binding sites as functional receptors (Oh, et al. 1993) was not fully substantiated. A large cell-surface protein termed the type V transforming growth factor-β receptor (TβRV) was later shown to bind IGFBP-3, but again its putative role in IGFBP-3 signaling was not well established (Leal, et al. 1997). This protein was subsequently determined to be identical to the low density lipoprotein receptor-related protein LRP1, also known as the α2-macroglobulin receptor, and its mediation of IGFBP-3 growth-inhibitory signaling was said to involve the dephosphorylation of insulin receptor substrate-2 (IRS-2) (Huang, et al. 2004). Another, unrelated report also described an inhibitory role for IGFBP-3 mediated through protein dephosphorylation (Ricort and Binoux 2002).
LRP1 is an endocytosing receptor and its impairment has been shown to result in extracellular accumulation of IGFBP-3, supporting a role for this receptor in IGFBP-3 internalization (Lee, et al. 2006). Other studies found that IGFBP-3 endocytosis requires a caveolin-binding structural motif and involves its binding to transferrin and internalization through the transferrin receptor (Lee, et al. 2004; Singh, et al. 2004). Interactions with β1 integrin and caveolin-1 have also been reported in other studies (Perks, et al. 2011). Since LRP1 can bind caveolin-1 and associate with caveolae (Zhang, et al. 2004), these mechanisms are not necessarily mutually exclusive. However, a dynamin 2-dependent endocytic pathway for IGFBP-3 has also been demonstrated in osteosarcoma cells (Micutkova, et al. 2012), but the kinetics of transferrin and IGFBP-3 uptake were found to be different, suggesting that uptake through the transferrin receptor is unlikely in these cells. How the various proposed cellular uptake mechanisms might inter-relate and mediate the regulation of cell signaling by IGFBP-3 remains to be elucidated.
IGFBP-3 inhibitory signaling through LRP1/TβRV was originally described as occurring without phosphorylation of the canonical TGFβ signaling intermediates, the Smad proteins (Leal, et al. 1999), but other studies showed that IGFBP-3 could activate Smad2 and Smad3 phosphorylation and required the type I and type 2 TGFβ receptors (TβRI and TβRII). In T47D breast cancer cells, that lack TβRII, restoration of this receptor by transfection sensitized the cells to Smad2 and Smad3 phosphorylation, and inhibition of cell proliferation, by IGFBP-3 (Fanayan, et al. 2000). TβRI was also phosphorylated in response to IGFBP-3, suggesting that IGFBP-3 required an intact TGFβ signaling pathway for growth inhibition of this cell line (Fanayan, et al. 2002). Subsequently Smad activation by IGFBP-3 has been demonstrated in a variety of other cell lines. In intestinal smooth muscle cells, IGFBP-3 was shown to stimulate TβRI and Smad2 phosphorylation, resulting in the inhibition of proliferation (Kuemmerle, et al. 2004). Similarly in human placental explants, IGFBP-3 inhibition of cytotrophoblast proliferation was shown to involve Smad2 activation and have a requirement for the TβRI/TβRII system (Forbes, et al. 2010).
Recently we also demonstrated that IGFBP-3 stimulates Smad2 activation in 3T3-L1 preadipocytes, possibly contributing to the inhibitory effect of IGFBP-3 on their adipogenic differentiation (de Silva, et al. 2012). This mechanism may be predicted to cross-talk with the PPARγ-dependent mechanism described above, since PPARγ ligands can oppose TGFβ signaling through Smad2 (Liu, et al. 2011) and conversely, loss of PPARγ in knockout mouse fibroblasts is associated with constitutive Smad signaling (Ghosh, et al. 2008). Interestingly, LRP1/TβRV is upregulated by the PPARγ agonist rosiglitazone (Moon, et al. 2012), and has also been implicated in the cross-talk between Smad signaling and PPARγ (Boucher, et al. 2007). Although IGFBP-3 clearly activates the Smad pathway in multiple cell types, a direct interaction between IGFBP-3 and the TβRI/TβRII receptors has not been demonstrated. The precise role (if any) of LRP1/TβRV and its adapter proteins such as GULP (Ma, et al. 2012), in IGFBP-3 activation of the TβR/Smad signaling pathway, is unknown.
Death receptor signaling
Cell death by apoptosis is defined as proceeding through either an intrinsic pathway, initiated by intracellular stimuli such as oxidative stress or DNA damage, or an extrinsic pathway, initiated by extracellular stress signals and generally mediated by ligation of a member of the death receptor family. These mechanisms require the activation of different proteases, including caspase-8 for death receptor-dependent extrinsic apoptosis and caspase-9 for caspase-dependent intrinsic apoptosis (Galluzzi, et al. 2012). IGFBP-3 is inducible by the tumor suppressor p53 (Buckbinder, et al. 1995) and has been demonstrated to induced apoptosis either directly (Butt, et al. 2000) or by potentiating other agents that activate the intrinsic pathway, such as ceramide (Gill, et al. 1997), chemotherapy drugs (Fowler, et al. 2000; Lee, et al. 2002), irradiation (Hollowood, et al. 2000) and other factors (Leibowitz, et al. 2013), although not necessarily in a p53-dependent manner (Butt et al. 2000; Rajah, et al. 1997). Apoptosis induced by IGFBP-3 can involve the activation of both caspase-8 and -9 (Butt, et al. 2005; Kim, et al. 2004), and under different experimental conditions either does (Butt et al. 2005; Jia, et al. 2010), or does not (Kim et al. 2004), cause the release of cytochrome c from the mitochondria, which is required for the formation of the apoptosome and the initiation of a caspase-9-dependent proteolytic cascade (Galluzzi et al. 2012).
Death receptor-mediated apoptosis initiated by IGFBP-3 was reported in MCF-7 breast cancer cells, the extrinsic pathway being defined by caspase-8 cleavage but no cytochrome c release or caspase-9 cleavage (Kim et al. 2004). Subsequently a novel cell “death receptor” involved in IGFBP-3 action was identified (Ingermann, et al. 2010) and designated as IGFBP-3R (a name previously given to LRP1/TβRV). This protein, encoded by the gene TMEM219 (transmembrane protein 219), contains 240 amino acids, i.e. is smaller than IGFBP-3 itself (264 amino acids), and is located in both the plasma membrane and cytoplasm of breast cancer cells. In contrast to members of the tumor necrosis factor (TNF) receptor family and other proteins designated as death receptors, it does not contain the characteristic motif known as a death domain, which is involved in formation of the “death-inducing signaling complex” (DISC) and activation of caspase-8 (Park 2011). However it was described by the authors as a death receptor because in the presence of IGFBP-3, it induces caspase-8-dependent apoptosis (Ingermann, et al. 2010). In prostate and breast cancer xenograft tumors in athymic mice, overexpression of IGFBP3-R caused some inhibition of tumor growth. In addition to its growth-inhibitory role in cancer cells, this protein has been shown to be involved in the inhibitory effect of IGFBP-3 on airway inflammation and hyper-responsiveness by opposing TNFα action, with a suggested role in asthma therapy (Lee, et al. 2011).
EGFR, IGF1R and sphingosine kinase signaling
In addition to extensively-documented growth-inhibitory effects of IGFBP-3, there is also abundant evidence that it can be growth-stimulatory. Exogenous IGFBP-3 was shown two decades ago to potentiate IGF-I-stimulated DNA synthesis and amino acid uptake by fibroblasts but the precise mechanism remained elusive (Conover, et al. 1996; De Mellow and Baxter 1988), although reported to involve Akt activation (Conover, et al. 2000). Subsequent studies have suggested that there may be many mechanisms through which IGFBP-3 can be growth-stimulatory rather than growth-inhibitory, one of which (the blockade of inhibitory atRA signaling) was mentioned previously.
A potentially growth-stimulatory role for IGFBP-3 in many cancer types is suggested by the observation that IGFBP-3 mRNA and/or protein levels are reported to be increased, inter alia, in squamous cell lung cancer (Kettunen, et al. 2004), melanoma (Xi, et al. 2006), clear cell renal cell carcinoma (Chuang, et al. 2008), and pancreatic cancer (Xue, et al. 2008). In breast cancer tissue, high IGFBP-3 expression has been associated in some studies with markers of poor prognosis, and/or worse overall survival (Rocha, et al. 1996; Sheen-Chen, et al. 2009; Vestey, et al. 2005). However, a relationship with overall or disease-free survival has not always been seen in breast (Ren, et al. 2007) or other cancers (Katsaros, et al. 2001), and there is evidence that in some cancers, for example head and neck (Papadimitrakopoulou, et al. 2006) and hepatoma (Aishima, et al. 2006), low tissue IGFBP-3 expression is associated with poorer patient outcome. Clearly, tissue IGFBP-3 expression is only one of a multitude of factors that might influence the survival of cancer patients, but it is of interest that a Kaplan-Meier plot (Györffy, et al. 2010) for women with basal-type breast cancers (that typically have high IGFBP-3 expression) shows a significantly lower probability of recurrence-free survival for patients whose tumors have high IGFBP-3 mRNA expression (n = 478, hazard ratio = 1.6, logrank P = 0.001) (Fig. 1a). In fact, the prognostic value of high IGFBP-3 expression in basal-type breast cancer appears to be at least as strong as that of epidermal growth factor receptor (EGFR) expression (Fig. 1b), an accepted marker in this molecular subtype (Burness, et al. 2010).
Fig. 1.

Kaplan-Meier plots showing the probability of recurrence-free survival for 478 patients with basal-type breast cancers, from a total of 2,977 breast cancer patients, that express either high or low IGFBP-3 mRNA (A) or EGFR mRNA (B), as indicated, autoselecting the best cut-off value. Data calculated using the KM Plotter online survival analysis tool (Györffy, et al. 2010)
The observation that expression of a constitutively active mutated c-Ha-Ras oncogene in mammary epithelial cells caused resistance to the growth-inhibitory effect of IGFBP-3 (Martin and Baxter 1999) led to the discovery that IGFBP-3 could potentiate ligand-dependent signaling through EGFR (Martin, et al. 2003). In these experiments the ERK and p38 MAP kinase pathways were activated, but not the Akt pathway, and the potentiation of EGF-stimulated DNA synthesis by IGFBP-3 was reflected in increased cell proliferation over 7 days. The related binding protein IGFBP-5 was slightly inhibitory rather than stimulatory, while IGFBP-3 mutated in the ALS-binding and nuclear localization domain (residues 228–232) was fully active in potentiating DNA synthesis. In these studies, no direct binding interaction between IGFBP-3 and EGFR was demonstrated (Martin et al. 2003). This growth-stimulatory effect of IGFBP-3 may depend on the tumor microenvironment, since it has been reported that in cells grown on fibronectin, it prevented, rather than stimulated, EGF-dependent cell proliferation (McIntosh, et al. 2010).
In a mouse xenograft model of human breast cancer, T47D cells expressing human IGFBP-3, although initially growth-inhibited in vitro, formed larger tumors in vivo than control cells with undetectable IGFBP-3 expression (Butt, et al. 2004), resembling the clinical situation where high-IGFBP-3 breast tumors in patients were larger that those with low IGFBP-3 expression (Rocha, et al. 1997). In the xenograft model, the faster-growing, IGFBP-3-expressing tumors showed higher staining for EGFR than control tumors, suggesting that IGFBP-3 expression might provide a selective advantage to cells with high EGFR expression. Consistent with the concept that the IGFBP-3-dependent growth stimulation was mediated through EGFR, only the IGFBP-3-expressing cells were growth-inhibited in cell culture by an EGFR kinase inhibitor (Butt et al. 2004). Interestingly, a more recent study using A431 epidermoid carcinoma cells similarly found that the development of resistance to the EGFR inhibitor gefitinib was associated with low IGFBP-3 expression, again supporting the idea that IGFBP-3 might be helping to drive cell growth through EGFR activation (Guix, et al. 2008).
A mechanistic explanation for these observations was suggested by a report that in endothelial cells, IGFBP-3 could stimulate the expression and activity of sphingosine kinase 1 (SphK1), an enzyme that phosphorylates the growth-inhibitory lipid, sphingosine, to a stimulatory form, sphingosine-1-phosphate (S1P) (Granata, et al. 2004). Testing this in breast epithelial cells demonstrated that SphK1 expression and activity was induced by IGFBP-3, and appeared to be the mechanism through which IGFBP-3 transactivates EGFR (Martin, et al. 2009). Silencing of SphK1, but not SphK2, by siRNA prevented EGFR transactivation, as did silencing of the S1P receptors S1P1 and S1P3, but not S1P2. The potentiatiation of EGFR signaling may be a “constitutive” role of endogenous IGFBP-3, since downregulation of endogenous IGFBP-3 markedly blunted the ability of EGF to stimulate tyrosine phosphorylation of EGFR (Martin et al. 2009). A similar phenomenon has been observed in four basal-type breast cancer cell lines (Martin and Baxter, unpublished data).
IGF1R is known to cross-talk with EGFR (van der Veeken, et al. 2009), and downregulating endogenous IGFBP-3 was similarly found to blunt the ability of a receptor-active, non-IGFBP-binding IGF-I analogue to activate IGF1R tyrosine phosphorylation. Indeed, all of the evidence linking IGFBP-3 to EGFR transactivation through activation of SphK1/S1P signaling similarly linked it to IGF1R transactivation (Martin et al. 2009). There appears to be a reciprocity in the cross-talk between IGF1R and EGFR activation by IGFBP-3, since small-molecule inhibitors of either receptor prevented IGFBP-3 from potentiating signaling through the other receptor. The ability of IGFBP-3 to potentiate IGF-I-dependent IGF1R signaling through SphK1 activation offers an explanation for the 25-year-old observation of this phenomenon in skin fibroblasts (De Mellow and Baxter 1988) and provides a powerful rationale for targeting the “IGFBP-3-SphK1 pathway” in conjunction with EGFR and/or IGF1R in patients whose tumors have high expression of EGFR and IGFBP-3, such as basal-type breast cancer.
DNA damage signaling
As discussed earlier, IGFBP-3 can potentiate the induction of apoptosis by ceramide (Gill et al. 1997) as well as other agents. In human umbilical vein endothelial cells (HUVECs), IGFBP-3 increased apoptosis induced by the DNA-damaging drug doxorubicin, but increased cell viability and inhibited apoptosis if the generation of ceramide in response to doxorubicin treatment was prevented by the drug fumonisin B1 (Granata et al. 2004). Ceramide, which is generated either by de novo synthesis or by the action of sphingomyelinases on the membrane lipid sphingomyelin, is a precursor of sphingosine and thus also of S1P (Wymann and Schneiter 2008). The balance between the non-phosphorylated sphingolipids ceramide and sphingosine, which oppose cell survival, and their pro-survival product S1P, may be important in determining whether cells survive or die in response to DNA damaging therapies such as doxorubicin (Young, et al. 2013). It is thus of interest that, in addition to stimulating sphingosine conversion to S1P, IGFBP-3 has been shown to inhibit an injury-induced increase in acid and neutral sphingomyelinases in mouse retina (Kielczewski, et al. 2011), thus potentially decreasing ceramide production and shifting the ceramide-S1P balance further towards the pro-survival S1P.
Doxorubicin (adriamycin) and etoposide, widely used in cancer treatment, exert their chemotherapeutic effects in part by inducing double strand breaks (DSB) in DNA, as does radiotherapy. This form of DNA damage is potentially the most hazardous to genomic integrity, in response to which cells initiate DNA repair processes but may, nevertheless, eventually undergo apoptotic death. DNA DSB damage can induce two main repair mechanisms: homologous recombination (HR), which uses a homologous DNA template to guide the repair process and is generally restricted to S and G2 phase, and non-homologous end-joining (NHEJ), more common in mammalian cells, which joins the broken DNA ends directly and can occur at any stage of the cell cycle (Brandsma and Gent 2012; Jackson and Bartek 2009). An early step in DNA DSB repair by NHEJ is the binding of the serine/threonine kinase, DNA-dependent protein kinase, catalytic subunit (DNA-PKcs) to the damaged DNA ends, followed by its autophosphorylation. There is also substantial evidence that EGFR translocates to the nucleus in response to DNA DSB damage, and forms part of the DNA-PK-dependent repair process (Dittmann, et al. 2005; Kriegs, et al. 2010).
We reported several years ago that DNA-PKcs could phosphorylate IGFBP-3, resulting in its enhanced nuclear entry and retention (Schedlich, et al. 2003). Since we had also shown that IGFBP-3 could activate EGFR through SphK1 in breast cancer cells suggesting that it might be involved in DNA repair, we investigated whether IGFBP-3 itself had a role in the activation of DNA repair by NHEJ in response to DNA-damaging chemotherapy. In the basal-type triple negative breast cancer cell line MDA-MB-468, which has high IGFBP-3 expression, endogenous IGFBP-3 was shown by proximity ligation assay (PLA) to complex with EGFR associated with lipid rafts, which decreased 2–4 h following treatment with etoposide. In parallel with the decline of the EGFR-IGFBP-3 complex in the plasma membrane, it increased in the cell nucleus, concomitant with increases in nuclear IGFBP-3-DNA-PKcs and EGFR-DNA-PKcs complexes (Lin, et al. 2012). The detection of these binary nuclear complexes, all demonstrated by both co-immunoprecipitation and PLA, suggests that all three proteins – IGFBP-3, EGFR and DNA-PKcs – probably form part of a single complex in response to DNA damage. Complex formation was prevented by the EGFR kinase inhibitor gefitinib, as shown in Fig. 2 for IGFBP-3 complexed with DNA-PKcs.
Fig. 2.

Four hours after treatment with 20 μM etoposide, complexes between IGFBP-3 and DNA-PKcs increase in MDA-MB-468 breast cancer cells, predominantly within the nucleus. This increase is blocked by the EGFR kinase inhibitor gefitinib. (A) IGFBP-3-DNA-PKcs interactions are visualized by proximity ligation assay. (B) Quantitation of nuclear and extranuclear complexes. The percentage of complexes detected within nuclei by confocal microscopy (%N) is indicated. *, P < 0.05 compared to control-treated cells. Adapted from (Lin, et al. 2012).
When endogenous IGFBP-3 was downregulated by two siRNAs, DNA-PKcs autophosphorylation was inhibited, as was the appearance of the nuclear EGFR-DNA-PKcs complex in response to etoposide. Importantly, when measured by a direct DNA end-joining assay, IGFBP-3 downregulation inhibited NHEJ activity in nuclear extracts, demonstrating that IGFBP-3 has a direct facilitating role in the repair of DNA DSB damage by NHEJ (Lin et al. 2012). Loss of SphK1 has been shown to make several cancer cell lines (breast, colon and non-small cell lung cancer) more sensitive to doxorubicin-induced DNA damage (Huwiler, et al. 2011), consistent with the idea that SphK1 signaling may promote the repair of damaged DNA, thus enhancing cell survival. Together these studies support a model in which IGFBP-3, through the activation of SphK1 and EGFR, has an enabling role in the DNA damage response by NHEJ, first complexing with EGFR in lipid rafts and then being co-transported with EGFR to the cell nucleus where it forms part of the EGFR-DNA-PKcs complex that is required for the DNA-PKc autophosphorylation response and the initiation of NHEJ. Targeting EGFR kinase activity as a therapeutic approach to sensitize cancers to radiotherapy is already widely used (Mehta 2012). The discovery of the involvement of IGFBP-3-SphK1 signaling in this process suggests that co-targeting this pathway may be of additional benefit.
GRP78 and autophagy
A recent unbiased search for intracellular IGFBP-3 binding partners found a strong interaction with the heat shock protein GRP78 (78 kDa glucose-regulated protein), also known as binding immunoglobulin protein (BiP) (Grkovic, et al. 2013). This interaction has also been detected in a yeast 2-hybrid screen (Li, et al. 2012). Because an important function of GRP78 is as an endoplasmic reticulum chaperone, coordinating the cellular response to protein misfolding (Luo and Lee 2013), the involvement of IGFBP-3 in this process was sought, but IGFBP-3 was not implicated in any pathway of unfolded protein response signaling (Grkovic et al. 2013). GRP78 has also been shown to be required for stress-induced autophagy (Li, et al. 2008), and an investigation of the possible role for the IGFBP-3-GRP78 interaction in this process in breast cancer cells revealed that IGFBP-3 could enhance the survival of cells subjected to glucose starvation and hypoxia in a GRP78-dependent manner (Grkovic et al. 2013). This effect was shown to depend on the induction of autophagy by its inhibition by 3-methyladenine, its prevention by downregulation of the autophagic pathway intermediates beclin-1 and Atg7, and an IGFBP-3-dependent increase in the autophagy marker LC3-II. Intriguingly, glucose deprivation was shown to cause a marked under-glycosylation of IGFBP-3. Since the interaction between IGFBP-3 and GRP78 is enhanced when the IGFBP-3 is underglycosylated (Grkovic et al. 2013), these finding imply that in solid tumors, where a deficit in nutrient and oxygen supply may occur commonly, IGFBP-3 may play a key role in mediating an autophagic survival response. This suggests that targeting the IGFBP-3-GRP78 interaction could be a valuable strategy in the therapy of solid tumors. Whether this IGFBP-3-dependent autophagic response to nutritional stress is also seen in response to genotoxic stress, and therefore contributes to the determination of cell fate after DNA-damaging therapy, will be an important area for future investigation.
Concluding comments
The past decade has seen considerable progress in understanding the many cellular roles of IGFBP-3. While its originally-discovered function as the major carrier of endocrine IGF-I and IGF-II in ternary complexes with ALS is vitally important in the regulation of somatic growth, the recognition that it has many ligands in addition to the IGFs and ALS, that mediate a multiplicity of biological functions, has provided not only new insights into the mechanisms of a variety of cellular processes, but also potential new targets for the treatment of cancer and other diseases. Acting at the crossroads between cell death and cell survival, IGFBP-3 may now be seen to act both as a “caretaker”, contributing to the repair of damaged DNA, as well as a “gatekeeper”, preventing cell replication and promoting cell death when genomic integrity is compromised (Kinzler and Vogelstein 1997). Its glycosylation-regulated interaction with GRP78, and its function in regulating the balance of bioactive sphingolipids, may well be important components of its contribution to these mechanisms (Fig. 3).
Fig. 3.
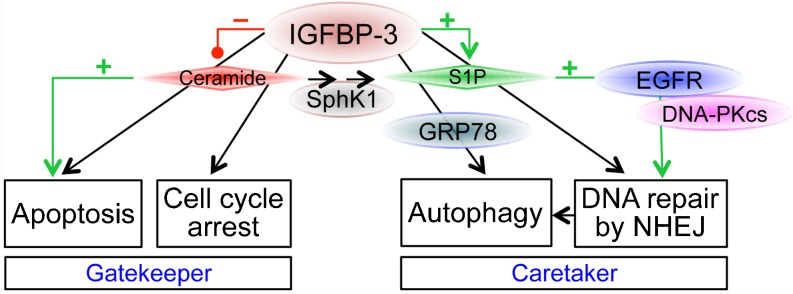
Proposed role of IGFBP-3 in regulating the balance between cell cycle arrest/cell death, and autophagy/cell survival. IGFBP-3 can promote apoptosis alone or in conjunction with ceramide and other agents, but suppresses ceramide generation by downregulating sphingomyelinases. By stimulating sphingosine kinase 1 (SphK1), IGFBP-3 can increase pro-survival sphingosine-1-phosphate (S1P), thus potentiating EGFR activation and cooperating with DNA-PKcs to permit DNA double-strand break repair by non-homologous end-joining (NHEJ). IGFBP-3 also promotes autophagy through interaction with GRP78. See text for details. By regulating both DNA damage repair and cell cycle arrest/apoptosis, IGFBP-3 has both caretaker and gatekeeper roles
It may be asked why it has taken so long to recognise how integrally IGFBP-3 is involved in such vital cell processes. A possible explanation is that, unlike genes such as the caretaker BRCA1 or the tumor suppressor TP53, no disease-causing mutations of the IGFBP3 gene have been identified. In the absence of this genetic evidence, detailed functional studies have been required to elucidate the pivotal place occupied by IGFBP-3 in several important biological processes. Exploiting this knowledge about IGFBP-3-regulated pathways may provide new opportunities for therapeutic intervention in a variety of diseases.
References
- Aishima S, Basaki Y, Oda Y, Kuroda Y, Nishihara Y, Taguchi K, Taketomi A, Maehara Y, Hosoi F, Maruyama Y, et al. High expression of insulin-like growth factor binding protein-3 is correlated with lower portal invasion and better prognosis in human hepatocellular carcinoma. Cancer Sci. 2006;97:1182–1190. doi: 10.1111/j.1349-7006.2006.00322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter RC. Circulating binding proteins for the insulinlike growth factors. Trends Endocrinol Metab. 1993;4:91–96. doi: 10.1016/1043-2760(93)90085-S. [DOI] [PubMed] [Google Scholar]
- Baxter RC. Insulin-like growth factor (IGF) binding proteins: Interactions with IGFs and intrinsic bioactivities. Am J Physiol. 2000;278:E967–E976. doi: 10.1152/ajpendo.2000.278.6.E967. [DOI] [PubMed] [Google Scholar]
- Boucher P, Li WP, Matz RL, Takayama Y, Auwerx J, Anderson RG, Herz J. LRP1 functions as an atheroprotective integrator of TGFbeta and PDFG signals in the vascular wall: implications for Marfan syndrome. PLoS One. 2007;2:e448. doi: 10.1371/journal.pone.0000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandsma I, Gent DC. Pathway choice in DNA double strand break repair: observations of a balancing act. Genome Integr. 2012;3:9. doi: 10.1186/2041-9414-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, Kley N. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature. 1995;377:646–649. doi: 10.1038/377646a0. [DOI] [PubMed] [Google Scholar]
- Burness ML, Grushko TA, Olopade OI. Epidermal growth factor receptor in triple-negative and basal-like breast cancer: promising clinical target or only a marker? Cancer J. 2010;16:23–32. doi: 10.1097/PPO.0b013e3181d24fc1. [DOI] [PubMed] [Google Scholar]
- Butt AJ, Firth SM, King MA, Baxter RC. Insulin-like growth factor-binding protein-3 modulates expression of Bax and Bcl-2 and potentiates p53-independent radiation-induced apoptosis in human breast cancer cells. J Biol Chem. 2000;275:39174–39181. doi: 10.1074/jbc.M908888199. [DOI] [PubMed] [Google Scholar]
- Butt AJ, Martin JL, Dickson KA, McDougall F, Firth SM, Baxter RC. Insulin-like growth factor binding protein-3 expression is associated with growth stimulation of T47D human breast cancer cells: the role of altered epidermal growth factor signaling. J Clin Endocrinol Metab. 2004;89:1950–1956. doi: 10.1210/jc.2003-030914. [DOI] [PubMed] [Google Scholar]
- Butt AJ, Dickson KA, Jambazov S, Baxter RC. Enhancement of tumor necrosis factor-alpha-induced growth inhibition by insulin-like growth factor-binding protein-5 (IGFBP-5), but not IGFBP-3 in human breast cancer cells. Endocrinology. 2005;146:3113–3122. doi: 10.1210/en.2004-1408. [DOI] [PubMed] [Google Scholar]
- Chan SS, Schedlich LJ, Twigg SM, Baxter RC. Inhibition of adipocyte differentiation by insulin-like growth factor-binding protein-3. Am J Physiol Endocrinol Metab. 2009;296:E654–663. doi: 10.1152/ajpendo.90846.2008. [DOI] [PubMed] [Google Scholar]
- Chuang ST, Patton KT, Schafernak KT, Papavero V, Lin F, Baxter RC, Teh BT, Yang XJ. Over expression of insulin-like growth factor binding protein 3 in clear cell renal cell carcinoma. J Urol. 2008;179:445–449. doi: 10.1016/j.juro.2007.09.106. [DOI] [PubMed] [Google Scholar]
- Clemmons DR. Metabolic actions of insulin-like growth factor-I in normal physiology and diabetes. Endocrinol Metab Clin North Am. 2012;41:425–443. doi: 10.1016/j.ecl.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover CA, Clarkson JT, Bale LK. Factors regulating insulin-like growth factor-binding protein-3 binding, processing, and potentiation of insulin-like growth factor action. Endocrinology. 1996;137:2286–2292. doi: 10.1210/en.137.6.2286. [DOI] [PubMed] [Google Scholar]
- Conover CA, Bale LK, Durham SK, Powell DR. Insulin-like growth factor (IGF) binding protein-3 potentiation of IGF action is mediated through the phosphatidylinositol-3-kinase pathway and is associated with alteration in protein kinase B/AKT sensitivity. Endocrinology. 2000;141:3098–3103. doi: 10.1210/en.141.9.3098. [DOI] [PubMed] [Google Scholar]
- Dawson MI, Xia Z. The retinoid X receptors and their ligands. Biochim Biophys Acta. 2012;1821:21–56. doi: 10.1016/j.bbalip.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mellow JS, Baxter RC. Growth hormone-dependent insulin-like growth factor (IGF) binding protein both inhibits and potentiates IGF-I-stimulated DNA synthesis in human skin fibroblasts. Biochem Biophys Res Commun. 1988;156:199–204. doi: 10.1016/S0006-291X(88)80824-6. [DOI] [PubMed] [Google Scholar]
- de Silva HC, Firth SM, Twigg SM, Baxter RC. Interaction between IGF binding protein-3 and TGFβ in the regulation of adipocyte differentiation. Endocrinology. 2012;153:4799–4807. doi: 10.1210/en.2011-1444. [DOI] [PubMed] [Google Scholar]
- Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, Chen DJ, Kehlbach R, Rodemann HP. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem. 2005;280:31182–31189. doi: 10.1074/jbc.M506591200. [DOI] [PubMed] [Google Scholar]
- Fanayan S, Firth SM, Butt AJ, Baxter RC. Growth inhibition by insulin-like growth factor-binding protein-3 in T47D breast cancer cells requires transforming growth factor-β (TGF-β) and the type II TGF-β receptor. J Biol Chem. 2000;275:39146–39151. doi: 10.1074/jbc.M006964200. [DOI] [PubMed] [Google Scholar]
- Fanayan S, Firth SM, Baxter RC. Signaling through the Smad pathway by insulin-like growth factor binding protein-3 in breast cancer cells: Relationship to transforming growth factor-β1 signaling. J Biol Chem. 2002;277:7255–7261. doi: 10.1074/jbc.M108038200. [DOI] [PubMed] [Google Scholar]
- Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev. 2002;23:824–854. doi: 10.1210/er.2001-0033. [DOI] [PubMed] [Google Scholar]
- Firth SM, Ganeshprasad U, Baxter RC. Structural determinants of ligand and cell-surface binding of insulin-like growth factor-binding protein-3. J Biol Chem. 1998;273:2631–2638. doi: 10.1074/jbc.273.5.2631. [DOI] [PubMed] [Google Scholar]
- Firth SM, Clemmons DR, Baxter RC. Mutagenesis of basic amino acids in the carboxyl-terminal region of insulin-like growth factor binding protein-5 affects acid-labile subunit binding. Endocrinology. 2001;142:2147–2150. doi: 10.1210/en.142.5.2147. [DOI] [PubMed] [Google Scholar]
- Forbes BE, McCarthy P, Norton RS. Insulin-like growth factor binding proteins: a structural perspective. Front Endocrinol (Lausanne) 2012;3:38. doi: 10.3389/fendo.2012.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes K, Souquet B, Garside R, Aplin JD, Westwood M. Transforming growth factor-{beta} (TGF{beta}) receptors I/II differentially regulate TGF{beta}1 and IGF-binding protein-3 mitogenic effects in the human placenta. Endocrinology. 2010;151:1723–1731. doi: 10.1210/en.2009-0896. [DOI] [PubMed] [Google Scholar]
- Fowler CA, Perks CM, Newcomb PV, Savage PB, Farndon JR, Holly JM. Insulin-like growth factor binding protein-3 (IGFBP-3) potentiates paclitaxel-induced apoptosis in human breast cancer cells. Int J Cancer. 2000;88:448–453. doi: 10.1002/1097-0215(20001101)88:3<448::AID-IJC18>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Fritton JC, Kawashima Y, Mejia W, Courtland HW, Elis S, Sun H, Wu Y, Rosen CJ, Clemmons D, Yakar S. The insulin-like growth factor-1 binding protein acid-labile subunit alters mesenchymal stromal cell fate. J Biol Chem. 2010;285:4709–4714. doi: 10.1074/jbc.M109.041913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Wei J, Wu M, Varga J. Constitutive Smad signaling and Smad-dependent collagen gene expression in mouse embryonic fibroblasts lacking peroxisome proliferator-activated receptor-gamma. Biochem Biophys Res Commun. 2008;374:231–236. doi: 10.1016/j.bbrc.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill ZP, Perks CM, Newcomb PV, Holly JM. Insulin-like growth factor-binding protein (IGFBP-3) predisposes breast cancer cells to programmed cell death in a non-IGF-dependent manner. J Biol Chem. 1997;272:25602–25607. doi: 10.1074/jbc.272.41.25602. [DOI] [PubMed] [Google Scholar]
- Granata R, Trovato L, Garbarino G, Taliano M, Ponti R, Sala G, Ghidoni R, Ghigo E. Dual effects of IGFBP-3 on endothelial cell apoptosis and survival: involvement of the sphingolipid signaling pathways. FASEB J. 2004;18:1456–1458. doi: 10.1096/fj.04-1618fje. [DOI] [PubMed] [Google Scholar]
- Grkovic S, O'Reilly VC, Han S, Hong M, Baxter RC Firth SM (2013) IGFBP-3 binds GRP78, stimulates autophagy and promotes the survival of breast cancer cells exposed to adverse microenvironments. Oncogene 32:2412–2420 [DOI] [PubMed]
- Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, Rinehart C, Seidel B, Yee D, Arteaga CL, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. 2008;118:2609–2619. doi: 10.1172/JCI34588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123:725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- Hollowood AD, Lai T, Perks CM, Newcomb PV, Alderson D, Holly JM. IGFBP-3 prolongs the p53 response and enhances apoptosis following UV irradiation. Int J Cancer. 2000;88:336–341. doi: 10.1002/1097-0215(20001101)88:3<336::AID-IJC3>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Huang SS, Leal SM, Chen CL, Liu IH, Huang JS. Identification of insulin receptor substrate proteins as key molecules for the TbetaR-V/LRP-1-mediated growth inhibitory signaling cascade in epithelial and myeloid cells. FASEB J. 2004;18:1719–1721. doi: 10.1096/fj.03-1377com. [DOI] [PubMed] [Google Scholar]
- Huwiler A, Kotelevets N, Xin C, Pastukhov O, Pfeilschifter J, Zangemeister-Wittke U. Loss of sphingosine kinase-1 in carcinoma cells increases formation of reactive oxygen species and sensitivity to doxorubicin-induced DNA damage. Br J Pharmacol. 2011;162:532–543. doi: 10.1111/j.1476-5381.2010.01053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikezoe T, Tanosaki S, Krug U, Liu B, Cohen P, Taguchi H, Koeffler HP. Insulin-like growth factor binding protein-3 antagonizes the effects of retinoids in myeloid leukemia cells. Blood. 2004;104:237–242. doi: 10.1182/blood-2003-07-2203. [DOI] [PubMed] [Google Scholar]
- Ingermann AR, Yang YF, Han J, Mikami A, Garza AE, Mohanraj L, Fan L, Idowu M, Ware JL, Kim HS, et al. Identification of a novel cell death receptor mediating IGFBP-3-induced anti-tumor effects in breast and prostate cancer. J Biol Chem. 2010;285:30233–30246. doi: 10.1074/jbc.M110.122226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iosef C, Gkourasas T, Jia CY, Li SS, Han VK. A functional nuclear localization signal in insulin-like growth factor binding protein-6 mediates its nuclear import. Endocrinology. 2008;149:1214–1226. doi: 10.1210/en.2007-0959. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Lee KW, Swerdloff R, Hwang D, Cobb LJ, Sinha Hikim A, Lue YH, Cohen P, Wang C. Interaction of insulin-like growth factor-binding protein-3 and BAX in mitochondria promotes male germ cell apoptosis. J Biol Chem. 2010;285:1726–1732. doi: 10.1074/jbc.M109.046847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsaros D, Yu H, Levesque MA, Danese S, Genta F, Richiardi G, Fracchioli S, Khosravi MJ, Diamandi A, Gordini G, et al. IGFBP-3 in epithelial ovarian carcinoma and its association with clinico-pathological features and patient survival. Eur J Cancer. 2001;37:478–485. doi: 10.1016/S0959-8049(00)00423-8. [DOI] [PubMed] [Google Scholar]
- Kettunen E, Anttila S, Seppanen JK, Karjalainen A, Edgren H, Lindstrom I, Salovaara R, Nissen AM, Salo J, Mattson K, et al. Differentially expressed genes in nonsmall cell lung cancer: expression profiling of cancer-related genes in squamous cell lung cancer. Cancer Genet Cytogenet. 2004;149:98–106. doi: 10.1016/S0165-4608(03)00300-5. [DOI] [PubMed] [Google Scholar]
- Kielczewski JL, Li Calzi S, Shaw LC, Cai J, Qi X, Ruan Q, Wu L, Liu L, Hu P, Chan-Ling T, et al. Free insulin-like growth factor binding protein-3 (IGFBP-3) reduces retinal vascular permeability in association with a reduction of acid sphingomyelinase (ASMase) Invest Ophthalmol Vis Sci. 2011;52:8278–8286. doi: 10.1167/iovs.11-8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Ingermann AR, Tsubaki J, Twigg SM, Walker GEOY. Insulin-like growth factor-binding protein 3 induces caspase-dependent apoptosis through a death receptor-mediated pathway in MCF-7 human breast cancer cells. Cancer Res. 2004;64:2229–2237. doi: 10.1158/0008-5472.CAN-03-1675. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386(761):763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- Kriegs M, Kasten-Pisula U, Rieckmann T, Holst K, Saker J, Dahm-Daphi J, Dikomey E. The epidermal growth factor receptor modulates DNA double-strand break repair by regulating non-homologous end-joining. DNA Repair (Amst) 2010;9:889–897. doi: 10.1016/j.dnarep.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Kuemmerle JF, Murthy KS, Bowers JG. IGFBP-3 activates TGF-beta receptors and directly inhibits growth in human intestinal smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G795–802. doi: 10.1152/ajpgi.00009.2004. [DOI] [PubMed] [Google Scholar]
- Lapierre LR, Hansen M. Lessons from C. elegans: signaling pathways for longevity. Trends Endocrinol Metab. 2012;23:637–644. doi: 10.1016/j.tem.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal SM, Liu Q, Huang SS, Huang JS. The type V transforming growth factor beta receptor is the putative insulin-like growth factor-binding protein 3 receptor. J Biol Chem. 1997;272:20572–20576. doi: 10.1074/jbc.272.33.20572. [DOI] [PubMed] [Google Scholar]
- Leal SM, Huang SS, Huang JS. Interactions of high affinity insulin-like growth factor-binding proteins with the type V transforming growth factor-beta receptor in mink lung epithelial cells. J Biol Chem. 1999;274:6711–6717. doi: 10.1074/jbc.274.10.6711. [DOI] [PubMed] [Google Scholar]
- Lee DY, Yi HK, Hwang PHOY. Enhanced expression of insulin-like growth factor binding protein-3 sensitizes the growth inhibitory effect of anticancer drugs in gastric cancer cells. Biochem Biophys Res Commun. 2002;294:480–486. doi: 10.1016/S0006-291X(02)00491-6. [DOI] [PubMed] [Google Scholar]
- Lee KW, Liu B, Ma L, Li H, Bang P, Koeffler HP, Cohen P. Cellular internalization of insulin-like growth factor binding protein-3: distinct endocytic pathways facilitate re-uptake and nuclear localization. J Biol Chem. 2004;279:469–476. doi: 10.1074/jbc.M307316200. [DOI] [PubMed] [Google Scholar]
- Lee KW, Ma L, Yan X, Liu B, Zhang XK, Cohen P. Rapid apoptosis induction by IGFBP-3 involves an insulin-like growth factor-independent nucleomitochondrial translocation of RXRalpha/Nur77. J Biol Chem. 2005;280:16942–16948. doi: 10.1074/jbc.M412757200. [DOI] [PubMed] [Google Scholar]
- Lee SH, Takahashi M, Honke K, Miyoshi E, Osumi D, Sakiyama H, Ekuni A, Wang X, Inoue S, Gu J, et al. Loss of core fucosylation of low-density lipoprotein receptor-related protein-1 impairs its function, leading to the upregulation of serum levels of insulin-like growth factor-binding protein 3 in Fut8-/- mice. J Biochem. 2006;139:391–398. doi: 10.1093/jb/mvj039. [DOI] [PubMed] [Google Scholar]
- Lee KW, Cobb LJ, Paharkova-Vatchkova V, Liu B, Milbrandt J, Cohen P. Contribution of the orphan nuclear receptor Nur77 to the apoptotic action of IGFBP-3. Carcinogenesis. 2007;28:1653–1658. doi: 10.1093/carcin/bgm088. [DOI] [PubMed] [Google Scholar]
- Lee YC, Jogie-Brahim S, Lee DY, Han J, Harada A, Murphy LJOY. Insulin-like growth factor-binding protein-3 (IGFBP-3) blocks the effects of asthma by negatively regulating NF-kappaB signaling through IGFBP-3R-mediated activation of caspases. J Biol Chem. 2011;286:17898–17909. doi: 10.1074/jbc.M111.231035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz BJ, Agostini-Dreyer A, Jetzt AE, Krumm CS, Cohick WS. IGF binding protein-3 mediates stress-induced apoptosis in non-transformed mammary epithelial cells. J Cell Physiol. 2013;228:734–742. doi: 10.1002/jcp.24220. [DOI] [PubMed] [Google Scholar]
- Li C, Harada A, Oh Y. IGFBP-3 sensitizes antiestrogen-resistant breast cancer cells through interaction with GRP78. Cancer Lett. 2012;325:200–206. doi: 10.1016/j.canlet.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008;15:1460–1471. doi: 10.1038/cdd.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MZ, Marzec KA, Martin JL Baxter RC. The role of insulin-like growth factor binding protein-3 in the breast cancer cell response to DNA-damaging agents. Oncogene. 2012 doi: 10.1038/onc.2012.538. [DOI] [PubMed] [Google Scholar]
- Liu B, Lee HY, Weinzimer SA, Powell DR, Clifford JL, Kurie JM, Cohen P. Direct functional interactions between insulin-like growth factor-binding protein-3 and retinoid X receptor-alpha regulate transcriptional signaling and apoptosis. J Biol Chem. 2000;275:33607–33613. doi: 10.1074/jbc.M002547200. [DOI] [PubMed] [Google Scholar]
- Liu Y, Dai B, Xu C, Fu L, Hua Z, Mei C. Rosiglitazone inhibits transforming growth factor-beta1 mediated fibrogenesis in ADPKD cyst-lining epithelial cells. PLoS One. 2011;6:e28915. doi: 10.1371/journal.pone.0028915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32:805–818. doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CI, Martin C, Ma Z, Hafiane A, Dai M, Lebrun JJ, Kiss RS. Engulfment protein GULP is regulator of transforming growth factor-beta response in ovarian cells. J Biol Chem. 2012;287:20636–20651. doi: 10.1074/jbc.M111.314997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JL, Baxter RC. Oncogenic ras causes resistance to the growth inhibitor insulin-like growth factor binding protein-3 (IGFBP-3) in breast cancer cells. J Biol Chem. 1999;274:16407–16411. doi: 10.1074/jbc.274.23.16407. [DOI] [PubMed] [Google Scholar]
- Martin JL, Baxter RC. Signalling pathways of insulin-like growth factors (IGFs) and IGF binding protein-3. Growth Factors. 2011;29:235–244. doi: 10.3109/08977194.2011.614237. [DOI] [PubMed] [Google Scholar]
- Martin JL, Ballesteros M, Baxter RC. Insulin-like growth factor-I (IGF-I) and transforming growth factor-beta 1 release IGF-binding protein-3 from human fibroblasts by different mechanisms. Endocrinology. 1992;131:1703–1710. doi: 10.1210/en.131.4.1703. [DOI] [PubMed] [Google Scholar]
- Martin JL, Weenink SM, Baxter RC. Insulin-like growth factor-binding protein-3 potentiates epidermal growth factor action in MCF-10A mammary epithelial cells. Involvement of p44/42 and p38 mitogen-activated protein kinases. J Biol Chem. 2003;278:2969–2976. doi: 10.1074/jbc.M210739200. [DOI] [PubMed] [Google Scholar]
- Martin JL, Lin MZ, McGowan EM, Baxter RC. Potentiation of growth factor signaling by insulin-like growth factor-binding protein-3 in breast epithelial cells requires sphingosine kinase activity. J Biol Chem. 2009;284:25542–25552. doi: 10.1074/jbc.M109.007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh J, Dennison G, Holly JM, Jarrett C, Frankow A, Foulstone EJ, Winters ZE, Perks CM. IGFBP-3 can either inhibit or enhance EGF-mediated growth of breast epithelial cells dependent upon the presence of fibronectin. J Biol Chem. 2010;285:38788–38800. doi: 10.1074/jbc.M110.177311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta VK. Radiotherapy and erlotinib combined: review of the preclinical and clinical evidence. Front Oncol. 2012;2:31. doi: 10.3389/fonc.2012.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micutkova L, Hermann M, Offterdinger M, Hess MW, Matscheski A, Pircher H, Muck C, Ebner HL, Laich A, Ferrando-May E, et al. Analysis of the cellular uptake and nuclear delivery of insulin-like growth factor binding protein-3 in human osteosarcoma cells. Int J Cancer. 2012;130:1544–1557. doi: 10.1002/ijc.26149. [DOI] [PubMed] [Google Scholar]
- Moon JH, Kim HJ, Kim HM, Yang AH, Lee BW, Kang ES, Lee HC, Cha BS. Upregulation of hepatic LRP1 by rosiglitazone: a possible novel mechanism of the beneficial effect of thiazolidinediones on atherogenic dyslipidemia. J Mol Endocrinol. 2012;49:165–174. doi: 10.1530/JME-12-0119. [DOI] [PubMed] [Google Scholar]
- Oh Y, Muller HL, Pham H, Rosenfeld RG. Demonstration of receptors for insulin-like growth factor binding protein-3 on Hs578T human breast cancer cells. J Biol Chem. 1993;268:26045–26048. [PubMed] [Google Scholar]
- Papadimitrakopoulou VA, Brown EN, Liu DD, El-Naggar AK, Jack Lee J, Hong WK, Lee HY. The prognostic role of loss of insulin-like growth factor-binding protein-3 expression in head and neck carcinogenesis. Cancer Lett. 2006;239:136–143. doi: 10.1016/j.canlet.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Park HH. Structural analyses of death domains and their interactions. Apoptosis. 2011;16:209–220. doi: 10.1007/s10495-010-0571-z. [DOI] [PubMed] [Google Scholar]
- Payet LD, Firth SM, Baxter RC. The role of the acid-labile subunit in regulating insulin-like growth factor transport across human umbilical vein endothelial cell monolayers. J Clin Endocrinol Metab. 2004;89:2382–2389. doi: 10.1210/jc.2003-031880. [DOI] [PubMed] [Google Scholar]
- Perks CM, Burrows C, Holly JM. Intrinsic, Pro-Apoptotic Effects of IGFBP-3 on Breast Cancer Cells are Reversible: Involvement of PKA, Rho, and Ceramide. Front Endocrinol (Lausanne) 2011;2:13. doi: 10.3389/fendo.2011.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajah R, Valentinis B, Cohen P. Insulin-like growth factor (IGF)-binding protein-3 induces apoptosis and mediates the effects of transforming growth factor-beta1 on programmed cell death through a p53- and IGF-independent mechanism. J Biol Chem. 1997;272:12181–12188. doi: 10.1074/jbc.272.18.12181. [DOI] [PubMed] [Google Scholar]
- Rajaram S, Baylink DJ, Mohan S. Insulin-like growth factor-binding proteins in serum and other biological fluids: regulation and functions. Endocr Rev. 1997;18:801–831. doi: 10.1210/er.18.6.801. [DOI] [PubMed] [Google Scholar]
- Ren Z, Shin A, Cai Q, Shu XO, Gao YT, Zheng W. IGFBP3 mRNA expression in benign and malignant breast tumors. Breast Cancer Res. 2007;9:R2. doi: 10.1186/bcr1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricort JM, Binoux M. Insulin-like growth factor-binding protein-3 activates a phosphotyrosine phosphatase. Effects on the insulin-like growth factor signaling pathway. J Biol Chem. 2002;277:19448–19454. doi: 10.1074/jbc.M200439200. [DOI] [PubMed] [Google Scholar]
- Rocha RL, Hilsenbeck SG, Jackson JG, Lee AV, Figueroa JA, Yee D. Correlation of insulin-like growth factor-binding protein-3 messenger RNA with protein expression in primary breast cancer tissues: detection of higher levels in tumors with poor prognostic features. J Natl Cancer Inst. 1996;88:601–606. doi: 10.1093/jnci/88.9.601. [DOI] [PubMed] [Google Scholar]
- Rocha RL, Hilsenbeck SG, Jackson JG, VanDenBerg CL, Weng C, Lee AV, Yee D. Insulin-like growth factor binding protein-3 and insulin receptor substrate-1 in breast cancer: correlation with clinical parameters and disease-free survival. Clin Cancer Res. 1997;3:103–109. [PubMed] [Google Scholar]
- Savkur RS, Burris TP. The coactivator LXXLL nuclear receptor recognition motif. J Pept Res. 2004;63:207–212. doi: 10.1111/j.1399-3011.2004.00126.x. [DOI] [PubMed] [Google Scholar]
- Schedlich LJ, Young TF, Firth SM, Baxter RC. Insulin-like growth factor-binding protein (IGFBP)-3 and IGFBP-5 share a common nuclear transport pathway in T47D human breast carcinoma cells. J Biol Chem. 1998;273:18347–18352. doi: 10.1074/jbc.273.29.18347. [DOI] [PubMed] [Google Scholar]
- Schedlich LS, Le Page SL, Firth SM, Briggs LJ, Jans DA, Baxter RC. Nuclear import of insulin-like growth factor binding protein-3 (IGFBP-3) and IGFBP-5 is mediated by the importin β subunit. J Biol Chem. 2000;275:23462–23470. doi: 10.1074/jbc.M002208200. [DOI] [PubMed] [Google Scholar]
- Schedlich LJ, Nilsen T, John AP, Jans DA, Baxter RC. Phosphorylation of insulin-like growth factor binding protein-3 by deoxyribonucleic acid-dependent protein kinase reduces ligand binding and enhances nuclear accumulation. Endocrinology. 2003;144:1984–1993. doi: 10.1210/en.2002-220798. [DOI] [PubMed] [Google Scholar]
- Schedlich LJ, O’Han MK, Leong GM, Baxter RC. Insulin-like growth factor binding protein-3 prevents retinoid receptor heterodimerization: implications for retinoic acid-sensitivity in human breast cancer cells. Biochem Biophys Res Commun. 2004;314:83–88. doi: 10.1016/j.bbrc.2003.12.049. [DOI] [PubMed] [Google Scholar]
- Schedlich LJ, Graham LD, O’Han MK, Muthukaruppan A, Yan X, Firth SM, Baxter RC. Molecular basis of the interaction between IGFBP-3 and retinoid X receptor: role in modulation of RAR-signaling. Arch Biochem Biophys. 2007;465:359–369. doi: 10.1016/j.abb.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Schedlich LJ, Muthukaruppan A, O’Han MK, Baxter RC. Insulin-like growth factor binding protein-5 interacts with the vitamin D receptor and modulates the vitamin D response in osteoblasts. Mol Endocrinol. 2007;21:2378–2390. doi: 10.1210/me.2006-0558. [DOI] [PubMed] [Google Scholar]
- Sheen-Chen SM, Zhang H, Huang CC, Tang RP. Insulin-like growth factor-binding protein-3 in breast cancer: analysis with tissue microarray. Anticancer Res. 2009;29:1131–1135. [PubMed] [Google Scholar]
- Siddle K. Molecular basis of signaling specificity of insulin and IGF receptors: neglected corners and recent advances. Front Endocrinol (Lausanne) 2012;3:34. doi: 10.3389/fendo.2012.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Charkowicz D, Mascarenhas D. Insulin-like growth factor-independent effects mediated by a C-terminal metal-binding domain of insulin-like growth factor binding protein-3. J Biol Chem. 2004;279:477–487. doi: 10.1074/jbc.M307322200. [DOI] [PubMed] [Google Scholar]
- Twigg SM, Kiefer MC, Zapf J, Baxter RC. Insulin-like growth factor-binding protein 5 complexes with the acid-labile subunit: Role of the carboxyl-terminal domain. J Biol Chem. 1998;273:28791–28798. doi: 10.1074/jbc.273.44.28791. [DOI] [PubMed] [Google Scholar]
- van der Veeken J, Oliveira S, Schiffelers RM, Storm G, van Bergen En Henegouwen PM, Roovers RC. Crosstalk between epidermal growth factor receptor- and insulin-like growth factor-1 receptor signaling: implications for cancer therapy. Curr Cancer Drug Targets. 2009;9:748–760. doi: 10.2174/156800909789271495. [DOI] [PubMed] [Google Scholar]
- Vestey SB, Perks CM, Sen C, Calder CJ, Holly JM, Winters ZE. Immunohistochemical expression of insulin-like growth factor binding protein-3 in invasive breast cancers and ductal carcinoma in situ: implications for clinicopathology and patient outcome. Breast Cancer Res. 2005;7:R119–129. doi: 10.1186/bcr963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9:162–176. doi: 10.1038/nrm2335. [DOI] [PubMed] [Google Scholar]
- Xi Y, Nakajima G, Hamil T, Fodstad O, Riker AJJ. Association of insulin-like growth factor binding protein-3 expression with melanoma progression. Mol Cancer Ther. 2006;5:3078–3084. doi: 10.1158/1535-7163.MCT-06-0424. [DOI] [PubMed] [Google Scholar]
- Xue A, Scarlett CJ, Jackson CJ, Allen BJ, Smith RC. Prognostic significance of growth factors and the urokinase-type plasminogen activator system in pancreatic ductal adenocarcinoma. Pancreas. 2008;36:160–167. doi: 10.1097/MPA.0b013e31815750f0. [DOI] [PubMed] [Google Scholar]
- Yakar S, Rosen CJ, Bouxsein ML, Sun H, Mejia W, Kawashima Y, Wu Y, Emerton K, Williams V, Jepsen K, et al. Serum complexes of insulin-like growth factor-1 modulate skeletal integrity and carbohydrate metabolism. FASEB J. 2009;23:709–719. doi: 10.1096/fj.08-118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Forbes BE, McNeil KA, Baxter RC, Firth SM. Role of N- and C-terminal residues of insulin-like growth factor (IGF)-binding protein-3 in regulating IGF complex formation and receptor activation. J Biol Chem. 2004;279:53232–53240. doi: 10.1074/jbc.M409345200. [DOI] [PubMed] [Google Scholar]
- Young MM, Kester M, Wang HG. Sphingolipids: regulators of crosstalk between apoptosis and autophagy. J Lipid Res. 2013;54:5–19. doi: 10.1194/jlr.R031278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Links PH, Ngsee JK, Tran K, Cui Z, Ko KW, Yao Z. Localization of low density lipoprotein receptor-related protein 1 to caveolae in 3T3-L1 adipocytes in response to insulin treatment. J Biol Chem. 2004;279:2221–2230. doi: 10.1074/jbc.M310679200. [DOI] [PubMed] [Google Scholar]