

Abstract
Reconstitution of membrane proteins in artificial membrane systems creates a platform for exploring their potential for pharmacological or biotechnological applications. Previously, we demonstrated amphiphilic block copolymers as promising building blocks for artificial membranes with long-term stability and tailorable structural parameters. However, the insertion of membrane proteins has not previously been realized in a large-area, stable, and solid-supported artificial membrane. Here, we show the first, preliminary model of a channel membrane protein that is functionally incorporated in a completely artificial polymer, tethered, solid-supported bilayer membrane (TSSBM). Unprecedented ionic transport characteristics that differ from previous results on protein insertion into planar, free-standing membranes, are identified. Our findings mark a change in understanding protein insertion and ion flow within natural channel proteins when inserted in an artificial TSSBM, thus holding great potential for numerous applications such as drug screening, trace analyzing, and biosensing.
Achieving highly controlled selectivity of passage of molecules and ions– the defining feature of cell membranes– in a stable, artificial membrane holds the promise of expanding our scientific understanding of living systems and has great potential for numerous applications. In cells, lipid membranes and their associated proteins are responsible for these key functions1,2,3. Therefore, various lipids have been used as model building blocks to generate membranes, either as vesicular structures in solution, called liposomes4, or as tethered, solid-supported bilayer membranes (TSSBM)5,6. However, without a long tether molecule to separate the lipid membranes from solid substrates, the thin water/hydrophilic layer between the lipid membrane and substrate (1–2 nm) is too small to prevent intense interaction and/or frictional coupling between incorporated membrane components (e.g. integral proteins) and the solid surface6,7,8, which leads to partial loss of functionality, or even to complete protein denaturation in the case of transmembrane proteins9.
Analogous to cell membranes, incorporating natural channel proteins in an artificial membrane matrix, is the key for controlling the passage of molecules and ions with high precision and specificity, just as in living systems. Amphiphilic block copolymers hold great potential over lipids10 as building blocks for such artificial membranes and as hosts for proteins, due to their long-term mechanical stability, tailorable structural parameters, and versatile chemical functionality11,12,13. In fact, natural proteins attached to polymer membranes have been widely studied for diagnosis, drug design and biotechnologies14,15. But merely immobilizing a protein onto a membrane surface cannot fulfil the function of a membrane channel protein. Previously, as a considerable step beyond surface attachment, we demonstrated the insertion of channel proteins into polymer vesicles and free-standing membranes16,17,18,19. Such systems, however, are either mechanically unstable or unsuitable for surface analytical techniques, thus obscuring measurements for extended insight or practical applications. TSSBM based on polymers as building blocks have increased durability and mechanical stability7,20, and by chemical modification with functional groups, significant additional advantages can be achieved. These include: i. stable immobilisation on a solid support, ii. longer membrane–support distance to allow insertion of membrane proteins without affecting their structure/functionality, and iii. simultaneous attachment/insertion of biomolecules to create multifunctional platforms. These properties make polymer TSSBM good candidates for protein incorporation, with resulting significance to biotechnological applications. However, to the best of our knowledge, the insertion of channel proteins with functions in a large-area, stable, polymer TSSBM has not been realized.
Here, we demonstrate a preliminary model of a channel membrane protein that is functionally incorporated in a completely artificial block copolymer TSSBM. A variation in electrical conductance during channel protein insertion and atomic force microscopy (AFM) images together establish the unequivocal functional incorporation of membrane proteins in a large-area, stable, polymer TSSBM. Unprecedented electrical characteristics distinctly differ from those of vesicle or free-standing membranes11,12,13 were identified. This behaviour of the artificial membrane originates from covalent bonding between the TSSBM and the Au substrate, which ensures a compact dielectric and a significant interface capacitance coupling between the electrolyte and the substrate.
Results
Preparation and characterization of the polymer TSSBM
One reason for the lack of a functional insertion of channel protein into a polymer TSSBM is the preparation of the polymer TSSBM. Until now, this has involved the use of surface-grafting, which features very densely packed polymer chains, or vesicle fusion process, which exhibits polycationic character; both methods exhibit limitations for protein insertion21,22,23,24. In the present work, we have combined surface chemistry and the self-assembly of amphiphilic block copolymers to produce a homogenous block copolymer TSSBM by consecutive Langmuir-Blodgett (LB) and Langmuir-Schaefer (LS) transfers25. The great advantage of LB and LS depositions is that each of the two methods allows control of layer density by the surface pressure of the molecular assembly. Combining both methods produces a TSSBM with a large-area, homogeneous, and defect-free hydrophilic-hydrophobic-hydrophilic structured bilayer, similar to a natural cell membrane (see Supplementary Fig. S1). Moreover, an LB-LS-transferred polymer TSSBM is more stable (more than two weeks in water; up to 12 h in air), compared to a free-standing polymer membrane (less than several hours in water)17. In the present study, the amphiphilic poly(butadiene)52-block-poly(ethylene oxide)29 (PB-PEO) that does not exhibit toxic effects on living cells26,27, was selected to serve as a suitable platform to probe the biomimetic potential of TSSBM for membrane protein reconstitution.
The PB-PEO polymer bilayer was transferred to the surface of a patterned gold electrode by using the above mentioned LB-LS transfer technique (Fig. 1a and Supplementary Fig. S2). This LB-LS-transferred, planar polymer TSSBM is coupled to a solid surface by means of its bottom polymer layer and gold/sulphur chemistry (using lipoic acid (LA)), while its upper layer attaches to this bottom layer by hydrophobic interaction (Fig. 1a). Note that the retained, upper layer fluidity benefits the reconstitution of peptides and proteins on this stability-enhanced polymer TSSBM.
Figure 1. Schematic representation of the TSSBM and conductance measurement across it.

(a) Hydroxyl-functionalized linear PB-PEO diblock copolymer (PB-PEO-OH) and lipoic acid functionalized linear PB-PEO diblock copolymer (PB-PEO-LA) were transferred onto gold substrates by the subsequent LB-LS technique to form a polymer TSSBM, which is suitable for protein insertion (b). (c) The setup to record electrical conductance across the TSSBM using a source-meter (S-M). The liquid chamber is defined by polydimethylsiloxane (PDMS). (d) Characteristic time course for conductance across the PB-PEO TSSBM before (black curve) and after (red curve) addition of αHL, at a voltage of 40 mV. Inset is an enlarged view of the stepwise increase in the characteristic time course of the conductance across the PB-PEO TSSBM with the addition of αHL.
The electrical conductance (G) across the membrane was measured in a two-electrode geometry, as shown in Fig. 1d. Compared to lipid bilayers (0.1–1 MΩ cm−2)28, we observed significant enhanced resistance (1/G) per area for polymer bilayers (> 10 MΩ cm−2), which can be attributed to tighter molecular packing and increased length of the hydrophobic region of the polymer chain.
There are principally two obstacles that can limit protein reconstitution in a polymer TSSBM. First, block copolymer membranes are usually thicker (> 10 nm17; our TSSBM membrane has a thickness of about 11.3 nm) than conventional lipid bilayers (2–5 nm)28, due to larger molecular size (Supplementary Fig. S3). Thus even when membrane proteins are inserted in a polymer membrane, they may still be too short to extend through the membrane completely. Second, the coupled layer of the polymer TSSBM is covalently immobilized on the solid surface, resulting in limited flexibility and conformational freedom, whilst a conformational change in the upper polymer layer remains possible. These limitations raise the question of whether, and to what extent, proteins can preserve their functions within a block copolymer TSSBM.
Reconstitution of natural channel proteins in polymer TSSBM
For protein reconstitution experiments, we used the well-characterized water soluble bacterial membrane polypeptide, α-haemolysin (αHL), as a preliminary model system that does not require detergent for stabilization. In vivo, αHL binds to a receptor on target cell membranes and oligomerizes to form heptameric transmembrane nanopores29. These channels (1–2 nm diameter)30 allow passive diffusion of small solutes such as ions, nutrients, and antibiotics across the membrane (Fig. 1b). Therefore, functional membrane incorporation of αHL can be monitored directly, both qualitatively and quantitatively, by conductance measurements across the TSSBM. The αHL was inserted by direct immersion of polymer TSSBM in the protein solution. We did not use the vesicle fusion technique, another way to form membranes on solid supports, because it does not allow generation of homogeneous and defect-free polymer TSSBM (one layer being covalently bound to the Au substrate). Obtaining a defect-free polymer TSSBM with high reproducibility is of key importance for electrical characterization of relatively large areas. To establish whether channel protein insertion has taken place, we introduced a characterisation method based on the measurement of the electrical conductance and ionic capacitance across a homogenous and defect-free TSSBM (Fig. 1c). Normally, the patch clamp technique is the conventional method for isolating single channel proteins and performing precise ion transport measurements in cells, in giant vesicles31, and even in planar membranes32. However, as the patch clamp method is based on the formation of a small section of free-standing membranes, it cannot be used for the characterisation of large-area TSSBM.
The characteristic time course for the change in conductance across the PB-PEO TSSBM with added αHL was compared to that of protein-free TSSBM (Fig. 1d). Initially, the conductance of the polymer TSSBM with added αHL was stable, and only slightly higher than the protein-free membrane. Interestingly, at less than 20 min after the addition of αHL, a significant change in membrane conductance occurred; it increased rapidly, then decreased slowly, forming a non-symmetrical peak. We attribute the increase in conductance to multiple, incremental αHL insertions, in agreement with previous measurements on lipid- or free-standing polymer membranes17,28. In natural lipid membranes, a single αHL channel contributes a conductance of about 0.8 nS under given buffer and temperature conditions33. Similar to natural lipid membranes, the conductance of our block copolymer membrane increased in a stepwise manner, at about 0.8 nS or 1.6 nS per step (inset of Fig. 1d), thereby suggesting successful one-by-one protein incorporation. At least 38 inserted proteins are implied by the conductance increase of 31 nS at the peak maximum, which corresponds to 420 αHL mm−2 (measured gold surface area 0.09 mm2). Flexibility and conformational freedom of the upper layer of the polymer bilayer molecules, important parameters for successful incorporation of αHL (see Supplementary Fig. S4) allowed TSSBM to adapt to the specific geometric and dynamic requirements of αHL insertion. When we applied an approaching force F of ~1.6 nN, corresponding to an effective approaching pressure 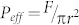 of ~300 kPa (r is the radium of the AFM tip), a characteristic peak in the force curve at a distance of about 9 nm appeared. This pressure is needed for the AFM tip to penetrate the upper polymer monolayer, and is therefore an indication of the stiffness/fluidity of the polymer membrane. In the case of a lipid membrane this peak has been reported for an approaching pressure of ~150 kPa34. Note that additional peaks due to later insertion also appeared randomly (see Supplementary Fig. S5) and we attribute them to the spatial adjustment of the upper layer polymer chains allowing supplementary protein insertion (within areas not previously occupied by proteins).
of ~300 kPa (r is the radium of the AFM tip), a characteristic peak in the force curve at a distance of about 9 nm appeared. This pressure is needed for the AFM tip to penetrate the upper polymer monolayer, and is therefore an indication of the stiffness/fluidity of the polymer membrane. In the case of a lipid membrane this peak has been reported for an approaching pressure of ~150 kPa34. Note that additional peaks due to later insertion also appeared randomly (see Supplementary Fig. S5) and we attribute them to the spatial adjustment of the upper layer polymer chains allowing supplementary protein insertion (within areas not previously occupied by proteins).
AFM was used to further confirm the incorporation of proteins into the polymer TSSBM. When a mushroom-shaped αHL is incorporated in a lipid membrane, its large, hydrophilic head, at a diameter of 6–7 nm and a height of 2–3 nm, protrudes from the membrane33. The AFM scan of pure PB-PEO TSSBM (Fig. 2a) indicates a smooth surface with an average roughness of about 0.9 nm. After incubation with αHLs, the AFM image of PB-PEO TSSBM indicates reconstitution (formed pores, Fig. 2b) in the polymer TSSBM. The observed head sizes of 5.5–7.3 nm and channel sizes of 1–2 nm (Fig. 2c) agree well with αHL inserted in lipid membranes29,30,33. The presence of non-circular, white areas may represent αHL monomers or possibly preformed oligomers that did not form channels and that are attached to, or partially inserted in the membrane (Fig. 2b).
Figure 2. Surface morphology of a PB-PEO TSSBM before and after insertion of αHL.
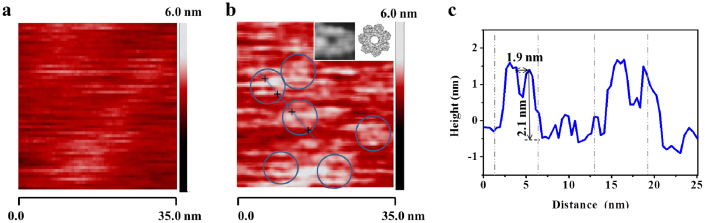
(a) AFM image of a pure PB-PEO TSSBM, (b) after insertion of αHL (insert is enlarged image of separate pores and reconstruction of the pore top obtained from the results of 3D homology modelling), (c) cross section along the blue line in (b).
In a free-standing membrane, the increased conductance caused by protein incorporation usually remains constant after insertion17,28. However, in the case of our polymer TSSBM, a subsequent decrease in conductance drew attention to a different behaviour. To investigate whether the decreasing conductance represented a specific phenomenon of protein incorporation in a solid-supported membrane, we tested a lipid (1, 2-diphytanoyl-sn-glycero-3-phosphocholine, DPhPC) solid-supported bilayer membrane (SSBM). The control shows that the variation in conductance of the lipid SSBM upon insertion of αHL was similar to that of the polymer TSSBM with inserted αHL (see Supplementary Fig. S6), but with an earlier incorporation time and more incorporated proteins (about 200 αHL at peak maximum). This observation that the lipid SSBM favours protein incorporation compared to the polymer TSSBM is consistent with previous reported studies on protein incorporation in polymer- and lipid free-standing membranes17,28. Extending beyond αHL, preliminary studies on the incorporation of water insoluble membrane proteins, such as outer membrane protein F (OmpF), in a polymer TSSBM show similar conductance behaviour (see Supplementary Fig. S7). For water insoluble proteins, it is necessary to add detergent to stabilize the proteins in solution, and this might affect the structure and the conductance of TSSBM. However, under the conditions of our experiment (2% n-octyl-oligo-oxyethylene as detergent), the addition of detergent did not affect significantly the behaviour of TSSBM.
Discussion
One question that remains is why the enhanced conductance, shown by both the polymer TSSBM and lipid SSBM resulting from incorporation of αHL channels, decreased. The mechanism that re-establishes cell resting membrane potential by counter-transport of potassium ions in the familiar sodium-potassium pump35 calls to mind our pattern of increasing and decreasing potential. As soon as channel protein insertion occurs, ions pass through the membrane via the channels and reach an equilibrium state according to the Donnan equation, which describes an equilibrium between two solutions separated by a thin, selective membrane35,36. In our case, the hydrophobic part of the block copolymer TSSBM (Fig. 3a), with protein incorporated, can function as a selective membrane for ion transport (Fig. 3b). With an applied voltage, Vappl·, and an ion concentration gradient, external ions pass through the membrane to the inner, hydrophilic part, which acts as a small reservoir for the ions. As the inside hydrophilic reservoir of TSSBM is very small, ions will accumulate and reach Donnan equilibrium in a very short time (Fig. 3c). A Donnan potential ΦD will be established across the membrane to exactly offset the applied voltage and the gradient potential of the ion concentration, thereby preventing any further net movement of ions.
Figure 3. Illustration of current variance during protein incorporation.

(a–c) Scheme for membrane protein incorporation in the block copolymer TSSBM (upper left) and the corresponding variance in the potential and current of the TSSBM (lower left). (d) Time dependent current measurement upon reversed voltage to the polymer TSSBMs before (black curve) and after (red curve) protein insertion. Inset shows the reproducibility of the measurement at a large time scale when the applied voltage is inverted periodically. (e) Normalized ionic capacitance ΔQCP/ΔQCP(ΦD = 20 mV) of the total inserted channel proteins against applied voltage ΦD (star points). The black line and red line are theoretical plots, according to a capacitor model and Donnan model, respectively. Inset, the normalized ionic capacitance ΔQCP/ΔQCP(ΦD = 40 mV) as function of PBS buffer concentration.
Here, based on the Donnan equation, we propose the first, general model for understanding conductance variation through a solid-supported, protein-inserted membrane. The Donnan potential of the membrane ΦD is given by:
where ΦDo, the ideal Donnan potential (without applied voltage), is defined as:
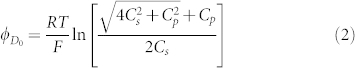 |
where Cs is the salt concentration in the external solution, and Cp is the concentration of hindered, large ions. Here, the PBS solution contains cations (Na+, H+) and anions (H2PO4−, HPO42−, PO43−, OH−). Without loss of generality, in our case, because αHL forms a pore that allows the diffusion of all ions in the external PBS solution, Cp equals 0, corresponding to an ideal Donnan potential ΦDo = 0. As a result, ΦD = Vappl. = 40 mV. In the Donnan equilibrium that applies in our case, the ratio of the concentration of each type of ion on the two sides of the TBBSM is:
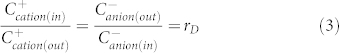 |
where rD is the Donnan equilibrium constant. ΦD can be calculated by the Nernst formula,
As a result, we can derive a Donnan equilibrium constant rD = 4.75. This means that, when ions accumulate in the channel pocket to a concentration of 4.75-fold more than that of the external solution, an ionic transport equilibrium is established (i.e. no net ions pass). It is clear now that the increased conductance upon channel protein insertion is caused by ion accumulation in the inner, hydrophilic part of the polymer membrane under the applied voltage. This ion accumulation, however, leads to an increased Donnan potential, which offsets the applied voltage, correspondingly preventing a net movement of ions, and thus lowering the conductance. Note here that because the external solution has a much larger volume than the internal hydrophilic part of the membrane, any variation in ion concentration on the outside can be neglected.
To further support the Donnan model described above, we conducted current measurements with reversed voltage applied to the polymer TSSBM, both before and after protein insertion. As shown in Fig. 3d, when the polarity of the applied voltage (at times indicated by the arrows) is inverted, the pulsed current increases first and then drops to a constant level. Such behaviour of the capacitance can be ascribed to the accumulation of ions, when inverting the applied voltage. The area of the peaks, corresponding to the amount of charges accumulated at the interface, can be integrated as Q1 and Q2, for the polymer TSSBM before and after αHL insertion, respectively. We found that Q2 (~32 nC) is larger than Q1 (~20 nC), which confirms that the proteins have been functionally incorporated in the artificial block copolymer TSSBM, and thereby enhanced the ionic capacitance of the membrane (as expected from the Donnan model). Note here that the appearance of sharp current peaks in a short time scale of several seconds upon inverting the applied voltage (Fig. 3d) is a capacitance effect. The time dependence of these peaks is different compared to that of the peaks corresponding to the conductance across TSSBM after protein insertion (Fig. 1d), which were broader in time (~100 s), and measured at a constant applied voltage of 40 mV. In addition, the peaks representing the current under normal voltage and under inverted periodic voltage have similar areas (see Supplementary Fig. S8). This demonstrates that protein insertion is quantitatively similar when the voltage is inverted periodically.
In addition, the Donnan model allows the study of the ionic capacitance of the inserted channel proteins as a function of the ion concentration in the bulk solution Cbulk and the applied voltage ΦD:
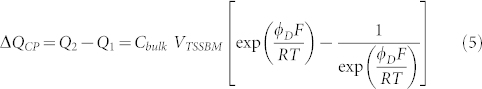 |
where VTSSBM, representing the volume of the inner, hydrophilic part of the polymer membrane, is a constant. In Fig. 3e the normalized ionic capacitance ΔQCP/ΔQCP(ΦD = 20 mV) of the channel protein is plotted against the applied voltage ΦD (star points). The black line represents the capacitor model, whilst the red line represents the Donnan model (exponential scale) (Eq.5). The experimental data clearly deviate from a pure capacitor behaviour (black line), but follow satisfactorily the anticipated behaviour according to the Donnan model (red line). The gradient of the normalized ionic capacitance ΔQCP/ΔQCP(ΦD = 40 mV) of the membrane at ΦD = 40 mV (inset of Fig. 3e) is proportional to the concentration of the solution, as expected from the Donnan model. These represent the first reported results of the ionic capacitance of inserted channel proteins systematically accessed in a TSSBM membrane. The Donnan model gives insight on the ionic transport behaviour of inserted channel proteins, which to the best of our knowledge, has not been yet reported in experiments using the patch clamp technique.
In summary, we have established the functional incorporation of channel proteins in a large-area, stable, polymer TSSBM by monitoring the variation in electrical conductance during channel protein insertion and AFM imaging. The unprecedented conductance variation, other than those of vesicle or free-standing membrane upon channel protein αHL insertion, is modelled by the Donnan potential caused by ion accumulation in the inner, hydrophilic part of the polymer membrane. This offsets the applied voltage and correspondingly prevents a net movement of the ions. This first example is in no way limiting, and intuitively leads to broader applications by substituting one membrane protein with another. For example, preliminary results indicate that the water insoluble membrane protein, OmpF, can also be successfully reconstituted in the polymer membrane. We believe that this newly introduced concept of membrane proteins functionally inserted in polymer TSSBM can be expected to serve as promising tool for biological studies, for example understanding protein insertion and ion flow within natural channel proteins, as well as for applications such as drug screening, trace analysing, and biosensing.
Methods
Materials
Hydroxyl-functionalized linear PB-PEO diblock copolymer (PB-PEO-OH) and lipoic acid functionalized linear PB-PEO diblock copolymer (PB-PEO-LA) were used. The polymers contain 52 PB and 29 PEO repeating units. Details on polymer synthesis, functionalization and characterization were reported earlier25.
αHL powder, purchased from Sigma-Aldrich, was dissolved in phosphate buffered saline (137 mM NaCl, 12 mM phosphate, 2.7 mM KCl, pH 7.4.) without detergent to yield a αHL solution of 0.5 mg mL−1.
Gold substrate preparation
Ultrasmooth template stripped gold surfaces (TSG) were prepared according to a procedure previously described37. 50 nm thin, gold films were deposited by electron-beam evaporation (0.8–1 Å s−1, 5 × 10−6 mbar) on clean silicon wafers (crysTec, Germany) and then glued to clean microcrown glass slides (Menzel, Germany) with epoxy glue (EPO-TEK 353 ND4, USA). For a patterned gold surface, a photoresistant layer (ma-N 415) was pre-deposited on silicon wafers, openings were then etched through the layer so that the target material could reach the surface of the substrate in those regions in which the final pattern was to be created. After gold was deposited over the whole area of the wafer, the remaining photoresistant layer that was not previously etched, together with the gold on top, was washed away by several applications of ethanol and water. After this separation, the gold remained only where it directly contacted the substrate.
Monolayer transfer
The first PB-PEO-LA monolayers were transferred onto TSG substrates by the Langmuir-Blodgett (LB) technique, using a KSV 5000 LB instrument (KSV Instruments, Finland). A Langmuir TeflonTM trough (area 1860 cm2) was placed on an antivibration table in a plastic cabinet. Prior to spreading the film, freshly cleaved TSG substrates were immersed in the subphase using a dipper. After compressing the film to the target pressure of 35 mN m−1, it was left for 15 min in order for the polymer chains to establish their most favourable orientation. Afterwards, a monolayer film was transferred at constant speed (0.3 m min−1) with a dipper upstroke.
Bilayer transfer
The second upper layers were transferred by the Langmuir-Schaefer (LS) technique. A compressed PB-PEO-OH film with a 35 mN m−1 target pressure was produced at the air-water interface. PB-PEO-LA coated slides were placed in the dipper horizontally above the floating monolayer. The substrate was lowered through the interface at constant dipper speed (50 mm min−1). The water surface was thoroughly cleaned and the gold slides were placed under water, into a crystallization dish.
When the membrane density of PB-PEO TSSBM lowered by transfer at the target pressure of 30 or 25 mN m−1, the defects increased and the resulting membrane conductance destabilized (Fig. S9).
Lipid SSBM were prepared as in the above process, with a target pressure of 35 mN m−1.
Surface plasmon resonance (SPR) spectroscopy
SPR measurements were performed using a home-built setup in the Kretschmann configuration with a He/Ne laser (λ = 633 nm). In scan mode, reflectivity was monitored as a function of the incident angle. In kinetic mode, reflectivity changes occurring at a fixed angle were recorded as a function of time. Spectra were analysed using a four-layer model, including the prism, gold, mono- or bilayer, and the surrounding medium (water or air).
Atomic force microscopy (AFM)
AFM contact mode imaging and force spectroscopy measurements were carried out in PBS, pH 7.4, at room temperature using a Multimode-Nanoscope IIIA controller (Veeco, Santa Barbara, CA) equipped with a 120 μm J-scanner and a standard liquid cell. Prior to each experiment, the system was allowed to thermally equilibrate for at least 1 h. Rectangular-shaped Si3N4 cantilevers with V-shaped tips were used (SNF-10, Olympus/OBL, Veeco).
Electrical measurements
Typically, conductance enhancement due to protein incorporation is in the nS range. Thus, the resistance of the host membrane should preferably be ≥ 1 GΩ28. For electrical measurements, microsized gold electrodes prepared by using a standard photolithography process were used together with a PDMS liquid chamber (Fig. S2). The gold wires were first attached to the TSSBM surface with silver paint, and the samples left for 10 min to stabilize. After that, a voltage of 40 mV was applied across the TSSBM with the liquid side at higher potential for several minutes before measurement in order to initialize the system until stable conductance of the membrane was achieved. The current was measured by a source-meter (Keithley 2636A) at a constantly applied 40 mV. As soon as a current map was obtained, the buffer solution was exchanged for a protein solution (about 1 ng of αHL). Again, the current was measured after 10 min stabilization. All devices were automatically controlled by a self-made LabView program.
Author Contributions
X.Z. worked on the preparation and characterization of polymer and lipid TSSBM. X.Z. and W.F. designed and conducted the electrical measurements. X.Z., W.F., C.P. and W.M. wrote the manuscript. C.P. and W.M. conceived the project.
Supplementary Material
Natural channel protein inserts and functions in a completely artificial, solid-supported bilayer membrane
Acknowledgments
The work was financially supported by Swiss National Science Foundation, NCCR Nanoscience, Swiss Nanosciene Institute, NRP 62 “Smart Materials”, and European Science Foundation on the project NANOCELL, this is gratefully acknowledged. The authors thank Prof. Christian Schönenberger from University of Basel for the access to the electric measurement, and Prof. Roderick Lim for the access to the SPR and AFM measurement. Authors thank Dr. B. A. Goodman for useful discussions and Mr Mark Inglin for editing the manuscript.
References
- Murata K. et al. Structural determinants of water permeation through aquaporin-1. Nature 407, 599–605 (2000). [DOI] [PubMed] [Google Scholar]
- Voulhoux R., Bos M. P., Geurtsen J., Mols M. & Tommassen J. Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 299, 262–265 (2003). [DOI] [PubMed] [Google Scholar]
- Engelman D. M. Membranes are more mosaic than fluid. Nature 438, 578–580 (2005). [DOI] [PubMed] [Google Scholar]
- Ginsberg L. Does Ca-2+ Cause Fusion or Lysis of Unilamellar Lipid Vesicles. Nature 275, 758–760 (1978). [DOI] [PubMed] [Google Scholar]
- Brian A. A. & Mcconnell H. M. Allogeneic Stimulation of Cyto-Toxic T-Cells by Supported Planar Membranes. P Natl Acad Sci-Biol 81, 6159–6163 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M. & Sackmann E. Polymer-supported membranes as models of the cell surface. Nature 437, 656–663 (2005). [DOI] [PubMed] [Google Scholar]
- Sackmann E. Supported membranes: Scientific and practical applications. Science 271, 43–48 (1996). [DOI] [PubMed] [Google Scholar]
- Tanaka M. & Sackmann E. Supported membranes as biofunctional interfaces and smart biosensor platforms. Phys Status Solidi A 203, 3452–3462 (2006). [Google Scholar]
- Sinner E. K. & Knoll W. Functional tethered membranes. Curr Opin Chem Biol 5, 705–711 (2001). [DOI] [PubMed] [Google Scholar]
- Hauser H., Stubbs M. & Phillips M. C. Ion Permeability of Phospholipid Bilayers. Nature 239, 342 (1972). [DOI] [PubMed] [Google Scholar]
- Bermudez H., Brannan A. K., Hammer D. A., Bates F. S. & Discher D. E. Molecular weight dependence of polymersome membrane structure, elasticity, and stability. Macromolecules 35, 8203–8208 (2002). [Google Scholar]
- Nardin C., Winterhalter M. & Meier W. Giant free-standing ABA triblock copolymer membranes. Langmuir 16, 7708–7712 (2000). [Google Scholar]
- Discher B. M. et al. Polymersomes: Tough vesicles made from diblock copolymers. Science 284, 1143–1146 (1999). [DOI] [PubMed] [Google Scholar]
- Roberts M. J., Bentley M. D. & Harris J. M. Chemistry for peptide and protein PEGylation. Adv Drug Deliver Rev 64, 116–127 (2012). [DOI] [PubMed] [Google Scholar]
- Higgins S. J. Conjugated polymers incorporating pendant functional groups - synthesis and characterisation. Chem Soc Rev 26, 247–257 (1997). [Google Scholar]
- Nardin C., Widmer J., Winterhalter M. & Meier W. Amphiphilic block copolymer nanocontainers as bioreactors. Eur Phys J E 4, 403–410 (2001). [Google Scholar]
- Meier W., Nardin C. & Winterhalter M. Reconstitution of channel proteins in (polymerized) ABA triblock copolymer membranes. Angew Chem Int Edit 39, 4599−+ (2000). [PubMed] [Google Scholar]
- Haefele T., Kita-Tokarczyk K. & Meier W. Phase behavior of mixed Langmuir monolayers from amphiphilic block copolymers and an antimicrobial peptide. Langmuir 22, 1164–1172 (2006). [DOI] [PubMed] [Google Scholar]
- Kumar M., Habel J. E. O., Shen Y. X., Meier W. P. & Walz T. High-Density Reconstitution of Functional Water Channels into Vesicular and Planar Block Copolymer Membranes. J Am Chem Soc 134, 18631–18637 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamm L. K. & Mcconnell H. M. Supported Phospholipid-Bilayers. Biophys J 47, 105–113 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhmatullina E. & Meier W. Solid-supported block copolymer membranes through interfacial adsorption of charged block copolymer vesicles. Langmuir 24, 6254–6261 (2008). [DOI] [PubMed] [Google Scholar]
- Rakhmatullina E., Mantion A., Burgi T., Malinova V. & Meier W. Solid-Supported Amphiphilic Triblock Copolymer Membranes Grafted from Gold Surface. J Polym Sci Pol Chem 47, 1–13 (2009). [Google Scholar]
- Dorn J., Belegrinou S., Kreiter M., Sinner E. K. & Meier W. Planar Block Copolymer Membranes by Vesicle Spreading. Macromol Biosci 11, 514–525 (2011). [DOI] [PubMed] [Google Scholar]
- Wang H. L. et al. Highly Permeable and Selective Pore-Spanning Biomimetic Membrane Embedded with Aquaporin Z. Small 8, 1185–1190 (2012). [DOI] [PubMed] [Google Scholar]
- Belegrinou S. et al. Biomimetic supported membranes from amphiphilic block copolymers. Soft Matter 6, 179–186 (2010). [Google Scholar]
- Lee J. C. M. et al. Preparation, stability, and in vitro performance of vesicles made with diblock copolymers. Biotechnol Bioeng 73, 135–145 (2001). [DOI] [PubMed] [Google Scholar]
- Vijayan K., Discher D. E., Lal J., Janmey P. & Goulian M. Interactions of membrane-active peptides with thick, neutral, nonzwitterionic bilayers. J Phys Chem B 109, 14356–14364 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D., Jeon T. J. & Schmidt J. Single molecule measurements of channel proteins incorporated into biomimetic polymer membranes. Nanotechnology 17, 3710–3717 (2006). [Google Scholar]
- Bhakdi S. et al. Staphylococcal alpha-toxin, streptolysin-O, and Escherichia coli hemolysin: Prototypes of pore-forming bacterial cytolysins. Arch Microbiol 165, 73–79 (1996). [DOI] [PubMed] [Google Scholar]
- Song L. Z. et al. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 274, 1859–1866 (1996). [DOI] [PubMed] [Google Scholar]
- Neher E. & Sakmann B. Single-Channel Currents Recorded from Membrane of Denervated Frog Muscle-Fibers. Nature 260, 799–802 (1976). [DOI] [PubMed] [Google Scholar]
- Cornell B. M., Alkhamici H., Brown L., Carne S. & Goodchild S. C. Ion Channel Proteins that Spontaneously Insert into Lipid Bilayer Membranes: An Impedance Spectroscopy Study Employing Tethered Membranes. Biophys J 102, 682a–683a (2012).22325292 [Google Scholar]
- Braha O. et al. Designed protein pores as components for biosensors. Chem Biol 4, 497–505 (1997). [DOI] [PubMed] [Google Scholar]
- Pera I., Stark R., Kappl M., Butt H. J. & Benfenati F. Using the atomic force microscope to study the interaction between two solid supported lipid bilayers and the influence of synapsin I. Biophys J. 87, 2446–2455 (2004). [DOI] [PMC free article] [PubMed]
- Skou J. C. The Influence of Some Cations on an Adenosine Triphosphatase from Peripheral Nerves. Biochim Biophys Acta 23, 394–401 (1957). [DOI] [PubMed] [Google Scholar]
- Donnan F. G. Theory of the balances of membranes and potential of membranes at the existence of non dialysing electrolytes - A contribution to physical chemical physiology. Z Elktrochem Angew P 17, 572–581 (1911). [Google Scholar]
- Naumann R. et al. Tethered lipid Bilayers on ultraflat gold surfaces. Langmuir 19, 5435–5443 (2003). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Natural channel protein inserts and functions in a completely artificial, solid-supported bilayer membrane