

Abstract
To determine whether DNA methylation (DNA-M) of the leptin receptor genotype (LEPR/LEPROT) links gestational smoking and leptin serum levels and BMI later in life, we focused on female offspring, 18 years of age, from the Isle of Wight Birth Cohort (IOWBC). Leptin binds to the leptin receptor encoded by the LEPR/LEPROT genotype. Using general linear models, we tested a two-stage model. First, we investigated whether single nucleotide polymorphisms (SNPs) acting as methylation quantitative trait loci (methQTLs) depending on gestational smoking were related to differentially methylated cytosine-phosphate-guanine (CpG) sites. In stage 2, we tested whether the selected CpG sites, in interaction with other SNPs (modifiable genetic variants, modGV), are associated with serum leptin and BMI (stage 2). Children from the IOWBC were followed from birth to age 18. Information on gestational smoking was gathered upon delivery. SNPs tagging LEPR and LEPROT genes were genotyped. Data on LEPR/LEPROT DNA-M and leptin were obtained from blood samples drawn at age 18; to determine BMI, height and weight were ascertained. Blood samples were provided by 238 girls. Of the 21 CpG sites, interactions between gestational smoking and SNPs were detected for 16 CpGs. Methylation of seven of the 16 CpGs were, in interaction with modGVs, associated with leptin levels at age 18 years. Two CpGs survived a multiple testing penalty and were also associated with BMI. This two-stage model may explain why maternal smoking has a long-term effect on leptin levels and BMI in girls at age 18 years.
Keywords: LEPR, LEPROT, leptin, CpG sites, in utero smoking exposure, rs12059300, BMI
Introduction
Leptin, a 16 kDa pleiotropic cytokine, is associated with obesity via appetite and energy expenditure regulation [1] and with wheezing, asthma, and allergic responses [2-5]. Associations with the latter outcomes motivated us to better understand which genetic, epigenetic, and environmental factors regulate leptin levels in the body. Previous studies have shown that a single nucleotide polymorphism (SNP), 2548 G>A (rs7799039), on the leptin (LEP) gene was associated with increased plasma and serum leptin levels in the body [6-10]. Given that leptin binds to the leptin receptor (which is encoded by the leptin receptor gene, LEPR) [11,12], it is important to also consider the role of the LEPR gene on circulating leptin levels. Studies in animals (cattle and pig) have detected associations between LEPR SNPs and serum leptin concentrations [13-17]. Previously the Q223R LEPR SNP (rs1137101) has been shown to be associated with increased leptin levels in Pacific Islanders [18], however two other studies observed no association between genetic variations of the LEPR gene and serum leptin levels [19,20]. Given these conflicting reports, there is a need for further investigation into the role that genetic variants may play in serum leptin levels. We focused on the LEPR and the leptin receptor overlapping transcript (LEPROT) genes.
Furthermore, there is a lack of understanding of the role of environmental factors that affect the association between genetic polymorphisms and leptin levels. In animal studies where rats were exposed to cigarette smoke in an enclosed space, a decrease in serum leptin levels were observed as compared to rats who were not exposed to cigarette smoke [21,22]. In humans, obese Japanese men who smoked were observed to have lower serum leptin levels than obese subjects who did not smoke [23].
Leptin is known to play a role in the control of the body’s fat stores [1] and serum leptin levels in humans have been associated with body mass index (BMI) [24,25]. Studies also support a relationship between maternal smoking during pregnancy and obesity in the offspring (both during infancy and later in life) [26-31]. These findings motivated us to look at whether serum leptin levels are associated with maternal smoking during pregnancy.
DNA methylation (DNA-M) can serve as a footprint for past environmental exposures [32,33], which, in conjunction with genetic variation, may influence leptin levels. Specifically, maternal smoking during pregnancy has been associated with differential DNA-M in epigenome-wide studies in Norwegian offspring [34]. Furthermore, it has been suggested that genetic variants, referred to as methylation quantitative trait loci (methQTLs), are considered to alter methylation levels [6,35-40].
Using data from a subsample of female offspring from the Isle of Wight Birth Cohort study, we tested a two-stage model explaining the combined influence of genetic variants and DNA-M of the LEPR/LEPROT gene and maternal smoking during pregnancy, on leptin levels at age 18 in girls [41]. In Stage 1, we tested, for the first time, whether maternal smoking during pregnancy interacts with LEPR/LEPROT SNPs to change the DNA-M status of specific CpG sites in the LEPR/LEPROT gene. Following this screening/filtering stage to identify LEPR/LEPROT CpG sites modified by maternal smoking, these were then tested for association with leptin and BMI. The possibility that altered DNA methylation could then lead to differential activity (effects on promoter or splicing [42,43]) of the gene led us to consider modifiable genetic variants (modGVs; e.g. SNPs whose effects on phenotype is modified by DNA methylation). In Stage 2, we therefore tested whether those same CpG sites on the LEPR/LEPROT, in interactions with modGVs in this locus, were associated with offspring serum leptin concentrations at age 18 years. To corroborate the leptin findings, we also tested the stage 2 model using offspring BMI at age 18 as the outcome.
Material and methods
Study population and characteristics
A whole population birth cohort was established on the Isle of Wight, UK, in 1989 to prospectively study the natural history of allergies from birth to 18 years of age. Ethics approvals were obtained from the Isle of Wight Local Research Ethics Committee (now the National Research Ethics Service Committee South Central – Southampton B). Of the 1,536 children born between January 1, 1989, and February 28, 1990, written informed consent was obtained from parents to enroll 1,456 newborns. Children were followed up at the ages of 1 (n=1,167), 2 (n=1,174), 4 (n=1,218), 10 (n=1,373), and 18 years (n=1,313). The IOW birth cohort has been described in detail elsewhere [44]. Detailed interviews and examinationswere completed for each child at each follow-up. When a visit was not possible, a telephone questionnaire was completed or a postal questionnaire was sent. Information on maternal smoking during pregnancy was obtained from mothers at the time recruitment (birth).
Leptin concentration and body mass index measurements
Leptin concentrations were obtained from blood samples collected at age 18 years. Aliquots of blood serum that were isolated from the blood samples were used in enzyme-linked immunosorbent assays from Biokit, S.A. (Barcelona, Spain). The assays were conducted according to the manufacturer’s kit instructions. Each sample, including standards and the blank, was assayed in duplicate. As part of the repeated follow ups, the original questionnaire-based information were updated and weight and height of the child were measured at age 18 years. Body mass index (BMI) was measured using the following formula: weight (kg)/height (m)^2.
LEPR/LEPROT genotyping and DNA methylation analysis
SNPs (n=21) that tagged the LEPR and neighboring LEPROT genes were identified using Tagger implemented in Haploview. We included the leptin receptor overlapping transcript (LEPROT) because it shares the same promoter and the first two exons as the LEPR gene. DNA was extracted from blood or saliva samples from cohort subjects (n=1,211). DNA samples were interrogated using Golden Gate Genotyping Assays (Illumina Inc, SanDiego, CA) on the Bead Xpress Veracode platform (Illumina, Inc, SanDiego, CA) per Illumina’s protocol [45,46]. In brief, samples were fragmented and hybridized to the pool of allele-specific primer sets. Following an extension/ligation reaction the samples were then hybridized to the Veracode bead pool and processed on the Bead Xpress reader. Data were analyzed using the genotyping module of the GenomeStudio Software package (Illumina, Inc, SanDiego, CA). DNA from each subject plus 37 replicate samples were analyzed. The quality threshold for allele determination was set at a GenCall score 0.25 (scores #0.25 were ‘‘no calls’’) with 98.3% retained for further analysis. Analysis of each locus included reclustering of genotyping data using our project data to define genotype cluster positions with additional manual reclustering to maximize both cluster separation and the 50th percentile of the distribution of the GenCall scores across all genotypes (50% GC score).
For measuring methylation levels, DNA was extracted from whole blood collected at age 18 years from 245 randomly selected female offspring [47]. One microgram of DNA was bisulfite-treated for cytosine to thymine conversion using the EZ 96-DNA methylation kit (Zymo Research, CA, USA), following the manufacturer’s standard protocol. Genome-wide DNA-M was assessed using the Illumina Infinium Human Methylation 450 Bead Chip (Illumina, Inc., CA, USA), which interrogates>484,000 CpG sites associated with approximately 24,000 genes. Arrays were processed using a standard protocol as described elsewhere [48], with multiple identical control samples assigned to each bisulphite conversion batch to assess assay variability and samples randomly distributed on microarrays to control against batch effects. The Bead Chips were scanned using a Bead Station, and the methylation level (beta value) calculated for each queried CpG locus using the Methylation Module of Bead Studio software.
Statistical analysis
To identify haplotype blocks, linkage disequilibrium analysis was performed on fourteen LEPR SNPs and seven LEPROT SNPs with Haploview 4.2 [49], using the Gabriel et al. method [50]. We then selected one SNP from each block that best presented the inherited haplotype block for further statistical analyses. After cleaning the DNA-M data, beta (β) values were presented as the proportion of methylated (M) over methylated (M) and unmethylated (U) sites (β=M/[c+M+U], with c being constant to prevent dividing by zero) were used to estimate the effect of DNA-M [51]. The methylation levels of thirty-one CpG sites spanning the LEPR and LEPROT gene regions were analyzed in this study.
To identify methQTLs, modeling was performed by using LEPR and LEPROT SNPs and their interaction with in utero smoking exposure to predict specific DNA-M (stage 1). Each CpG site was modeled against 10 SNPs (one from each haplotype block and the five SNPs that were not in any blocks, Figure 1), with each SNP interacting with in utero smoking exposure. The most parsimonious model was determined via backward elimination using the 10% rule, first by removing interaction terms followed by individual SNPs. This process filters those CpG sites that were statistically significantly affected by the combined effect of intrauterine smoke exposure and SNPs identified as methQTLs (1st filtering process). Bonferonni adjustment for multiple testing was applied to the interaction terms of genetic variants with smoking, the main focus of stage 1. In the second step of the two-stage model, the selected CpG sites were then tested on whether they modified the association that other LEPR/LEPROT SNPs had with leptin serum concentrations at age 18 (2nd filtering process). In this step, our focus was again on the interaction effects and Bonferroni adjustment was employed to correct for multiple testing among the tests for interaction effects between CpG sites and genetic variants. For both statistical analyses, the GLM procedure was used in SAS 9.2 (SAS, Gary, NC, USA). The knowledge that BMI plays a role in serum leptin levels [7,52] allowed us to corroborate our leptin findings by repeating the stage 2 analysis with two CpG sites (cg11807188 and cg03050981) that survived the multiple testing penalty by testing offspring BMI at age 18 as an outcome.
Figure 1.
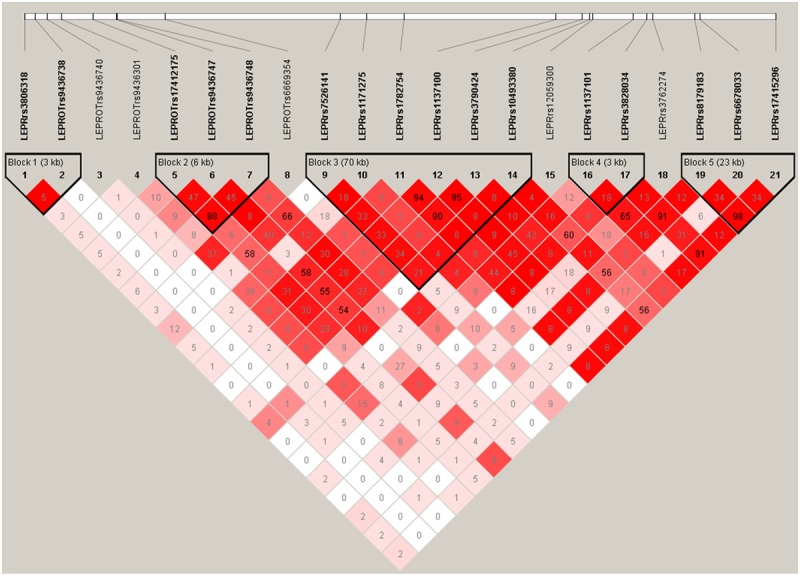
Linkage disequilibrium of LEPR and LEPR single nucleotide polymorphisms.
Results
Population characteristics
There was no statistically significant difference between female offspring among the whole cohort and the 245 who were randomly selected for DNA methylation analysis (in terms of gestational smoke exposure, leptin concentrations, and allele proportions in LEPR and LEPROT genes, Table 1). Of the 21 SNPs that were analyzed, one LEPR SNP in the 5’UTR region preceded the overlapping LEPROT gene segment, which contained three LEPROT SNPs in the intron region, followed by four LEPROT SNPs in the 3’UTR region (Table 2). The remaining LEPR SNPs spanned the 5’UTR (located downstream of the LEPROT SNPs), intron, coding, and 3’UTR regions. In six SNPs (rs17412175, rs9436743, rs7526141, rs1137101, rs3762274, and rs6678033) the heterozygous genotype had the greatest frequency. Allele and genotype frequencies were similar to those previously reported for the Caucasian population and no deviation from Hardy-Weinberg equilibrium was observed. The haplotype analysis revealed 5 blocks and 5 SNPs that did not have strong linkage disequilibrium with other SNPs (were not in a block with other SNPs, Figure 1).
Table 1.
Population characteristics of offspring
| Sub population with methylation data | Whole cohort population | P-Value | ||
|---|---|---|---|---|
| SNP | N (%) | N (%) | ||
|
| ||||
| rs3806318 | AA | 116 (50.7) | 294 (51.1) | 0.77 |
| AG | 92 (40.2) | 234 (40.7) | ||
| GG | 21 (9.2) | 47 (8.2) | ||
| rs9436738 | AA | 1 (0.4) | 5 (0.9) | 0.95 |
| AG | 54 (23.1) | 130 (22.4) | ||
| GG | 179 (76.5) | 445 (76.7) | ||
| rs9436740 | AA | 15 (6.5) | 50 (8.7) | 0.38 |
| AT | 89 (38.7) | 224 (38.8) | ||
| TT | 126 (54.8) | 303 (52.5) | ||
| rs9436301 | CT | 140 (59.6) | 342 (57.2) | 0.85 |
| TT | 86 (36.6) | 216 (37.5) | ||
| CC | 9 (3.8) | 23 (5.4) | ||
| rs17412175 | AA | 53 (22.9) | 132 (21.1) | 0.77 |
| AT | 114 (49.4) | 276 (48.7) | ||
| TT | 64 (27.7) | 169 (30.2) | ||
| rs9436747 | AA | 32 (13.7) | 83 (15.1) | 0.67 |
| AG | 97 (41.6) | 247 (44.1) | ||
| GG | 104 (44.6) | 248 (40.8) | ||
| rs9436748 | AA | 120 (20.9) | 120 (18.7) | 0.79 |
| AC | 269 (47.0) | 269 (48.6) | ||
| CC | 184 (32.1) | 184 (32.7) | ||
| rs6669354 | AA | 412 (78.3) | 412 (78.7) | 0.72 |
| AC | 104 (19.8) | 104 (19.5) | ||
| CC | 10 (1.9) | 10 (1.9) | ||
| rs7526141 | AA | 135 (23.4) | 135 (22.2) | 0.73 |
| AG | 276 (47.8) | 276 (48.2) | ||
| GG | 166 (28.8) | 166 (29.6) | ||
| rs1171275 | AA | 11 (1.9) | 11 (2.8) | 0.96 |
| AG | 169 (29.3) | 169 (29.0) | ||
| GG | 397 (68.8) | 397 (68.2) | ||
| rs1782754 | AA | 300 (51.6) | 300 (50.8) | 0.50 |
| AG | 235 (40.5) | 235 (40.7) | ||
| GG | 46 (7.9) | 46 (8.4) | ||
| rs1137100 | AA | 296 (51.4) | 296 (51.3) | 0.34 |
| AG | 238 (41.3) | 238 (40.3) | ||
| GG | 42 (7.3) | 42 (8.4) | ||
| rs3790424 | AA | 292 (51.3) | 292 (50.7) | 0.38 |
| AG | 230 (40.4) | 230 (40.0) | ||
| GG | 47 (8.3) | 47 (9.4) | ||
| rs10493380 | AA | 390 (68.1) | 390 (69.5) | 0.60 |
| AC | 167 (29.1) | 167 (27.8) | ||
| CC | 16 (2.8) | 16 (2.7) | ||
| rs12059300 | AA | 21 (3.6) | 21 (3.2) | 0.38 0.60 |
| AG | 190 (32.6) | 190 (32.7) | ||
| GG | 372 (63.8) | 372 (64.1) | ||
| rs1137101 | AA | 169 (29.4) | 3169 (30.2) | 0.46 |
| AG | 292 (50.8) | 292 (48.5) | ||
| GG | 114 (19.8) | 114 (21.4) | ||
| rs3828034 | AA | 390 (68.1) | 390 (67.8) | 0.76 |
| AG | 163 (28.5) | 163 (28.8) | ||
| GG | 20 (3.5) | 20 (3.4) | ||
| rs3762274 | AA | 218 (38.9) | 218 (37.8) | 0.31 |
| AG | 265 (47.3) | 265 (47.2) | ||
| GG | 77 (13.8) | 77 (15.0) | ||
| rs8179183 | CC | 18 (3.1) | 18 (3.2) | 0.85 |
| CG | 162 (28.2) | 162 (28.0) | ||
| GG | 395 (68.7) | 395 (68.8) | ||
| rs6678033 | CC | 76 (13.2) | 76 (14.0) | 0.65 |
| AG | 261 (45.5) | 261 (46.8) | ||
| GG | 237 (41.3) | 237 (39.2) | ||
| rs17415296 | AA | 19 (3.3) | 19 (3.3) | 0.55 |
| AC | 166 (28.5) | 166 (28.6) | ||
| CC | 398 (68.3) | 398 (68.2) | ||
| In utero smoking exposure (Yes) | 47 (19.3) | 188 (25.3) | 0.055 | |
|
| ||||
| Variable | N (Median; 5,95%) | N (Median; 5,95%) | ||
|
| ||||
| Leptin at age 18 years (ng/ml) | 239 (13.1; 2.8, 54.6) | 265 (13.1; 2.4, 54.6) | 0.85 | |
| BMI at age 18 years (kg/m²) | 240 (22.9; 19.1, 32.9) | 499 (22.9; 18.3, 33.7) | 0.89 | |
Table 2.
Location of LEPR and LEPROT SNPs
| SNP | Alleles | Gene | Location | Coordinate |
|---|---|---|---|---|
| rs3806318 | A, G | LEPR | 5’UTR | 65885357 |
| rs9436738 | A, G | LEPROT | Intron | 65888560 |
| rs9436740 | A, T | LEPROT | Intron | 65891901 |
| rs9436301 | T, C | LEPROT | Intron | 65895927 |
| rs17412175 | A, T | LEPROT | 3’UTR | 65904886 |
| rs9436747 | A, G | LEPROT | 3’UTR | 65911607 |
| rs9436748 | A, C | LEPROT | 3’UTR | 65911672 |
| rs6669354 | A, C | LEPROT | 3’UTR | 65925349 |
| rs7526141 | A, G | LEPR | 5’UTR | 65975275 |
| rs1171275 | A, G | LEPR | 5’UTR | 65982633 |
| rs1782754 | A, G | LEPR | 5’UTR | 65993348 |
| rs1137100 | A, G | LEPR | Coding | 66036441 |
| rs3790424 | A, G | LEPR | Intron | 66044013 |
| rs10493380 | A, C | LEPR | Intron | 66046117 |
| rs12059300 | A, G | LEPR | Intron | 66047072 |
| rs1137101 | A, G | LEPR | Coding | 66058513 |
| rs3828034 | A, G | LEPR | Intron | 66062325 |
| rs3762274 | A, G | LEPR | Intron | 66064113 |
| rs8179183 | C, G | LEPR | Coding | 66075952 |
| rs6678033 | A, G | LEPR | Intron | 66077624 |
| rs17415296 | A, C | LEPR | 3’UTR | 66099013 |
The measurement of methylation of CpG sites (cg) found on the LEPR and LEPROT genes revealed that the methylation sites were either highly methylated (mean β>0.65) or lowly methylated (mean β≤0.2, Table 3). Given that the LEPROT gene shares the same promoter and first two exons of the LEPR gene, the body of the LEPROT gene is between the 5’UTR methylated segments (Table 3). Low methylation levels were observed from the transcription start site (TSS) 1500 to the beginning of the 5’UTR region (cg26876444). Higher methylation was observed from cg15466952 to cg14199090 (all in the 5’UTR region). Low methylation again was observed in the next cluster of five CpG sites (cg08234308 to cg03514351), followed by high methylation going into sites on the body and 3’UTR region of the gene (Table 3).
Table 3.
Distribution of methylation on CpG sites on the LEPR gene
| Gene | CPG site | Location | Coordinate | Mean Methylation | 5% | 95% |
|---|---|---|---|---|---|---|
| LEPROT; LEPR | cg03853587 | TSS1500 | 65885364 | 0.12 | 0.078 | 0.17 |
| LEPROT; LEPR | cg25307371 | TSS1500 | 65885547 | 0.079 | 0.05 | 0.096 |
| LEPROT; LEPR | cg10062258 | TSS1500 | 65885702 | 0.033 | 0.02 | 0.049 |
| LEPROT; LEPR | cg23055818 | TSS1500 | 65885868 | 0.077 | 0.049 | 0.11 |
| LEPROT; LEPR | cg07342512 | TSS1500 | 65886002 | 0.06 | 0.04 | 0.087 |
| LEPROT; LEPR | cg14976592 | TSS200 | 65886160 | 0.11 | 0.067 | 0.17 |
| LEPROT; LEPR | cg13202122 | TSS200 | 65886182 | 0.041 | 0.021 | 0.068 |
| LEPROT; LEPR | cg07921092 | Exon 1, 5’UTR | 65886266 | 0.026 | 0.01 | 0.044 |
| LEPROT; LEPR | cg27502791 | Exon 1, 5’UTR | 65886279 | 0.051 | 0.035 | 0.066 |
| LEPROT; LEPR | cg08610741 | Exon 1, 5’UTR | 65886304 | 0.023 | 0.01 | 0.041 |
| LEPROT; LEPR | cg13446852 | 5’UTR | 65886635 | 0.052 | 0.04 | 0.065 |
| LEPROT; LEPR | cg08922075 | 5’UTR, Body | 65886896 | 0.092 | 0.058 | 0.13 |
| LEPROT; LEPR | cg26876444 | 5’UTR | 65887413 | 0.23 | 0.16 | 0.31 |
| LEPROT; LEPR | cg15466952 | 5’UTR | 65889855 | 0.67 | 0.56 | 0.74 |
| LEPROT; LEPR | cg01933519 | 5’UTR | 65891704 | 0.89 | 0.86 | 0.91 |
| LEPR | cg03050981 | 5’UTR | 65906670 | 0.69 | 0.6 | 0.79 |
| LEPR | cg11807188 | 5’UTR | 65924844 | 0.75 | 0.69 | 0.8 |
| LEPR | cg23688719 | 5’UTR | 65935654 | 0.81 | 0.76 | 0.86 |
| LEPR | cg11228758 | 5’UTR | 65976172 | 0.81 | 0.76 | 0.85 |
| LEPR | cg26342890 | 5’UTR | 65988782 | 0.82 | 0.76 | 0.86 |
| LEPR | cg06558372 | 5’UTR | 65990448 | 0.9 | 0.88 | 0.93 |
| LEPR | cg14199090 | 5’UTR | 65990564 | 0.87 | 0.84 | 0.9 |
| LEPR | cg08234308 | 5’UTR | 65991176 | 0.067 | 0.044 | 0.095 |
| LEPR | cg00630958 | 5’UTR | 65991461 | 0.12 | 0.089 | 0.16 |
| LEPR | cg16987305 | 5’UTR | 65991596 | 0.034 | 0.016 | 0.057 |
| LEPR | cg03401738 | 5’UTR | 65991664 | 0.11 | 0.088 | 0.13 |
| LEPR | cg03514351 | 5’UTR | 65991765 | 0.15 | 0.11 | 0.18 |
| LEPR | cg16265717 | 5’UTR | 65993149 | 0.88 | 0.85 | 0.91 |
| LEPR | cg07959978 | 5’UTR | 65994091 | 0.82 | 0.77 | 0.86 |
| LEPR | cg21683619 | Body | 66070348 | 0.7 | 0.59 | 0.77 |
| LEPR | cg03607891 | Body, 3’UTR | 66099615 | 0.88 | 0.83 | 0.92 |
The CpG sites are presented in the order they appear on the LEPR and LEPROT genes from 5’ to 3’. TSS1500=1500 base pairs from the transcription start site. TSS200=200 base pairs from the transcription start site.
MethQTL analysis and DNA methylation predicting serum leptin concentrations
Of the 31 methylation sites that were modeled, 16 sites were found to be influenced by adjacent SNPs (methQTLs) in interaction with maternal smoking during pregnancy (Table 4). Nine of these methQTLs were found in the intron region of the LEPR and LEPROT genes, three were in the flanking 5’UTR region, one was in the 3’UTR region, and 4 were found in the coding region. Fourteen of the 16 CpG sites with methQTLs were located in the promoter region of the LEPR and LEPORT genes, one at the end of the promoter region and into the body and one on the body of the gene (Table 4).
Table 4.
Interaction effect of prenatal exposure to smoking with methylation quantitative trait loci for LEPR and LEPROT genes
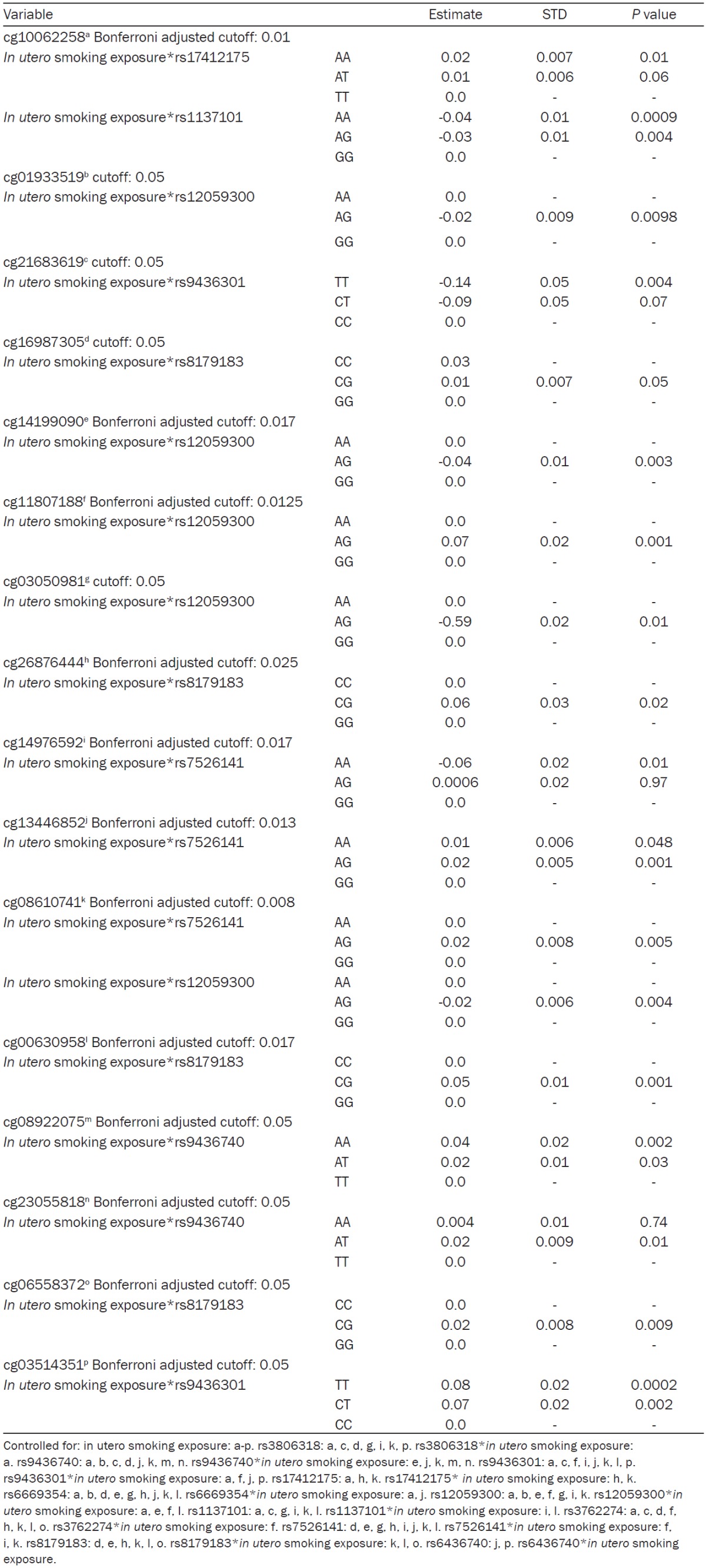
|
Among the CpG sites in the promoter region, five of them had rs12059300 (located in the intron region) as a methQTL (Table 4). In these sites, with the exception of site cg11807188, girls, who were exposed to smoking and had the heterozygous genotype of rs12059300, presented a lower proportion of methylation than those children who had the GG or AA genotype.
Two methQTLs were detected in the coding region of the genes, of which one (rs1137101) influenced methylation at the TSS1500 region of LEPR and LEPROT, which was 84,356 base pairs (bp) downstream from the CpG site (cg10062258, Table 4). This was the only methQTL in the coding region that had a negative association with DNA-M. In four other CpG sites in the promoter region, all downstream from cg10062258, children with the heterozygous genotype (AG) for the rs8179183 SNP and exposure to smoking during pregnancy had higher methylation than those with the AA genotype and mothers who did not smoke during pregnancy (Table 4).
The methQTL that was detected in the 3’UTR region (rs174121475), in interaction with in utero smoking exposure, was associated with increased DNA-M in the TSS1500 region (cg10062258, Table 4). This is the same CpG site observed to have decreased methylation with at methQTL located in the coding segment of the gene and in interaction with maternal smoking during pregnancy. Lastly, the methQTL in the flanking 5’UTR region (rs7526141) tended to be positively associated (in interaction with maternal smoking during pregnancy) with DNA-M at the 5’UTR region (Table 4). However, when considering DNA-M upstream in the TSS200 region, a negative association was observed (Table 4).
In most of the models we observed that, although the main effects of both gestational smoking and SNPs were not strongly associated with DNA-M at the promoter region, their interactions had a stronger and statistically significant influence even after adjustment for multiple testing. In the first filtering process, representing stage 1 of the 2-stage model, we identified sixteen CpG sites of 21 potential CpGs that were differentially methylated by the interaction of maternal smoking during pregnancy and the child’s LEPR/LEPROT genotype.
Addressing stage 2, we then tested these sixteen CpG sites to determine whether the smoking fingerprint changed the role of other SNPs in the LEPR and LEPROT genes on their association with leptin concentrations at age 18 years. SNPs that identified as being modified are named modifiable genetic variants (modGV). Again, modifying effects (interactions) of all tag SNPs with these 16 CpG sites were tested. The most parsimonious model yielded thirty-two tests (i.e.-thirty two SNP-CpG interactions). Applying Bonferroni adjustment for these thirty-two tests resulted in a p-value cut off point of 0.00156. We found that two of the sixteen CpG sites, in interaction with modGVs, were statistically significantly associated with serum leptin concentrations at age 18 years (Table 5). Both of these CpG sites were in the 5’UTR region of the gene and both interacted with rs12059300 (located in the intron region) to influence serum leptin levels. The CpG sites cg11807188 and cg03050981 were upstream from rs12059300 by 122,228 and 140,402 base pairs respectively. In those with the AA genotype of rs12059300, serum leptin levels were observed to decrease as proportion of methylation at cg11807188 and cg03050981 increased as compared to those with the GG genotype. Additionally, for cg03050981, those with the AG genotype showed a similar pattern as those with the AA genotype (Table 5).
Table 5.
Methylation of cg11807188 and cg03050981 associated with leptin concentrations (ng/mL) and BMI (kg/m²) at age 18 years
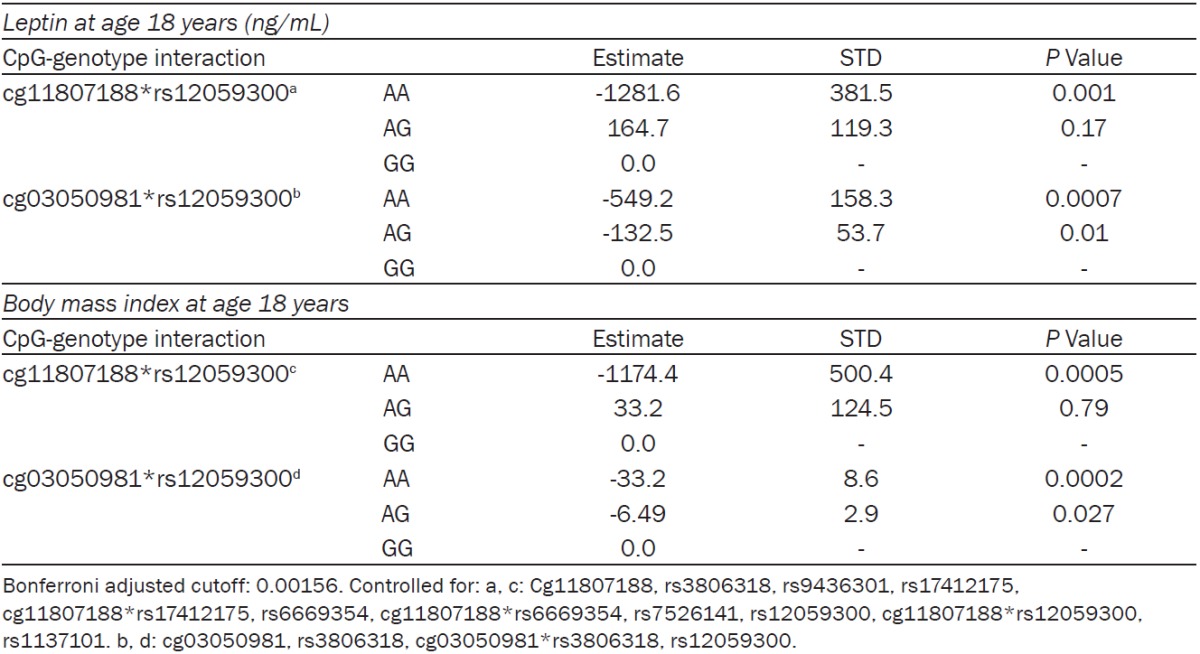
|
The serum leptin levels from those with the AA genotype of rs12059300 ranged from 2.90 ng/mL to 60.6 ng/mL; decreasing as the proportion of methylation at cg03050981 increased (Figure 2). Those with the AG genotype were observed to have a similar pattern although the difference was not as dramatic. Regarding methylation at cg11807188, those with the AA genotype had serum leptin concentrations ranging from 3.29 ng/mL to 78.1 ng/mL; the leptin levels decreased as proportion of methylation at that site increased. Leptin levels for those with the AG and GG genotype remained constant across different methylation levels (Figure 3).
Figure 2.
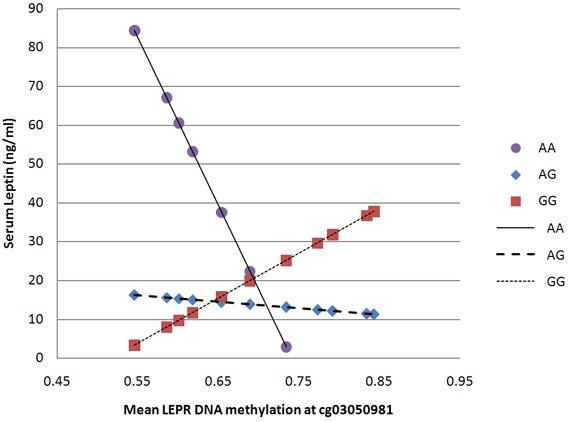
Association of methylation at cg03050981 with serum leptin concentrations across different rs12059300 genotypes.
Figure 3.
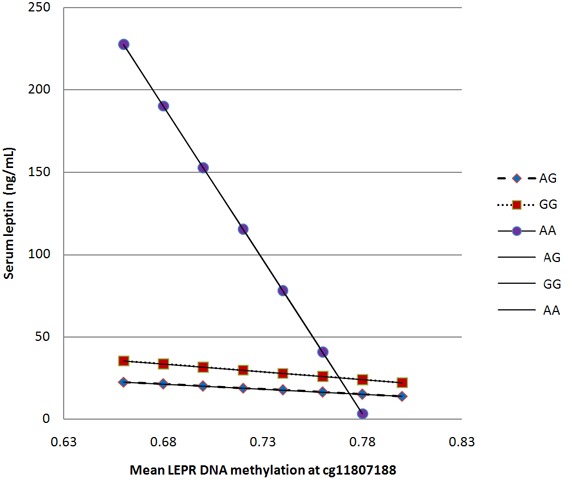
Association of methylation at cg11807188 with serum leptin concentrations across different rs12059300 genotypes.
Previous studies have suggested that body mass index (BMI) plays a role in serum leptin levels [7,51]. We therefore repeated the stage 2 analysis with the same two statistically significant CpG sites in interaction with rs12059300 predicting standardized BMI levels at age 18 years. We found the same significant associations. BMI was observed to decrease as proportion of methylation increased in those with the AA genotype as compared to those with the GG genotype (Table 5).
Discussion
We detected seven methQTLs working in interaction with in utero smoking exposure to influence DNA methylation at 16 sites on the LEPR and LEPROT genes (Table 4). Two of these same CpG sites were found to modify the effect of LEPR and LEPPROT SNPs on serum leptin concentration levels at age 18 years (Table 5). This is the first study to identify SNPs (methQTLs) on the leptin receptor gene that in interaction with in utero smoking exposure change the DNA methylation of sixteen CpG sites, which in turn modifies the association that genetic variants (SNP) had on serum leptin concentrations. This association was then corroborated with BMI as an outcome of the stage 2 analysis.
The probability for selection bias influencing our analysis is minimal since the samples from the 245 female offspring were randomly selected. Furthermore, there were no differences observed between the subset population and the female population from the whole cohort. However, the small sample size in our analysis is a potential weakness of our study. Regarding the exclusion of male offspring in the analysis, given that boys and girls in our study have significantly different leptin levels, especially following puberty (1.2 ng/mL and 13.1 ng/mL in boys and girls, respectively; data not shown), this same analysis needs to be done in the future separately in boys. A strength of our study is the use of the Infinium Human Methylation 450 array to obtain DNA methylation profiles, which has been reported to have strong reproducibility and validity [45,46]. Additionally, the SNPs that were detected to be associated with DNA-M levels in this study do not overlap with the probes on the methylation array, suggesting that our findings are actual methQTLs and not technical artifacts caused by differential probe binding.
The distance between the methylation site and the SNPs used in our analysis is sometimes greater than 10,000 bp (Tables 1 and 2). Methylation in the promoter region of the LEPR gene is influenced by SNPs either in the same region or downstream. This is plausible biologically as well as within the confines of cis-acting regulation [6,36].
We focused on the LEPR gene because it codes for the leptin receptor, which is known to bind to leptin [53,54]. Our analysis shows that two of the methylation sites that are influenced by a methQTL in interaction with a prenatal environmental exposure, are in turn associated with leptin concentrations in the body at age 18 years. This suggests a potential pathway from a prenatal smoke exposure altering DNA methylation, and the DNA methylation subsequently influencing a phenotypic outcome in adulthood. Given that the environmental factor in this case is a modifiable risk factor (maternal smoking during pregnancy), our findings support other studies that have tested intervention methods for obesity [55] and point to the development of further efforts.
Our findings fit into a two-stage model [41]: Stage 1 addresses SNPs that make the DNA at some CpG sites more or less prone to DNA methylation conditional of exposure (maternal smoking during pregnancy). Stage 2 emphasizes that these CpG sites then regulate the activity of other genetic variants of the gene, either masking it or facilitating more penetrance.
Given that leptin levels and BMI are correlated, we attempted to corroborate our findings in stage 2 using BMI at age 18 as the final outcome. The two SNP - DNA methylation interactions found for leptin were also statistically significantly associated with BMI. We know that in utero exposure to maternal smoking is associated with offspring BMI into late childhood [26-30] but the mechanism behind this relationship is not understood. Our findings linking maternal smoking during pregnancy with leptin suggest an intermediary role of leptin in providing a possible explanation. Future studies will address the leptin gene and whether we can find similar effects in boys and in the offspring of the birth cohort.
Conclusion
A two-stage model consisting of genetic variants in the LEPR/LEPROT loci, gestational smoke exposure, and DNA methylation provides an explanation of how maternal smoking during pregnancy can be linked to increased serum leptin levels and BMI at age 18 years. Results of the first stage demonstrate that DNA methylation which sets the offspring at a higher risk of increased leptin and BMI can be avoided by reducing maternal smoking during pregnancy. The second stage, explaining serum leptin concentrations and BMI, shows strong interactions between DNA methylation and genetic variants suggesting that the study of DNA methylation and genetic variants is much more powerful than the single analyses of genetic variants.
Acknowledgements
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases under Award Number R01 AI091905-01 (PI: Wilfried Karmaus) and R01 AI061471 (PI: Susan Ewart). The 10-year follow-up of this study was funded by National Asthma Campaign, UK (Grant No 364) and the 18-year follow-up by NIH/NHLBI R01 HL082925-01 (PI: S. Hasan Arshad). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors gratefully acknowledge the cooperation of the children and parents who participated in this study, and appreciate the hard work of Mrs. Sharon Matthews and the Isle of Wight research team in collecting data and Nikki Graham for technical support. We thank the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant reference 090532/Z/09/Z and MRC Hub grant G0900747 91070) for the generation of the methylation data.
References
- 1.de Luis DA, Perez Castrillon JL, Duenas A. Leptin and obesity. Minerva Med. 2009;100:229–236. [PubMed] [Google Scholar]
- 2.Guler N, Kirerleri E, Ones U, Tamay Z, Salmayenli N, Darendeliler F. Leptin: does it have any role in childhood asthma? J Allergy Clin Immunol. 2004;114:254–259. doi: 10.1016/j.jaci.2004.03.053. [DOI] [PubMed] [Google Scholar]
- 3.Bruno A, Chanez P, Chiappara G, Siena L, Giammanco S, Gjomarkaj M, Bonsignore G, Bousquet J, Vignola AM. Does leptin play a cytokine-like role within the airways of COPD patients? Eur Respir J. 2005;26:398–405. doi: 10.1183/09031936.05.00092404. [DOI] [PubMed] [Google Scholar]
- 4.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 5.La Cava A, Matarese G. The weight of leptin in immunity. Nat Rev Immunol. 2004;4:371–379. doi: 10.1038/nri1350. [DOI] [PubMed] [Google Scholar]
- 6.Zhang D, Cheng L, Badner JA, Chen C, Chen Q, Luo W, Craig DW, Redman M, Gershon ES, Liu C. Genetic control of individual differences in gene-specific methylation in human brain. Am J Hum Genet. 2010;86:411–419. doi: 10.1016/j.ajhg.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sahin DS, Tumer C, Demir C, Celik MM, Celik M, Ucar E, Gunesacar R. Association with Leptin Gene c. -2548 G>A Polymorphism, Serum Leptin Levels, and Body Mass Index in Turkish Obese Patients. Cell Biochem Biophys. 2013 Mar;6:243–7. doi: 10.1007/s12013-012-9427-1. [DOI] [PubMed] [Google Scholar]
- 8.Cieslak J, Bartz M, Stachowiak M, Skowronska B, Majewska KA, Harasymczuk J, Stankiewicz W, Fichna P, Switonski M. Effect of three common SNPs in 5’-flanking region of LEP and ADIPOQ genes on their expression in Polish obese children and adolescents. Mol Biol Rep. 2012;39:3951–3955. doi: 10.1007/s11033-011-1174-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ben Ali S, Kallel A, Ftouhi B, Sediri Y, Feki M, Slimane H, Jemaa R, Kaabachi N. Association of G-2548A LEP polymorphism with plasma leptin levels in Tunisian obese patients. Clin Biochem. 2009;42:584–588. doi: 10.1016/j.clinbiochem.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 10.Riestra P, Garcia-Anguita A, Viturro E, Schoppen S, de Oya M, Garces C. Influence of the leptin G-2548A polymorphism on leptin levels and anthropometric measurements in healthy Spanish adolescents. Ann Hum Genet. 2010;74:335–339. doi: 10.1111/j.1469-1809.2010.00586.x. [DOI] [PubMed] [Google Scholar]
- 11.Mistrik P, Moreau F, Allen JM. BiaCore analysis of leptin-leptin receptor interaction: evidence for 1:1 stoichiometry. Anal Biochem. 2004;327:271–277. doi: 10.1016/j.ab.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 12.Denver RJ, Bonett RM, Boorse GC. Evolution of leptin structure and function. Neuroendocrinology. 2011;94:21–38. doi: 10.1159/000328435. [DOI] [PubMed] [Google Scholar]
- 13.Uemoto Y, Kikuchi T, Nakano H, Sato S, Shibata T, Kadowaki H, Katoh K, Kobayashi E, Suzuki K. Effects of porcine leptin receptor gene polymorphisms on backfat thickness, fat area ratios by image analysis, and serum leptin concentrations in a Duroc purebred population. Anim Sci J. 2012;83:375–385. doi: 10.1111/j.1740-0929.2011.00963.x. [DOI] [PubMed] [Google Scholar]
- 14.Thompson DB, Norman RA, Ravussin E, Knowler WC, Bennett P, Bogardus C. Association of the GLN223ARG polymorphism in the leptin receptor gene with plasma leptin levels, insulin secretion and NIDDM in Pima Indians. Diabetologia. 1997;40:A177. [Google Scholar]
- 15.Liefers SC, Veerkamp RF, te Pas MF, Delavaud C, Chilliard Y, van der Lende T. A missense mutation in the bovine leptin receptor gene is associated with leptin concentrations during late pregnancy. Anim Genet. 2004;35:138–141. doi: 10.1111/j.1365-2052.2004.01115.x. [DOI] [PubMed] [Google Scholar]
- 16.Amills M, Villalba D, Tor M, Mercade A, Gallardo D, Cabrera B, Jimenez N, Noguera JL, Sanchez A, Estany J. Plasma leptin levels in pigs with different leptin and leptin receptor genotypes. J Anim Breed Genet. 2008;125:228–233. doi: 10.1111/j.1439-0388.2007.00715.x. [DOI] [PubMed] [Google Scholar]
- 17.Kuehn LA, Nonneman DJ, Klindt JM, Wise TH. Genetic relationships of body composition, serum leptin, and age at puberty in gilts. J Anim Sci. 2009;87:477–483. doi: 10.2527/jas.2008-0936. [DOI] [PubMed] [Google Scholar]
- 18.Furusawa T, Naka I, Yamauchi T, Natsuhara K, Kimura R, Nakazawa M, Ishida T, Inaoka T, Matsumura Y, Ataka Y, Nishida N, Tsuchiya N, Ohtsuka R, Ohashi J. The Q223R polymorphism in LEPR is associated with obesity in Pacific Islanders. Hum Genet. 2010;127:287–294. doi: 10.1007/s00439-009-0768-9. [DOI] [PubMed] [Google Scholar]
- 19.Su PH, Yang SF, Yu JS, Chen SJ, Chen JY. Study of leptin levels and gene polymorphisms in patients with central precocious puberty. Pediatr Res. 2012;71:361–367. doi: 10.1038/pr.2011.69. [DOI] [PubMed] [Google Scholar]
- 20.Kim EY, Chin HM, Park SM, Jeon HM, Chung WC, Paik CN, Jun KH. Susceptibility of gastric cancer according to leptin and leptin receptor gene polymorphisms in Korea. J Korean Surg Soc. 2012;83:7–13. doi: 10.4174/jkss.2012.83.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomoda K, Kubo K, Nishii Y, Yamamoto Y, Yoshikawa M, Kimura H. Changes of ghrelin and leptin levels in plasma by cigarette smoke in rats. J Toxicol Sci. 2012;37:131–138. doi: 10.2131/jts.37.131. [DOI] [PubMed] [Google Scholar]
- 22.Ypsilantis P, Politou M, Anagnostopoulos C, Tsigalou C, Kambouromiti G, Kortsaris A, Simopoulos C. Effects of Cigarette Smoke Exposure and Its Cessation on Body Weight, Food Intake and Circulating Leptin, and Ghrelin Levels in the Rat. Nicotine Tob Res. 2013 Jan;15:206–12. doi: 10.1093/ntr/nts113. [DOI] [PubMed] [Google Scholar]
- 23.Nagayasu S, Suzuki S, Yamashita A, Taniguchi A, Fukushima M, Nakai Y, Nin K, Watanabe N, Nagasaka S, Yabe D, Nishimura F. Smoking and adipose tissue inflammation suppress leptin expression in Japanese obese males: potential mechanism of resistance to weight loss among Japanese obese smokers. Tob Induc Dis. 2012;10:3. doi: 10.1186/1617-9625-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al Maskari MY, Alnaqdy AA. Correlation between Serum Leptin Levels, Body Mass Index and Obesity in Omanis. Sultan Qaboos Univ Med J. 2006;6:27–31. [PMC free article] [PubMed] [Google Scholar]
- 25.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 26.Birbilis M, Moschonis G, Mougios V, Manios Y; Healthy Growth Study’ group. Obesity in adolescence is associated with perinatal risk factors, parental BMI and sociodemographic characteristics. Eur J Clin Nutr. 2013;67:115–121. doi: 10.1038/ejcn.2012.176. [DOI] [PubMed] [Google Scholar]
- 27.Karaolis-Danckert N, Buyken AE, Sonntag A, Kroke A. Birth and early life influences on the timing of puberty onset: results from the DONALD (DOrtmund Nutritional and Anthropometric Longitudinally Designed) Study. Am J Clin Nutr. 2009;90:1559–1565. doi: 10.3945/ajcn.2009.28259. [DOI] [PubMed] [Google Scholar]
- 28.Abbassi V. Growth and normal puberty. Pediatrics. 1998;102:507–511. [PubMed] [Google Scholar]
- 29.Timmermans SH, Mommers M, Gubbels JS, Kremers SP, Stafleu A, Stehouwer CD, Prins MH, Penders J, Thijs C. Maternal smoking during pregnancy and childhood overweight and fat distribution: the KOALA Birth Cohort Study. Pediatr Obes. 2013 doi: 10.1111/j.2047-6310.2012.00141.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 30.Haga C, Kondo N, Suzuki K, Sato M, Ando D, Yokomichi H, Tanaka T, Yamagata Z. Developmental trajectories of body mass index among Japanese children and impact of maternal factors during pregnancy. PLoS One. 2012;7:e51896. doi: 10.1371/journal.pone.0051896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi Y, De Groh M, Morrison H. Perinatal and early childhood factors for overweight and obesity in young canadian children. Can J Public Health. 2013;104:e69–74. doi: 10.1007/BF03405658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med. 2009;180:462–467. doi: 10.1164/rccm.200901-0135OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, Huang Z, Hoyo C, Midttun O, Cupul-Uicab LA, Ueland PM, Wu MC, Nystad W, Bell DA, Peddada SD, London SJ. 450K Epigenome-Wide Scan Identifies Differential DNA Methylation in Newborns Related to Maternal Smoking during Pregnancy. Environ Health Perspect. 2012;120:1425–1431. doi: 10.1289/ehp.1205412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, Johnson R, Zielke HR, Ferrucci L, Longo DL, Cookson MR, Singleton AB. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shoemaker R, Deng J, Wang W, Zhang K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 2010;20:883–889. doi: 10.1101/gr.104695.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, Gilad Y, Pritchard JK. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011;12:R10. doi: 10.1186/gb-2011-12-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12:529–541. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E, Li K, Murty VV, Schupf N, Vilain E, Morris M, Haghighi F, Tycko B. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet. 2008;40:904–908. doi: 10.1038/ng.174. [DOI] [PubMed] [Google Scholar]
- 40.Hellman A, Chess A. Extensive sequence-influenced DNA methylation polymorphism in the human genome. Epigenetics Chromatin. 2010;3:11. doi: 10.1186/1756-8935-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karmaus W, Ziyab AH, Everson T, Holloway JW. Epigenetic mechanisms and models in the origins of asthma. Curr Opin Allergy Clin Immunol. 2013;13:63–69. doi: 10.1097/ACI.0b013e32835ad0e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malousi A, Kouidou S. DNA hypermethylation of alternatively spliced and repeat sequences in humans. Mol Genet Genomics. 2012;287:631–642. doi: 10.1007/s00438-012-0703-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oberdoerffer S. A conserved role for intragenic DNA methylation in alternative pre-mRNA splicing. Transcription. 2012;3:106–109. doi: 10.4161/trns.19816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurukulaaratchy RJ, Fenn MH, Waterhouse LM, Matthews SM, Holgate ST, Arshad SH. Characterization of wheezing phenotypes in the first 10 years of life. Clin Exp Allergy. 2003;33:573–578. doi: 10.1046/j.1365-2222.2003.01657.x. [DOI] [PubMed] [Google Scholar]
- 45.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, Fan JB, Shen R. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 46.Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011;6:692–702. doi: 10.4161/epi.6.6.16196. [DOI] [PubMed] [Google Scholar]
- 47.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bibikova M, Fan JB. GoldenGate assay for DNA methylation profiling. Methods Mol Biol. 2009;507:149–163. doi: 10.1007/978-1-59745-522-0_12. [DOI] [PubMed] [Google Scholar]
- 49.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 50.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 51.Kuan PF, Wang S, Zhou X, Chu H. A statistical framework for Illumina DNA methylation arrays. Bioinformatics. 2010;26:2849–2855. doi: 10.1093/bioinformatics/btq553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blum WF, Englaro P, Hanitsch S, Juul A, Hertel NT, Muller J, Skakkebaek NE, Heiman ML, Birkett M, Attanasio AM, Kiess W, Rascher W. Plasma leptin levels in healthy children and adolescents: dependence on body mass index, body fat mass, gender, pubertal stage, and testosterone. J Clin Endocrinol Metab. 1997;82:2904–2910. doi: 10.1210/jcem.82.9.4251. [DOI] [PubMed] [Google Scholar]
- 53.Prokop JW, Duff RJ, Ball HC, Copeland DL, Londraville RL. Leptin and leptin receptor: Analysis of a structure to function relationship in interaction and evolution from humans to fish. Peptides. 2012;38:326–336. doi: 10.1016/j.peptides.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hansel NN, Gao L, Rafaels NM, Mathias RA, Neptune ER, Tankersley C, Grant AV, Connett J, Beaty TH, Wise RA, Barnes KC. Leptin receptor polymorphisms and lung function decline in COPD. Eur Respir J. 2009;34:103–110. doi: 10.1183/09031936.00120408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moleres A, Campion J, Milagro FI, Marcos A, Campoy C, Garagorri JM, Gomez-Martinez S, Martinez JA, Azcona-Sanjulian MC, Marti A. Differential DNA methylation patterns between high and low responders to a weight loss intervention in overweight or obese adolescents: the EVASYON study. FASEB J. 2013 Jun;27:2504–12. doi: 10.1096/fj.12-215566. [DOI] [PubMed] [Google Scholar]