

Abstract
Polymerases ϵ and δ are the main enzymes that replicate eukaryotic DNA. Accurate replication occurs through Watson–Crick base pairing and also through the action of the polymerases' exonuclease (proofreading) domains. We have recently shown that germline exonuclease domain mutations (EDMs) of POLE and POLD1 confer a high risk of multiple colorectal adenomas and carcinoma (CRC). POLD1 mutations also predispose to endometrial cancer (EC). These mutations are associated with high penetrance and dominant inheritance, although the phenotype can be variable. We have named the condition polymerase proofreading-associated polyposis (PPAP). Somatic POLE EDMs have also been found in sporadic CRCs and ECs, although very few somatic POLD1 EDMs have been detected. Both the germline and the somatic DNA polymerase EDMs cause an ‘ultramutated’, apparently microsatellite-stable, type of cancer, sometimes leading to over a million base substitutions per tumour. Here, we present the evidence for POLE and POLD1 as important contributors to the pathogenesis of CRC and EC, and highlight some of the key questions in this emerging field. Copyright © 2013 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd
Keywords: DNA polymerase, exonuclease, proofreading, germline mutation, somatic mutation, colorectal cancer, colorectal adenomas, polyposis, endometrial cancer
Introduction
In comparison with other cancers, there exist a relatively large number of syndromes in which a high lifetime risk of colorectal cancer (CRC) is caused by inheriting a mutation in a single gene. The specific Mendelian CRC syndromes (and their mutant genes) are familial adenomatous polyposis (APC), Lynch syndrome/HNPCC (mismatch repair genes MSH2, MLH1, MSH6, PMS2), Peutz–Jeghers syndrome (LKB1/STK11), juvenile polyposis (SMAD4, BMPR1A), MUTYH-associated polyposis (the base excision repair gene MUTYH), and hereditary mixed polyposis (GREM1). All of these conditions, except MUTYH-associated polyposis, are dominantly inherited, although a recessive version of HNPCC exists, in which both copies of one of the mismatch repair (MMR) genes are mutated. Each syndrome differs in its clinical features, but in most cases, there is a primary predisposition to multiple (10s–1000s) adenomas or other benign polyps, with a secondary CRC risk, probably owing to progression of the benign lesions. The exception is Lynch syndrome, in which there is usually a small excess of adenomas and the primary predisposition is to CRC.
An ongoing question for several years has been whether there are any more high-penetrance CRC predisposition genes to be found. There certainly exist patients whose clinical features and family history make them a priori likely to carry a high-penetrance CRC predisposition allele, but who have no mutations in the known Mendelian CRC genes. One such group of patients comprises individuals with hyperplastic polyposis syndrome (HPPS), although the hypothetical HPPS gene(s) has not yet been identified. Another set of patients likely to carry high-penetrance CRC mutations has multiple adenomas. Typically, these tumours number 10–100 at presentation or after a few years of screening. Patients may present before the age of 60 and they have often developed one or more CRCs. Some of these individuals come from extensive CRC pedigrees, although others have no significant family history of colorectal tumours.
In this article, we summarize the recent identification of DNA polymerase ϵ and δ mutations in familial colorectal cancer cases, many of whom have multiple adenomas. In common with other genes such as APC and SMAD4, the polymerases are additionally somatically mutated in a recently-reported subset of sporadic CRCs 1. There is also accruing evidence that, like the mismatch repair genes, POLE and POLD1 mutations play roles in endometrial carcinogenesis.
High-penetrance germline DNA polymerase ϵ and δ mutations cause colorectal and endometrial cancers
Using a combination of whole-genome sequencing of highly-selected multiple adenoma patients, linkage analysis, and studies of loss of heterozygosity (LOH) in tumours, followed by replication in a large set of familial CRC cases, we recently identified two specific germline mutations that caused carriers to develop multiple colorectal adenomas and CRC 2. These mutations were in two related DNA polymerase genes: POLE (p.Leu424Val) and POLD1 (p.Ser478Asn). Neither mutation was present in nearly 7000 UK controls or in public databases of controls. Subsequently, we found an additional, probably pathogenic mutation, POLD1 p.Pro327Leu, in a further multiple adenoma patient.
Both mutations show dominant inheritance and confer high-penetrance predisposition to multiple colorectal adenomas, large adenomas, early-onset CRC, or multiple CRCs (Figure 1). For this reason, we have called the disease polymerase proofreading-associated polyposis (PPAP). The phenotype varies among carriers: some have tens of adenomas that do not always appear to progress rapidly to cancer, whereas others have a small number of large adenomas or carcinomas. The histological features of the tumours are unremarkable. They are mostly conventional adenomas and carcinomas that occur throughout the large bowel. There is currently no evidence that mutation carriers are at risk of upper-gastrointestinal tumours, but female carriers of POLD1 p.Ser478Asn have a greatly increased risk of endometrial cancer (EC). Overall, the PPAP phenotype overlaps with the phenotypes associated with germline mutations in APC, MUTYH, and the MMR genes. POLE p.Leu424Val, POLD1 p.Ser478Asn, and POLD1 p.Pro327Leu all map within the proofreading (exonuclease) domain of the respective enzyme, suggesting that deficient proofreading repair during DNA replication is the cause of our patients' tumours. It is of note that POLD1 in particular additionally participates in both MMR and base excision repair. However, the families' tumours are microsatellite-stable. Although the reasons for this are currently unclear, it is possible to speculate that the strand slippage that results in insertion–deletion mutations does not involve a mispaired, single-strand intermediate that is recognized by the polymerase proofreading domain, and, moreover, that polymerase proofreading has a minor role in mismatch repair.
Figure 1.
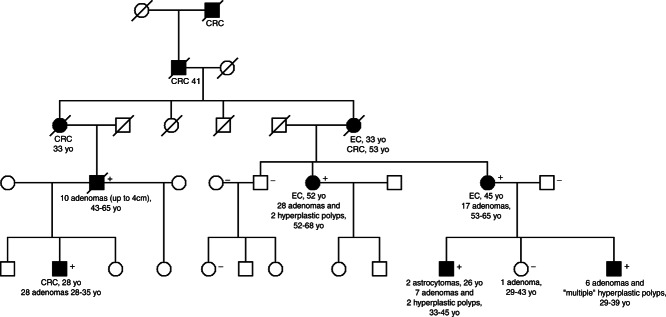
Pedigree of a POLD1 p.Ser478Asn family. Shading denotes those affected with multiple (> 5) colorectal adenomas (adenomas) and/or early-onset colorectal cancer (CRC). In addition, three women developed endometrial carcinoma (EC). + denotes individuals tested and found to be gene carriers, and − denotes tested non-carriers. Ages denote the time interval over which colorectal polyps developed or the time at which cancer occurred. Note that one non-gene carrier developed a very small colorectal adenoma by age 43 years and that one carrier developed two astrocytomas, raising the possibility that POLD1 mutations also predispose to this tumour type.
Somatic polymerase ϵ mutations in sporadic colorectal and endometrial cancers
Separately from the work that discovered germline POLE and POLD1 mutations in colorectal cancer patients, POLE was highlighted as a somatically mutated gene in CRC by The Cancer Genome Atlas (TCGA) exome sequencing project 1. A set of cancers with a very large number of coding mutations (over 50 per 106 bases), in the absence of MMR defects or microsatellite instability (MSI), was also identified, and this set of cancers overlapped with the POLE-mutant set. Subsequently, it was found that almost all of the hypermutant, MS-stable cancers had POLE exonuclease domain mutations (EDMs) 2,3. The seven POLE EDMs in the TCGA cohort, out of a total of 226 CRCs (3%), were all missense changes, although the germline p.Leu424Val change was absent. Two recurrent changes were found, p.Val411Leu and p.Ser459Phe. These data are consistent with those from another CRC exome sequencing project 4 that found two of 74 (3%) cancers to have acquired POLE p.Pro286Arg mutations. Recently released, but unpublished data from the TCGA have confirmed codons 286, 411, and 459 as mutation hotspots 5. Interestingly, POLE residue 286 is homologous to the probably pathogenic germline mutation that we have reported at residue 327 in POLD1. However, there is no good evidence of pathogenic, somatic POLD1 EDMs.
The TCGA 5 and we ourselves 6 have found somatic POLE EDMs in ECs. These occur at a slightly higher frequency (∼7%) than that in CRCs. As was the case for CRCs, POLD1 EDMs were very rare common, seen in just one tumour in each cohort (approximately 0.5%). The POLE mutation spectra of the ECs showed overlap with those of the CRCs, with p.Pro286Arg and p.Val411Leu particularly frequent. p.Ser297Phe was also found in two ECs. In addition, a somatic p.Leu424Val change – the mutation present in the germline of CRC cases – was present in two ECs, and one cancer possessed a mutation of a residue that forms the exonuclease catalytic site (p.Asp275Val) (Figure 2). Like CRCs, the POLE-mutant ECs tended to be MSI-negative and had an ultramutator phenotype. Figure 2 shows the locations of the somatic and germline POLE and POLD1 mutations within the exonuclease domain.
Figure 2.
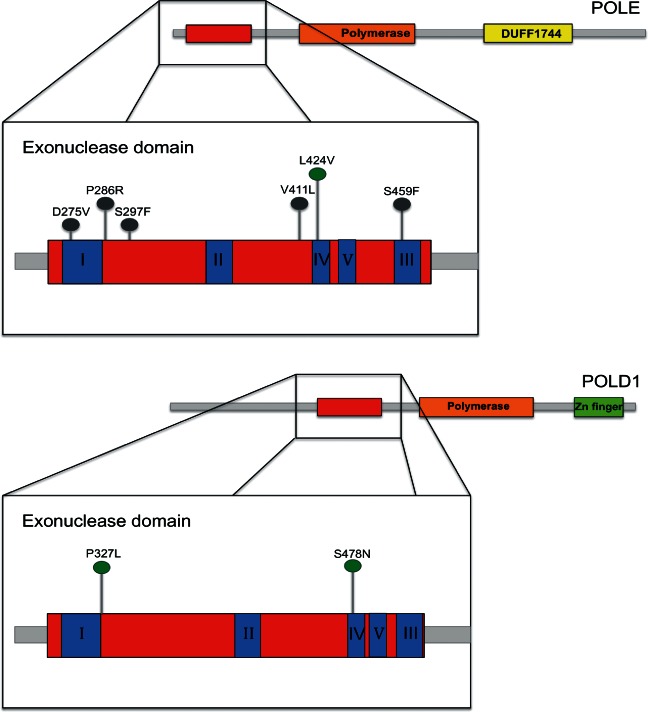
The structure of POLE and POLD1 demonstrating the position of key mutations. Conserved exo motifs I–V within the exonuclease domain are highlighted in blue. Green circles denote germline mutations; grey circles denote somatic mutations.
The normal roles of polymerase ϵ and δ
POLE and POLD1 are related B family polymerases. They form the major catalytic and proofreading subunits of the Polϵ and Polδ enzyme complexes that respectively synthesize the leading and lagging strands in DNA replication 7,8. Their proofreading (exonuclease) function detects and removes misincorporated bases in the daughter strand through failed complementary pairing with the parental strand. The high fidelity of DNA replication is in part due to very low error rates in dNTP incorporation by the polymerase (10−4 to 10−5) and in part due to proofreading by the exonuclease domain, which improves this fidelity approximately 100-fold. POLE and POLD1 have greatest homology (23% identity, 37% similarity) over their exonuclease domains (residues 268–471 of POLE and 304–517 of POLD1). Both genes are ubiquitously expressed and show high levels of evolutionary conservation, especially in the exonuclease domain. A number of yeast mutants exist in the homologues of POLE and POLD1, and these models have shown that mutator phenotypes can result from outside in the polymerase domain 9–14. Other variants actually have improved DNA repair capacity 15, although they also have lower processivity (rate of synthesis of the daughter strand).
The Polϵ and Polδ enzymes are both heterotetramers in higher eukaryotes. The accessory subunits (POLE2/3/4 and POLD2/3/4) are involved in regulating synthesis and in binding co-factors such as PCNA. It is of note that a common polymorphism within POLD3 has been found to be associated with a modestly increased risk of CRC in the general northern European population 16. Whether this acts in a similar way to POLD1 mutations is currently unclear.
POLD1 – and perhaps also POLE – is thought to play an additional role in new strand synthesis as part of the processes of base excision repair and MMR. POLE is involved in break-induced replication, a form of double-strand break repair in which the homologous chromosome is used as a template, resulting in copy-neutral LOH. Whether these aspects of DNA polymerase function are important for tumour development is unknown, as is the explanation for the near-absence of somatic POLD1 EDMs. At the very least, it is striking that defects in at least three pathways involved in the repair of base pair-level mutations can predispose to colorectal tumours.
How do POLE and POLD1 mutations cause tumorigenesis?
The most common somatic and germline mutations in POLE and POLD1 have been mapped onto a hybrid structure of yeast DNA polymerase (3iay) and T4 polymerase (Figure 3). The residues equivalent to the two germline mutations (POLE p.Leu424Val and POLD1 p.Ser478Asn) pack together at the interface between two helices that form the base of the exonuclease active site. Mutations are predicted to distort the packing of the helices; this will in turn be propagated to the active site, affecting nuclease activity 2. The residue equivalent to the most common somatic POLE mutation at amino acid 286 localizes to the DNA binding pocket adjacent to the exonuclease active site, with its side chain very close to the nascent single-stranded DNA, and substitutions at this site are predicted significantly to perturb the structure of the DNA binding pocket. Structural analysis also shows that amino acid 297 interacts with exonuclease catalytic site residue 275, and mutation here would probably alter the active site conformation. Interestingly, POLE residue 411, whilst conserved, is not predicted to interact with DNA or catalytic site residues, and the effects on tumorigenesis may be through secondary effects on the binding pocket. Although the structural data do not explain the recurrent nature of some POLE mutations, they strongly suggest that the POLE and POLD1 mutations impair polymerase proofreading. For most of these mutations, moreover, studies in model organisms including T4 bacteriophage, yeast, and mice have confirmed these effects 2 #path4185-bib-0010 #path4185-bib-0017"/>. For example, the p.Pro123Leu mutation in the T4 bacteriophage is at the equivalent residue to human Pro286 and produces a strong mutator phenotype 18.
Figure 3.
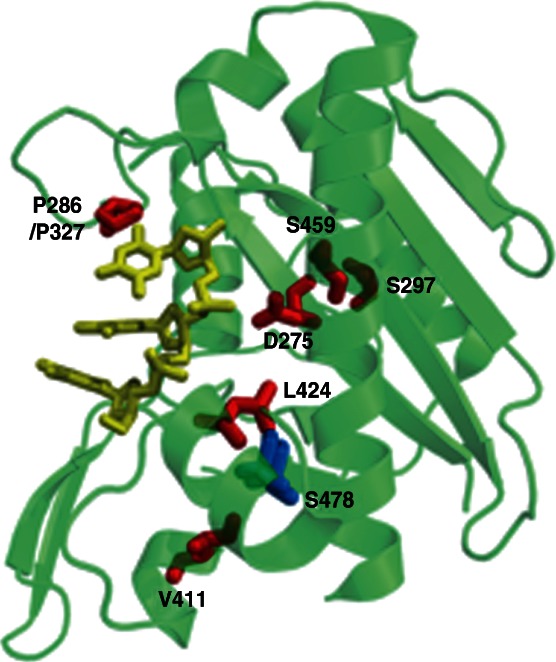
Pymol-generated image of POLE and POLD1 EDMs on a composite structure of yeast Polδ (PDB 3IAY) and the ssDNA component (yellow) of the T4 polymerase complex (PDB 1NOY). Mutant amino acids are shown in red (POLE) or blue (POLD1). The key mutations generally cluster around the active site (D275) close to the ssDNA, an exception being V411 which lies some distance away and may act through affecting the positions of other residues closer to the active site.
Hypermutation is thus a very plausible consequence of EDM POLE and POLD1 mutations. Whether this is the only tumour-promoting consequence of these mutations remains unclear. It will also be intriguing to determine whether proofreading deficiency has any effect on polymerase processivity, since negative effects on this function may be selectively deleterious for the cell.
Pathways of tumorigenesis
Exome sequencing data from CRCs and ECs with somatic POLE EDMs show that the coding regions alone of these tumours have acquired a mean of about 5000 somatic base substitutions 1,5. All types of base substitution are increased in frequency compared with cancers without EDMs, and C:G → T:A changes generally remain the most common. However, the mutation spectrum is changed, with a particular increase in the proportion of G:C → T:A and A:T → C:G transversions. Although there is considerable variation in the number of mutations among cancers with EDMs, there is some evidence that specific mutations have different effects on the mutation spectrum. For example, cancers that carry p.Pro286Arg show a much stronger bias towards transversions than cancers with p.Val411Leu. Interestingly, the exome sequence data show that the somatic mutations resulting from deficient exonuclease proofreading tend to occur at sites flanked by an A base on the positive DNA strand, rather than by T, G or C. The causes for this observation are currently unknown, although one possibility is the ability of mutations to be corrected by the MMR machinery.
POLE and POLD1 are not classical tumour suppressor genes, as loss-of-function mutations appear not to be pathogenic. Instead, there is loss of a specific function, proofreading, that is unlikely to be achieved through protein-truncating mutations. Furthermore, it is not clear whether ‘two hits’ at POLE or POLD1 are required for tumourigenesis. In Pole-mutant mice, a mutator phenotype and increased frequency of tumour formation are only seen when Pole mutations are homozygous 17. In humans, some, but not all, tumours from patients with germline POLE or POLD1 mutations show LOH, although data on other forms of ‘second hit’ are lacking in these tumours. LOH has not been reported in cancers with somatic POLE mutations, although a few of these tumours have protein-truncating mutations that could act as ‘second hits’. From a functional perspective, a Polϵ or Polδ protein with a heterozygous EDM would probably cause an increase in mutation frequency because 50% of polymerase activity would be error-prone. However, it is conceivable that this error rate is insufficient to overwhelm the other repair systems such as MMR.
A further unanswered question is when POLE and POLD1 mutations occur during tumourigenesis. For colorectal tumours with either somatic or germline POLE and POLD1 EDMs, the mutation spectrum of the APC gene shows deficiency of frameshift (insertion–deletion) changes compared with other CRCs. Moreover, some APC mutations that are generally rare in CRCs are relatively common in POLE- and POLD1-mutant tumours, an example being the Arg1114X change. These data suggest that the POLE and POLD1 mutations occur before APC mutations. However, at least in sporadic CRCs with POLE EDMs, the frequency of pathogenic mutations in the other known driver genes is low, perhaps suggesting that they follow an atypical pathway of tumorigenesis.
POLE and POLD1 mutations outside the exonuclease domain
POLE and POLD1 are both quite large genes, with protein-coding regions of about 6.6 and 3.3 kb, respectively. Inevitably, therefore, they will acquire many ‘passenger’ mutations in cancers. The causal role of EDMs has been deduced as a result of finding (i) germline mutations in PPAP and (ii) the so-called ‘ultramutator’, MSI-negative phenotype in sporadic cancers. No such clues exist in support of a pathogenic role for the non-exonuclease domain POLE and POLD1 mutations that are found in 3–4% of CRCs and ECs. In fact, the majority of these non-EDM changes occur in MSI-positive cancers and most seem likely to be passengers.
Future prospects
Evidently, the identification of germline and somatic POLE and POLD1 mutations that cause CRC and EC is only the first stage in understanding how those changes act and, if possible, exploiting them for cancer prevention and therapy. Perhaps the immediate priority is to determine the full Polϵ and Polδ mutation spectra, to determine which mutations are pathogenic, and then to understand their effects. The somatic mutation spectrum of POLE and POLD1 in the common cancers will be increasingly well described in the coming months as further large-scale sequencing programmes come to fruition. It will also be important to test POLE as a prognostic marker. In the germline, similar mutation profiling efforts will undoubtedly be performed, one scenario being that there exist a number of germline Polϵ and Polδ variants with different magnitudes of effect on risk. Given that common polymorphisms have been addressed by CRC and EC GWAS, the uncharacterized germline risk variants are likely to be individually uncommon (<5% allele frequency) and some may not necessarily be in the exonuclease domains of POLE and POLD1. It will be necessary to obtain evidence for their effects using a variety of functional assays. A particularly interesting issue is why germline POLE or POLD1 mutations can cause cancer, yet only POLE is somatically mutated. This has a parallel in the MMR genes, of which four can be mutated in Lynch syndrome, but only MLH1 plays a role somatically. It may be that POLD1 has an as yet unidentified essential function that precludes its somatic mutation or that the effects of somatic mutation are too weak (or indeed too strong) to be effective other than in the germline setting. Finally, the discovery of a new type of CRC and EC based on POLE mutations rather than the established classifiers of MSI and chromosomal instability raises the prospect of future similar discoveries, leading to an increasingly refined classification of cancer based on DNA analysis, which is potentially more robust than analysis of gene or protein expression.
Acknowledgments
In this report, we have made use of data from the TCGA project on colorectal and endometrial cancer, much of which is currently unpublished, and we acknowledge with thanks the investigators of those projects and the NIH. We are grateful to David Church for assistance with Figure 3. SEWB is in receipt of a Research Bursary from Cancer Research UK. We also acknowledge core funding to the Wellcome Trust Centre for Human Genetics from the Wellcome Trust (090532/Z/09/Z).
Author contribution statement
Both authors wrote and revised the manuscript and had final approval of submitted and published versions.
References
- 1.The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487::330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature Genet. 2012;45:136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donehower LA, Creighton CJ, Schultz N, et al. MLH1-silenced and non-silenced subgroups of hypermutated colorectal carcinomas have distinct mutational landscapes. J Pathol. 2013;229:99–110. doi: 10.1002/path.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seshagiri S, Stawiski EW, Durinck S, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–664. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Church DN, Briggs SEW, Palles C, et al. DNA polymerase ϵ and δ exonuclease domain mutations in endometrial cancer. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt131. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pursell ZF, Isoz I, Lundstrom EB, et al. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McElhinny SA, Gordenin DA, Stith CM, et al. Division of labor at the eukaryotic replication fork. Mol Cell. 2008;30:137–144. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simon M, Giot L, Faye G. The 3′ to 5′ exonuclease activity located in the DNA polymerase delta subunit of Saccharomyces cerevisiae is required for accurate replication. EMBO J. 1991;10:2165–2170. doi: 10.1002/j.1460-2075.1991.tb07751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrison A, Johnson AL, Johnston LH, et al. Pathway correcting DNA-replication errors in Saccharomyces cerevisiae. EMBO J. 1993;12:1467–1473. doi: 10.1002/j.1460-2075.1993.tb05790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrison A, Bell JB, Kunkel TA, et al. Eukaryotic DNA polymerase amino acid sequence required for 3′–5′ exonuclease activity. Proc Natl Acad Sci U S A. 1991;88:9473–9477. doi: 10.1073/pnas.88.21.9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kokoska RJ, Stefanovic L, DeMai J, et al. Increased rates of genomic deletions generated by mutations in the yeast gene encoding DNA polymerase delta or by decreases in the cellular levels of DNA polymerase delta. Mol Cell Biol. 2000;20:7490–7504. doi: 10.1128/mcb.20.20.7490-7504.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pavlov YI, Shcherbakova PV, Kunkel TA. In vivo consequences of putative active site mutations in yeast DNA polymerases alpha, epsilon, delta, and zeta. Genetics. 2001;159:47–64. doi: 10.1093/genetics/159.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daee DL, Mertz TM, Shcherbakova PV. A cancer-associated DNA polymerase delta variant modeled in yeast causes a catastrophic increase in genomic instability. Proc Natl Acad Sci U S A. 2010;107:157–162. doi: 10.1073/pnas.0907526106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Venkatesan RN, Hsu JJ, Lawrence NA, et al. Mutator phenotypes caused by substitution at a conserved motif A residue in eukaryotic DNA polymerase delta. J Biol Chem. 2006;281:4486–4494. doi: 10.1074/jbc.M510245200. [DOI] [PubMed] [Google Scholar]
- 16.Dunlop MG, Dobbins SE, Farrington SM, et al. Common variation near CDKN1APOLD3 and SHROOM2 influences colorectal cancer risk. Nature Genet. 2012;44:770–776. doi: 10.1038/ng.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Albertson TM, Ogawa M, Bugni JM, et al. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci U S A. 2009;106:17101–17104. doi: 10.1073/pnas.0907147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reha-Krantz LJ. Amino acid changes coded by bacteriophage T4 DNA polymerase mutator mutants. Relating structure to function. J Mol Biol. 1988;202:711–724. doi: 10.1016/0022-2836(88)90552-9. [DOI] [PubMed] [Google Scholar]