

Abstract
Mutations in PARK8, encoding leucine-rich repeat kinase 2 (LRRK2), are a frequent cause of Parkinson's disease (PD). Nonetheless, the physiological role of LRRK2 remains unclear. Here, we demonstrate that LRRK2 participates in canonical Wnt signaling as a scaffold. LRRK2 interacts with key Wnt signaling proteins of the β-catenin destruction complex and dishevelled proteins in vivo and is recruited to membranes following Wnt stimulation, where it binds to the Wnt co-receptor low-density lipoprotein receptor-related protein 6 (LRP6) in cellular models. LRRK2, therefore, bridges membrane and cytosolic components of Wnt signaling. Changes in LRRK2 expression affects pathway activity, while pathogenic LRRK2 mutants reduce both signal strength and the LRRK2–LRP6 interaction. Thus, decreased LRRK2-mediated Wnt signaling caused by reduced binding to LRP6 may underlie the neurodegeneration observed in PD. Finally, a newly developed LRRK2 kinase inhibitor disrupted Wnt signaling to a similar extent as pathogenic LRRK2 mutations. The use of LRRK2 kinase inhibition to treat PD may therefore need reconsideration.
INTRODUCTION
Parkinson's disease (PD) is a progressive movement disorder characterized by the degeneration of dopaminergic neurons of the ‘substantia nigra pars compacta’ and is the second most common neurodegenerative disease worldwide (1–3). Although typically idiopathic, genetic linkage has led to the identification of PD-causing mutations in PARK genes. Interest in PARK8 is particularly strong, since PARK8 mutations account for up to 40% of PD cases in some populations, and elicit symptoms and brain pathologies resembling idiopathic PD (1–3). PARK8 encodes leucine-rich repeat kinase 2 (LRRK2), a 2527 amino acid protein with two distinct enzymatic activities, namely serine/threonine kinase activity and GTPase activity, the latter conferred by a RocCOR (Ras in complex; C-terminal of Roc) tandem domain. The combination of these enzymatic activities has inevitably suggested a possible function for LRRK2 in signal transduction (1,2,4,5). However, despite a growing body of data linking LRRK2 to various cellular functions including autophagy and endocytosis, much remains unknown about the role of this protein. In particular, the precise cellular mechanisms by which LRRK2 mutations elicit neurodegeneration are still a mystery.
Wnt (Wingless/Int) pathways are evolutionarily conserved signaling cascades (6–8). Activation of the well-defined canonical Wnt pathway leads to nuclear accumulation of the transcriptional co-factor β-catenin and resultant changes in transcription (6–8). Under basal conditions, β-catenin is retained in a cytoplasmic multi-protein complex known as the β-catenin destruction complex (BDC). Here, β-catenin is phosphorylated by glycogen synthase kinase-3β (GSK3β) triggering β-catenin ubiquitination and degradation (6–8). However, when cells are stimulated by the binding of an extracellular Wnt ligand to frizzled (Fz) receptors, the BDC is recruited to membranes via interaction with a second cytosolic complex, dishevelled (DVL) polymers. DVL proteins are believed to interact with the intracellular face of Fz receptors, while the BDC associates with a second transmembrane protein, the Wnt signaling co-receptor low-density lipoprotein receptor-related protein 6 (LRP6). These aggregates of cytosolic protein complexes and membrane-localized receptors have been described as ‘signalsomes’ and their formation as a crucial step in the transduction of canonical Wnt signals. Within the signalsome, β-catenin phosphorylation is inhibited by mechanisms involving the subsequent internalization of the signalsome complex, resulting in the sequestration of GSK3β into multi-vesicular bodies (6–9). No longer phosphorylated and targeted for degradation, β-catenin is free to enter the nucleus and modulate downstream transcription (Fig. 1A).
Figure 1.

LRRK2 associates with the BDC and DVL proteins. (A) Overview of canonical Wnt signaling and potential interactions with LRRK2. (B) LRRK2 co-immunoprecipitates from mouse brain cytoplasm with components of the BDC and DVL proteins. Immunoprecipitations with anti-LRRK2 antibody (MJFF2). Anti-LRRK2 IP confirmed by western blotting with a second anti-LRRK2 antibody (NeuroMab N138/6). LRRK2 and co-complexed proteins are present in MJFF2 eluates and cell lysate, but not IgG control. (C) siRNA-mediated knockdown of LRRK2 increased basal and Wnt3a-induced TOPflash activity in SH-SY5Y cells. For each treatment condition, values are normalized to control siRNA to show the effect of LRRK2 knockdown. siRNA to Axin1 used as a positive control. P-values relative to siRNA control are shown. (D) Knockdown of LRRK1 and/or LRRK2 enhances DVL1-mediated Wnt signaling in HEK293 cells. P-values relative to siRNA control are shown.
The importance of canonical Wnt signaling in embryonic development and carcinogenesis is well described, but growing evidence suggests a role for this pathway in the function of mature neurons. For example, Wnt ligands are now well established as modulators of synaptic plasticity (10–17), while N-methyl-d-aspartate receptor stimulation has been reported to cause β-catenin activation (18). Wnt signaling has also been linked to neurodegenerative disease—indeed, dysregulated Wnt signaling has been suggested as a unifying hypothesis underlying Alzheimer's disease (19). Despite this, few connections between Wnt signaling and PD exist, although there are some clues that this might be a fruitful area of study. For example, Parkin, encoded by PARK2, has been described as a repressor of β-catenin activation (20), while we have reported a protein–protein interaction between overexpressed LRRK2 and the DVL proteins DVL1–3 (21). LRRK2 has also been described as a GSK3β interactor in Drosophila (22), although it was not reported if this represents the small fraction of total cellular GSK3β that is involved in Wnt signaling.
Here, we demonstrate a functional role for LRRK2 in the canonical Wnt pathway. Through interactions spanning DVL, BDC and LRP6 proteins, we present data indicating a scaffolding role for LRRK2 at the heart of canonical Wnt signaling. Our investigation has allowed LRRK2 to be placed physically and functionally into a well-defined signaling cascade for the first time. The following results suggest an intimate relationship between the canonical Wnt pathway and late-onset PD.
RESULTS
LRRK2 interacts with the BDC in vivo
We investigated the association of endogenous LRRK2 with Wnt signaling proteins by immunoprecipitation with anti-LRRK2 and anti-DVL antibodies from the cytosolic fraction of mouse brain. Since these experiments used tissue from animals that had not been treated to stimulate the canonical Wnt pathway, it can be assumed that these samples are representative of basal Wnt activity. These immunoprecipitations confirmed interaction between LRRK2 and DVL proteins in vivo (Fig. 1B; Supplementary Material, Fig. S1A). Importantly, LRRK2 was also found to exist in complex with multiple components of the BDC, including β-catenin, GSK3β and Axin1 (Fig. 1B). In addition, LRRK2 associated with β-arrestin—another protein implicated in canonical Wnt signaling (23) (Supplementary Material, Fig. S1B). The relative enrichment of GSK3β in LRRK2 murine brain immunoprecipitates in comparison with GSK3α, a protein not considered to play a major role in Wnt signaling, and the failure to co-immunoprecipitate cytosolic Rab5b, confirmed the specificity of these assays. Thus, these assays implicated endogenous LRRK2 in two distinct Wnt signaling complexes in the murine brain: DVL protein complexes and the BDC.
In some of the following experiments, the LRRK2 homolog LRRK1 was used to establish similarities and differences between these proteins in Wnt signaling. However, it is important to note that sequence variations in the LRRK1 gene have not been found to segregate with PD (24,25). In addition, the introduction of pathogenic LRRK2 missense mutations into equivalent positions in LRRK1 demonstrated that LRRK2 mutants are more prone to form inclusion bodies in transfected cells and are more toxic than the equivalent, artificial changes in LRRK1 (26).
The effect of LRRK2 knockdown on canonical Wnt activity was investigated to probe the functional role of LRRK2 in the BDC. These experiments were performed in dopaminergic SH-SY5Y cell culture, a standard in vitro model for PD research, which has previously been shown to respond to treatment with the Wnt ligand, Wnt3a (27). Using the TOPflash reporter (28) to measure β-catenin activity, the siRNA-mediated knockdown of LRRK2 reproducibly increased basal and Wnt3a-stimulated canonical Wnt activity, although to a lesser extent than the knockdown of the core BDC component Axin1 (Fig. 1C). The specificity of these siRNAs for LRRK2 and Axin1 was demonstrated in HEK293 cells, which unlike SH-SY5Y cells express LRRK2 at readily detectible levels. Interestingly, and consistent with the TOPflash data, siRNAs targeting LRRK2 or Axin1 induced an increase in lower-molecular-weight β-catenin species, likely caused by decreased β-catenin ubiquitination (Supplementary Material, Fig. S3). In addition, we observed an increase in LRRK2 protein levels upon Axin1 knockdown. This is consistent with a destabilization of the BDC upon Axin1 knockdown, allowing LRRK2 to leave the BDC and to bind to DVL proteins. The co-expression of LRRK2 and DVL was shown previously to increase LRRK2 protein levels (21). LRRK2 knockdown also increased TOPflash activity in a second in vitro model of canonical Wnt pathway activation, transient transfection of HEK293 cells with DVL1 (Supplementary Material, Fig. S1C). Moreover, similar data were obtained using siRNA probes to LRRK1, suggesting some redundancy of function between LRRK proteins (Fig. 1D). Thus, our data support the idea that LRRK2 is a component of the BDC and that LRRK2 knockdown compromises this complex, leading to β-catenin stabilization and Wnt signaling activation.
LRRK2 enhances canonical Wnt signaling at membranes
The role of LRRK2 in canonical Wnt signaling was also investigated in overexpression studies. When averaged across all experiments, LRRK2 overexpression had no effect on basal TOPflash activity relative to the control FOPflash reporter (Supplementary Material, Fig. S2A). Furthermore, ectopic LRRK2 had no reproducible effect on Wnt3a-stimulated TOPflash activity (Supplementary Material, Fig. S2B). Thus, the overexpression of LRRK2 alone was insufficient to significantly affect the canonical Wnt pathway. However, when this signal transduction cascade was activated by transient transfection of any of the three human DVL proteins, LRRK2 co-expression caused a statistically significant enhancement of reporter activity (Fig. 2A). Increased canonical Wnt activity was also observed when LRRK2 was co-transfected with LRP6 or the combination of LRP6 and Fz5 (Fig. 2B). Analogous results could be obtained using LRRK1 (Fig. 2C; Supplementary Material, Fig. S4). Since both LRRK proteins contain a kinase domain with homology to the MAPKKK superfamily (4), these observations raised the concern that the enhanced activation of canonical Wnt signaling might be a common and likely non-specific observation upon the overexpression of other kinases with MAPKKK homology. Importantly, the MAPKKK MEKK3 had the opposite effect to LRRK protein overexpression on Wnt signaling, repressing rather than activating DVL1-mediated TOPflash activation (Fig. 2D). This indicates that the effects of LRRK1 and LRRK2 are specific and unlikely to be an artifact of overexpressing proteins with MAPKKK homology.
Figure 2.

LRRK2 enhances DVL and LRP6-driven Wnt signaling. Overexpressed LRRK2 enhances the TOPflash activation elicited by overexpressed (A) DVL proteins or (B) LRP6 and LRP6 plus Fz5 in SH-SY5Y cells. In both figures, the fold effect of LRRK2 is included as inset graphs. (C) An analogous effect on DVL1-mediated Wnt signaling is seen with LRRK1. (D) In contrast, MEKK3 suppresses DVL1-mediated TOPflash activation, indicating that the effect of LRRK1 and LRRK2 is not a generic effect caused by the overexpression of proteins with MAPKKK domains. In all figures, P-values for the effect of LRRK1, LRRK2 or MEKK3 overexpression relative to the appropriate vector control are shown.
The observation that either LRRK protein could significantly enhance canonical Wnt signaling upon co-expression with additional Wnt pathway components suggested that the co-transfected proteins modify the behavior of LRRK1 and LRRK2. Consistent with this suggestion, overexpressed DVL proteins form polymeric structures (29) and we have previously reported that LRRK2 is efficiently recruited to these bodies, most likely via the direct association of the DVL DEP domain with the LRRK2-RocCOR domain (21). Intriguingly, DVL polymers have been suggested to at least partially localize to membranous compartments (9,29), while LRP6 is a transmembrane protein and present at cell membranes. We therefore hypothesized that overexpressed DVL and LRP6 could recruit LRRK2 to membranes and that membrane-associated LRRK2 exerted a positive effect on Wnt signaling. To test this idea, two LRRK2 expression constructs were generated with membrane-targeting mutations: the first containing amino acids 1–12 of c-Src to confer N-terminal myristoylation (30), the second encoding a T2524C amino acid substitution to create a CAAX motif conferring C-terminal prenylation (31). Supporting a role for LRRK2 in Wnt signaling at membranous compartments, both membrane-targeted LRRK2 mutants enhanced DVL1-stimulated TOPflash reporter activity to a greater extent than wild-type LRRK2 (Fig. 3A). In contrast, no increased activation was observed using the combination of Fz5 and LRP6 to activate canonical Wnt signaling (Fig. 3B), suggesting that either Fz5 or LRP6 is sufficient to confer the maximal membrane relocalization of wild-type LRRK2. Confocal microscopy revealed that the co-transfection of LRRK2 with any of three Fz receptors caused a limited redistribution of LRRK2 to membranes and some co-localization with Fz receptors (Supplementary Material, Fig. S5). However, the co-expression with LRP6 elicited a marked redistribution of LRRK2, with considerable co-localization between the two proteins (Fig. 4A).
Figure 3.

LRRK2 facilitates DVL1-mediated Wnt signaling at membranes. (A) Membrane-targeting of LRRK2 by myristoylation or prenylation potentiates the enhancement of DVL1-driven TOPflash activation elicited by wild-type LRRK2. (B) In contrast, membrane-targeting of LRRK2 has no significant effect on the ability of this protein to enhance LRP6 and Fz5-driven Wnt activation. In both figures, P-values for the effect of membrane targeting are shown.
Figure 4.
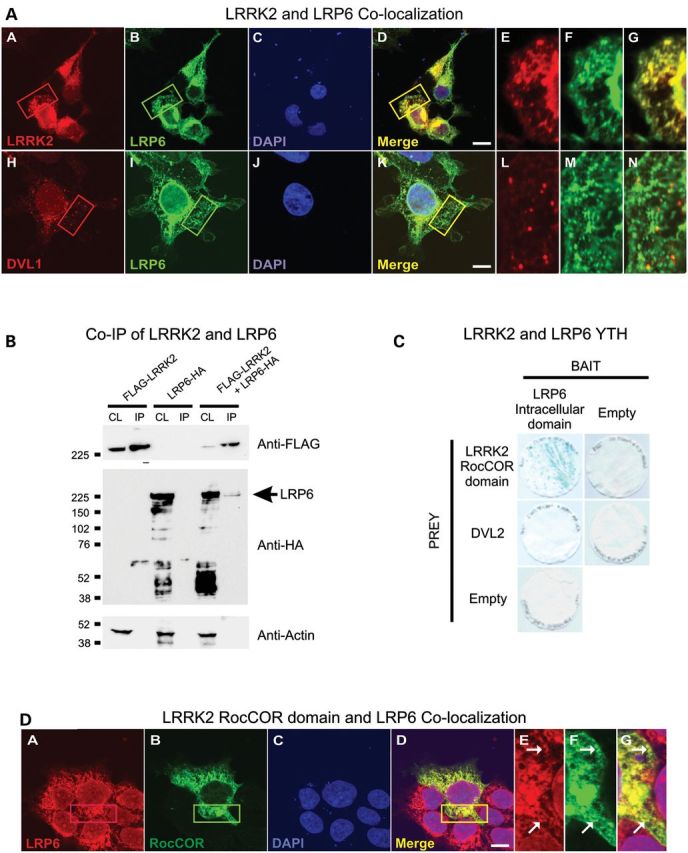
LRRK2 binds directly to the intracellular domain of LRP6. (A) Myc-tagged LRRK2 (red) and HA-tagged LRP6 (green) show almost complete co-localization in HEK293 cells (a–g). Almost no co-localization was seen between FLAG-tagged DVL1 (red) and HA-tagged LRP6 (green) (h–n). (B) FLAG-tagged LRRK2 and HA-tagged LRP6 co-immunoprecipitate from HEK293 cells. (C) The intracellular domain of LRP6 binds the LRRK2-RocCOR domain but not DVL2 in yeast. X-gal freeze-fracture assays indicate protein–protein interactions in blue. All negative controls show no color change. (D) Consistent with a requirement for the LRRK2-RocCOR domain for the LRP6–LRRK2 interaction the myc-tagged LRRK2-RocCOR domain (green) co-localizes with HA-tagged LRP6 similar to full-length protein in HEK293 cells (A, a–g). In (A) and (D), DNA staining with 4’,6-diamidino-2-phenylindole (blue) is shown, scale bar: 10 μm.
LRRK2 interacts with LRP6 and is recruited to membranes by Wnt ligands
Since the high degree of co-localization between LRRK2 and LRP6 suggested a potential physical interaction, we confirmed that overexpressed LRRK2 was able to co-immunoprecipitate LRP6 (Fig. 4B) but not Fz1, Fz4 or Fz5 receptors. To determine whether the interaction was direct, yeast two-hybrid (YTH) assays were performed using the intracellular domain of LRP6 as bait and various portions of LRRK2 as prey. This experiment revealed a strong direct interaction between LRP6 and the LRRK2-RocCOR tandem domain (Fig. 4C), but not between LRP6 and additional LRRK2 domains (Roc, COR, kinase and WD40; data not shown). In agreement with this observation, the RocCOR tandem domain co-localized with LRP6 in a similar manner to full-length LRRK2 (Fig. 4D). Taken together, these data indicate that LRRK2 can interact with the intracellular domain of LRP6 in cells and that this binding is likely to be direct, via the LRRK2-RocCOR domain.
A number of Wnt signaling proteins, including DVL proteins, have been reported to translocate to membranous compartments in response to acute stimulation with Wnt ligands (29). Indeed, the recruitment of the BDC to juxtamembrane protein aggregates is considered a key step in the stabilization of β-catenin (6–9). Thus, the interaction between LRRK2 and BDC in mouse brain cytoplasm and the recruitment of LRRK2 to intracellular membranes following LRP6 overexpression raised the possibility that the cellular distribution of LRRK2 might be regulated by Wnt stimulation. To this end, HEK293 cells, which unlike SH-SY5Y cells express LRRK2 at readily detectible levels, were treated with recombinant Wnt3a for 0, 30 and 60 min and the localization of endogenous LRRK2 to membrane fractions was determined. These experiments revealed a strong increase in the level of LRRK2 present at membranes at both time points following Wnt3a treatment (Fig. 5, Supplementary Material, Fig. S6).
Figure 5.

LRRK2 is recruited to membranes by Wnt3a. Acute treatment of HEK293 cells with recombinant Wnt3a increases the amount of endogenous LRRK2 present in crude membrane fractions. (A) A representative experiment. (B and C) Quantifications of the relative levels of membrane-associated LRRK2 at each time point from four independent experiments, normalized to (B) calnexin and (C) Rab5b. (D) Confocal images showing HEK293 cells expressing LRRK2 under basal conditions (A–D) and after activation with Wnt3a (E–H), scale bar: 10 μm.
The enhancement of DVL1-driven canonical Wnt signaling is weakened by PARK8 mutations and LRRK2 kinase inhibition
An increase in the catalytic activity of LRRK2, in particular LRRK2 kinase activity, has been suggested as the underlying cause of the neurodegeneration observed in PARK8 patients. Therefore, we investigated the importance of LRRK2 kinase and GTPase activities in canonical Wnt signaling. Kinase activity was first investigated using a pharmacological inhibitor of LRRK2, LRRK2-IN-1 (32). This compound markedly reduced the TOPflash activation elicited by DVL1 or a combination of LRRK2 and DVL1 (Fig. 6B). In agreement with a requirement for LRRK2 kinase activity for canonical Wnt signal activation, the effect of LRRK2 on DVL1-mediated Wnt signaling was also decreased by two artificial kinase-dead mutations (Fig. 6C). The importance of GTP binding was investigated in parallel using a K1347A substitution to block guanyl nucleotide binding. This mutant showed a similar deficiency in the ability to increase Wnt signaling to that of the kinase-dead constructs (Fig. 6C). Interestingly, introduction of the analogous mutation into LRRK1 did not appear to affect the function of this protein in identical experiments (Fig. 6D), while enzymatic inhibition did not appear to compromise the effect of LRRK proteins on Fz5 and LRP6-mediated TOPflash activation (Supplementary Material, Fig. S7A and B). Thus, our data suggest the importance of LRRK2, but not LRRK1, catalytic activity for Wnt signaling activation.
Figure 6.
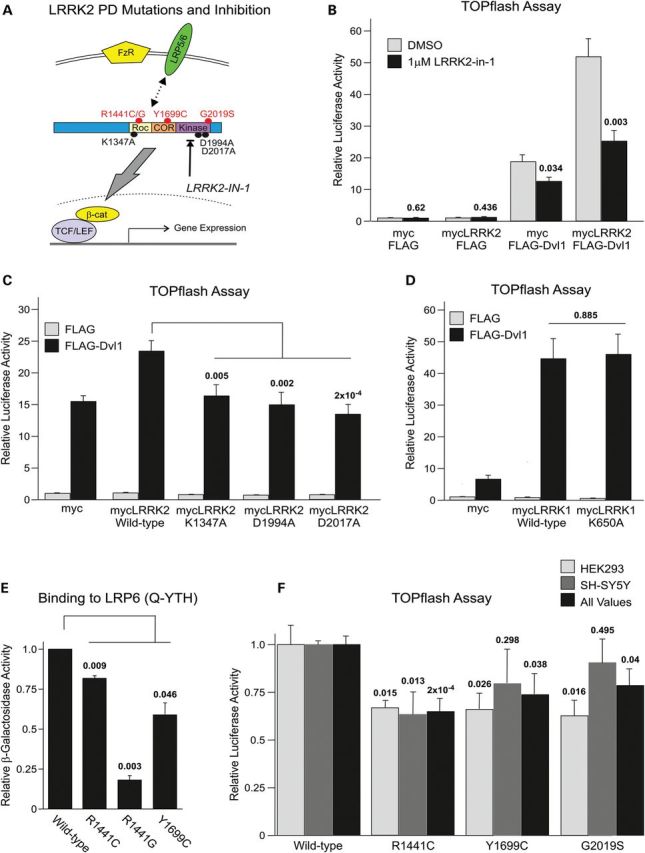
Pathogenic PARK8 mutations and loss of kinase or GTPase activity impair LRRK2 function in Wnt signaling. (A) Schematic of the pathological PARK8 mutations and strategies employed to inhibit kinase and GTPase function. (B) The LRRK2 kinase inhibitor, LRRK2-IN-1 inhibits TOPflash activation elicited by DVL1 or DVL1 and LRRK2. (C) The LRRK2-mediated enhancement of DVL1-driven TOPflash activation is inhibited by kinase-dead (D1994A and D2017A) or GTP/GDP-binding site mutations (K1347A). (D) An analogous GTP/GDP-binding mutation to LRRK1 has no effect on DVL1-driven Wnt signaling. (E) Pathogenic PARK8 mutations weaken the binding of the LRRK2-RocCOR tandem domain to LRP6. (F) PARK8 mutations weaken LRRK2-mediated enhancement of DVL1-driven TOPflash activation in two cell lines.
Numerous papers have investigated the influence of familial PARK8 mutations on the interaction of LRRK2 with accessory proteins, including some implicated in canonical Wnt signaling. For example, the G2019S kinase domain mutation increases the binding of LRRK2 to GSK3β (22). Furthermore, Roc domain mutations at R1441 increase the binding of LRRK2 to DVL1, while the Y1699C COR domain mutation weakens interaction with all three DVL proteins (21). Intriguingly, the binding of the LRRK2-RocCOR tandem domain to LRP6 is weakened by R1441C, R1441G and Y1699C mutations (Fig. 6E). In light of these observations and the requirements for LRRK2 kinase and GTPase activity (Fig. 6B and C), we determined the effect of pathogenic PARK8 mutations on canonical Wnt activity. In the absence of stimulation or when LRP6 and Fz5 were used to activate canonical Wnt signaling, no statistically significant differences of any of the mutations in comparison with wild-type LRRK2 were found (Supplementary Material, Fig. S7A). However, in co-transfection experiments with DVL1 performed in SH-SY5Y or HEK293 cells, all mutations tested (R1441C, Y1699C and G2019S) decreased TOPflash activity relative to wild-type LRRK2 (Fig. 6F).
DISCUSSION
Taken together, our data support a central role for LRRK2 in canonical Wnt signaling (summarized as a schematic in Fig. 7). In particular, our data are consistent with a model where under basal cell conditions without measurable canonical Wnt signaling activity, LRRK2 is associated with the BDC. Following Wnt stimulation, LRRK2 is recruited to the plasma membrane and directly associates with the intracellular domain of LRP6. LRRK2 also interacts with DVL proteins and proteins of the BDC, which are also recruited to the cell membrane after Wnt signal stimulation. Therefore, LRRK2 is able to assist in the formation of LRP6 signalosomes at the cell membrane. As such, despite the observed importance of GTP-binding and kinase activity, the role of LRRK2 is likely to be primarily as a scaffold for Wnt signaling complexes. This scaffold function takes place under basal conditions in the cytoplasmic BDC and after signal activation in association with LRP6 at membranes. LRP6 signalosomes are then internalized into the endosomal system, leading to the sequestration of GSK3β and other Wnt signaling components into multi-vesicular bodies. While it remains to be determined whether efficient intracellular trafficking of LRP6 signalosomes requires LRRK2, it is intriguing to note that both LRRK proteins are implicated in endocytosis (33–36). The interaction of LRRK2 with DVL proteins, β-arrestin and GSK3β also suggests the possibility that LRRK2 participates in non-canonical Wnt signaling, although this possibility requires further investigation.
Figure 7.
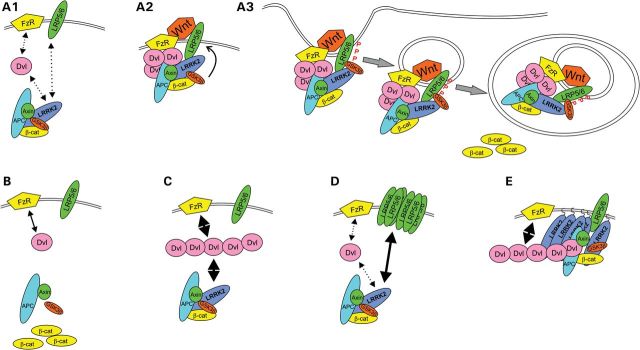
Schematic summary of the main study findings. (A1) In the basal state, LRRK2 is associated with the BDC. The distribution of BDCs between membrane and cytosol is in dynamic equilibrium dependent on interactions between Fz receptors (FzR) and DVL proteins, between DVL proteins and LRRK2 and between LRP5/LRP6 co-receptors and LRRK2. (A2) Following stimulation with Wnt ligand, DVL proteins are polymerized, thereby recruiting BDCs to the membrane via interaction with LRRK2 and additional BDC proteins. LRRK2 also interacts with the intracellular domain of LRP5/LRP6, thus bridging DVL proteins, the BDC and LRP5/LRP6, and facilitating LRP5/LRP6 phosphorylation by GSK3β, and the formation of LRP5/LRP6 signalosomes. (A3) Signalosomes containing phosphorylated LRP5/LRP6 and GSK3β are internalized into the endosomal system and ultimately sequestered into multi-vesicular bodies. Sequestration of GSK3β prevents phosphorylation of newly synthesized β-catenin allowing the protein to accumulate in the cytoplasm and activate β-catenin-dependent transcription in the nucleus. Whether LRRK2 is sequestered into MVBs with LRP6 and GSK3β or recycled to the cytosol remains to be determined. Note that LRRK2 is involved in multiple steps and interacts with multiple Wnt components. By altering strength of interaction with these proteins, PARK8 mutations are expected to compromise canonical Wnt activity stimulated by Wnt ligands, and perhaps also to alter basal β-catenin activity. (B) The primary effect of loss of LRRK2 is disruption of the BDC, leading to β-catenin stabilization. (C) Overexpressed DVL protein polymerizes and is sufficient to recruit BDCs to the membrane via increased interactions with FzRs and LRRK2. Presumably, the overexpression of LRRK2 further increases the strength of the LRRK2-DVL interaction. (D) Overexpressed LRP6 also increases the membrane recruitment of BDC via binding to LRRK2. (E) Membrane-targeting of LRRK2 ensures the DVL-LRRK2 interaction takes place at membranes, thereby maximizing the membrane recruitment of BDCs.
Importantly, PARK8 mutations to the LRRK2 Roc, COR and kinase domains all weaken the activation of canonical Wnt signaling elicited by the overexpression of DVL1 and LRRK2 (Fig. 6F), although whether these mutations disrupt Wnt signaling in vivo remains to be determined. However, given that LRRK2 interactions with LRP6 (Fig. 6E), DVLs (21) and GSK3β (22) are all altered by pathogenic PARK8 mutations, it seems unlikely that β-catenin activation will remain unaffected. Such in vivo studies might be difficult however, since PARK8 mutations are not fully penetrant—in fact, the most prevalent G2019S mutation is thought to have a penetrance of around 30% at the age of seventy (2)—suggesting that observed effects on Wnt signaling might only be obvious in aged animals or following additional insults, e.g. challenge with agents causing oxidative stress or co-existing genetic variables.
Wnt ligands, Wnt antagonists and β-catenin modulators are expressed in overlapping patterns throughout the developing central nervous system (37,38), demonstrating the redundancy of function for some as well as specific function for other Wnt signaling components. The pattern of Wnt β-catenin activity in the adult brain is also complex mirroring suggested Wnt signaling functions in neuronal maintenance, including synapse formation, synaptic plasticity and cell proliferation, and is further complicated by the observation that the expression of some Wnt components is age- and sex-dependent (39–41). The most evident Wnt-responsive tissues in the adult brain include the dentate gyrus, hippocampus, sensory telencephalic cortex, several thalamic nuclei, collicula and cerebellar cortex (40). Importantly, Wnt signaling pathways have also been demonstrated to be of fundamental importance for the biology of dopaminergic neurons in the ventral midbrain (42–45). For example, impaired dopaminergic development and altered midbrain morphology is seen upon the disruption of Wnt1 (46) and Lrp6 (47) genes encoding two proteins that function specifically within the canonical Wnt pathway. A recent study also suggested an interaction between dopamine D2 receptors and β-catenin in the adult brain (48).
LRRK2 is also a ubiquitously expressed protein, found in relative abundance in most brain regions, including the substantia nigra, thalamus, striatum, cortex, olfactory tubercle, nucleus accumbens, hippocampus and cerebellum (49–53). Interestingly, LRRK2 was also shown to be present in the subventricular zone (SVZ), suggesting a role for LRRK2 in adult neurogenesis (53,54). Deregulated Wnt signaling has also been shown to play an important role in impaired adult neurogenesis seen in PD. In particular, reactive astrocytes and microglia were shown to protect dopaminergic neurons in animal models of PD by activating canonical Wnt signaling and promoting neurogenesis from adult SVZ neuroprogenitor cells, by a mechanism based on the interplay between inflammation and canonical Wnt signaling (55–57). Investigation of an interaction between LRRK2 and Wnt signaling contributing to adult neurogenesis in the SVZ would be of great interest to further research into the pathogenesis of PD and the identification of therapeutic targets.
The impairment in canonical Wnt signaling activation caused by PARK8 mutations in our study (Fig. 6F) is in good accord with decreased Wnt signaling observed in neurodegeneration causing Alzheimer's disease and frontotemporal dementia (58). Moreover, a general requirement for Wnt signaling for the development of dopaminergic neurons and negative effects of decreased Wnt signaling on neuronal survival is well established (42,47,59–61). However, other studies show that increases in Wnt signaling might also lead to neurodegeneration, as observed in PARK2 early onset PD and JNPL3 frontotemporal dementia mouse models (20,62). Thus, it is likely that neuronal Wnt signaling is subject to regulation within defined boundaries to permit normal neuronal and synaptic function without eliciting cell cycle reentry leading to cell death in postmitotic neurons. Thus, investigations into the effects of PARK8 mutations on Wnt signaling in patients with familial and idiopathic PD are a future priority.
Dysregulated canonical Wnt signaling represents an intriguing candidate mechanism for PD pathogenesis. The importance of Wnt signaling for basic neuronal functions is well established, not least synaptic plasticity (10,12–18,63) and the control of microtubule stability (64–67). It is important to note that not all of these events require β-catenin-dependent transcriptional regulation, indicating both genomic and non-genomic actions. Moreover, the development of ventral mid-brain dopaminergic neurons appears particularly dependent on canonical Wnt signaling (42,47,59–61). Thus, dopaminergic neurons could be particularly sensitive to mild perturbations in canonical Wnt signaling that would elicit a slow but progressive loss of neuronal function ultimately ending in cell death. Furthermore, dysregulated Wnt signaling has the potential to explain the link between PD and variations in MAPT (encoding the microtubule-binding protein, tau) linked to PD in numerous genomic linkage studies (68). Tau is well described as a GSK3β substrate, and this phosphorylation event can be modulated by Wnt ligands (19,69). Moreover, tau phosphorylation is a pathological hallmark of Alzheimer's disease (19,69) and frontotemporal dementia and has been strongly linked to PARK8 mutations and increased LRRK2 expression in both PD patients and animal models (22,68,70,71). Thus, in addition to suggesting an intriguing mechanism for PD pathogenesis, our data also suggest tantalizing links to Alzheimer's disease and frontotemporal dementia.
Our observations are also likely to have implications beyond neurodegeneration. For example, constitutive activation of β-catenin is well described in numerous types of cancer and we note that an increased risk of non-skin cancer has been observed for individuals with PARK8 mutations (72,73). Moreover, the capacity of the LRRK2-IN-1 compound to inhibit canonical Wnt signaling (Fig. 6B) suggests the possibility of targeting LRRK2 kinase activity in the treatment of tumors linked to elevated β-catenin activation. Unfortunately, LRRK2 kinase inhibitors showed an inhibitory effect on Wnt signaling that was similar to that seen with pathogenic LRRK2 mutations. This suggests that the usefulness of LRRK2 inhibitors for the treatment of PD might be limited. In fact, LRRK2 inhibitors could have detrimental effects on disease progression and cognitive ability, since decreased Wnt signaling was observed in Alzheimer's disease (19).
In conclusion, our data show LRRK2 to be a central component of canonical Wnt signaling. We propose that dysregulated Wnt signaling is a new potential pathomechanism leading to PARK8 PD and suggest that alterations in Wnt signaling pathways might also be a common cause of idiopathic PD. Our data also suggest that the targeting of LRRK2 kinase activity might not be the most suitable approach for the treatment of neurodegeneration. In contrast, controlled Wnt signaling activation—e.g. with small molecules targeting LRP6—appears a more promising therapeutic approach for PD.
MATERIALS AND METHODS
Plasmids, cloning and siRNA
pRK5-Flag-DVL1-3, pRK5-mycLRRK2, pDS-LRRK2-RocCOR and pACT2-DVL expression plasmids have been described previously (21). pACT2-RocCOR, pACT2-Roc and pACT2-COR were made by inserting polymerase chain reaction (PCR) fragments encoding amino acids 1288–1844, 1288–1516 and 1516–1844 into the BamHI and XhoI sites in pACT2. All mutant derivatives of these plasmids were created by site-directed mutagenesis. pRK5-mycLRRK2CAAX was also made by site-directed mutagenesis to introduce a T2524C mutation, thereby creating a C-terminal CAAX prenylation motif. pRK5-myr-mycLRRK2 was generated from pRK5-mycLRRK2 by PCR using a forward primer encoding the first 12 amino acids of v-Src upstream of the myc tag. pRK5-3xFLAG-LRRK2 was generated by the PCR amplification of the triple-FLAG-tagged LRRK2 insert from pCHMWS-3flagLRRK2 (a kind gift from Jean-Marc Taymans, Katholieke Universiteit Leuven, Belgium) and insertion into pRK5. pEGFP-N1-Fzd1, -Fzd4 and -Fzd5 (74) were kindly provided by Yosuke Funato and Hiroaki Miki (Osaka University, Osaka, Japan). TOPflash and FOPflash were obtained from Addgene; pTK-RL is from Promega. To make pRK5-LRP6-2xHA, human LRP6 was amplified from cDNA using primers designed to introduce two HA epitopes at the LRP6 C terminus and cloned into the pRK5 vector. pYTH16-LRP6-ICD was made by PCR cloning the intracellular domain of LRP6 (amino acids 1416–1613) and insertion into the pYTH16 vector in frame with the Gal4 DNA-binding domain. All constructs were verified by DNA sequencing.
siRNAs were synthesized by Eurofins and designed to recognize published sequences. siRNAs used were as follows: axin CGAGAGCCAUCUACCGAAA (75); GFP GCUACGUCCAGGAGCGCAC (Eurofins); LRRK1#A GGAAUCACUCACUGACUAC (76); LRRK1#B CAGAGAUUCUUCCUUUAUA (76); LRRK2#1 CGUCGACUUAUACGUGUAA (77); LRRK2#2 GAAUUUCAUCAUAAGCUAA (77); LRRK2#3 AUUAUUCUCUCCUCUUGUA*; Fz5 UUGUAAUCCAUGCAGAGGA (75); luciferase GL3 CUUACGCUGAGUACUUCGA (Eurofins); non-specific control AGGUAGUGUAAUCGCCUUG (Eurofins). The asterisk denotes the previously undescribed siRNA targeting the human LRRK2 3′-untranslated region.
Cell culture and transfection
All mammalian cells were grown at 37°C in 5% CO2. Cells for immunofluorescence experiments were seeded onto poly-d-lysine-coated coverslips. HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) plus 100 U/ml penicillin G and 100 μg/ml streptomycin and were transfected using Lipofectamine LTX (Invitrogen) at a 2:1 μl transfection reagent to μg DNA ratio. SH-SY5Y cells were cultured in a 1:1 mixture of DMEM and F12 Ham's media supplemented with 10% FBS plus penicillin and streptomycin and were transfected using FuGENE 6 (Roche) at a 2.5:1 μl transfection reagent to μg DNA ratio.
Immunofluorescence staining and imaging
Cells were transfected with 500 ng of the relevant plasmid per coverslip. Forty-eight hours post-transfection, cells were fixed with 4% (w/v) paraformaldehyde and stained with antibodies to myc and FLAG (Sigma) or HA (Covance) as described previously (21). Alexa-488, -546 and -633 conjugated secondary antibodies were from Invitrogen. 4’,6-diamidino-2-phenylindole stain (Invitrogen) was performed at a 300 nm concentration for 5 min. Confocal microscopy was performed using a Zeiss LSM 710, with an AxioObserver.Z1 microscope. Fluorescence excited by a 30 mW 405 nm diode laser, a 25 mW Argon ion laser (488 nm line), a 25 mW 565 nm diode laser and a 5 mW 633 nm HeNe laser was detected separately. All images were taken at room temperature with a 63× objective with immersion oil, using Zen software.
YTH assays
Experiments were performed as described previously (21), except that the Y190 strain (Clontech) was used, and cells were grown in the presence of 10 mm 3-amino-1,2,4-triazole. For quantitative experiments, sample means were calculated from at least three replicates per experiment.
Luciferase assays
Unless indicated otherwise, TOPflash assays were performed in human dopaminergic SH-SY5Y cells. Except for siRNA-mediated knockdown experiments, assays were performed in a 6-well plate format, using three wells per experimental condition. Five hundred nanograms of pRK5-FLAG-DVL plasmids was used per well in Figure 2A; in subsequent experiments, a mixture of 200 ng of DVL and 300 ng of pRK5-FLAG was used. All other plasmid DNAs were used at 500 ng/well, except pRL-TK (50 ng). siRNA experiments were performed in 24-well plates using 100 ng of TOPflash or FOPflash and 10 ng of pRL-TK, plus 10 pmol of each siRNA per well. Cells were extracted 24 h post-transfection using Passive Lysis Buffer (Promega). Luciferase assays were performed using a Dual Luciferase Reporter Assay kit (Promega) and Turner Instruments 20/20 luminometer. Values obtained (relative luciferase units) are the ratio of luciferase and Renilla from each well, to adjust for variations in transfection efficiency.
Biochemistry of mouse brains
Wild-type male CD1 mice for co-immunoprecipitation experiments were obtained in house. Brains were snap-frozen in liquid nitrogen immediately after removal and stored at −80°C prior to extraction. To isolate mouse brain cytosol, brains were first homogenized into Tris-buffered saline [50 mm Tris, pH 7.5, 150 mm NaCl supplemented with 1× complete protease inhibitor cocktail (Roche) and 1× Halt phosphatase inhibitor cocktail (Pierce)] using a dounce homogenizer and then clarified by centrifugation at 14 000g for 10 min. All steps were performed at 4°C. Anti-LRRK2 immunoprecipitation was performed on cytosolic extracts, using a Dynabeads Co-immunoprecipitation kit (Invitrogen) coupled with rabbit anti-LRRK2 antibody (MJFF2; Epitomics) or non-specific rabbit IgG (Sigma).
Biochemistry of in vitro cell lines
In vitro biochemistry was performed in HEK293 cells due to ease of transfection and because these cells express endogenous LRRK2 to a much greater degree than SH-SY5Y cells. Cells were grown in 10 cm dishes and, where indicated, transfected with 4 μg of each plasmid 36 h prior to lysis. Wnt3a treatment was performed at a final concentration of 100 ng/ml for the indicated incubation time, using recombinant human Wnt3a protein (R&D Systems). For Co-IP, cells were extracted in a buffer containing 20 mm Tris, pH 7.5, 50 mm NaCl, 2 mm ethylenediaminetetraacetic acid, 1% Triton X-100 and 1× complete protease inhibitor cocktail (Roche). Lysates were clarified by centrifugation at 14 000g for 10 min at 4°C, and supernatants retained. For Co-IP, 65 μl was retained as whole cell lysate, the remainder was adjusted to 250 mm NaCl, and 40 μl anti-FLAG-M2 affinity gel added, prior to incubation at 4°C with rotation. After 4 h beads were washed four times in extraction buffer containing 250 mm NaCl and bound protein eluted by incubation with 15 ng/μl 3xFLAG peptide (Sigma) for 30 min at room temperature. For membrane translocation studies, cells were extracted according to the published protocols (78). The relative localization of LRRK2 to membrane fractions was determined as the ratio of LRRK2 to Calnexin or to membrane-bound Rab5b in each sample, with images acquired from the same gel.
Western blot
Eluates and cell lysate samples were denatured by the addition of 4× LDS sample loading buffer and 10× sample reducing agent (both Invitrogen) followed by heating to 99°C for 10 min. Samples were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis using 4–12% Bis–Tris pre-cast gels (Invitrogen) and transferred to polyvinylidene difluoride membrane (Millipore). Membranes were rinsed in PBS containing 0.1% Tween-20 (PBS-T), and then blocked by incubation for 30 min in PBS-T containing 5% non-fat dry milk powder. Primary antibody incubations were performed overnight at 4°C with gentle shaking, with all antibodies diluted in blocking buffer to the following concentrations: anti-myc, anti-FLAG (both Sigma) 1:2000; anti-GFP (AbCam) 1:3000; anti-HA (Covance) 1:5000; anti-actin (Sigma) 1:6000. Horseradish peroxidase-conjugated secondary antibodies (1:2000 in blocking buffer; Santa Cruz) were incubated with membranes for 1 h at room temperature. Protein bands were visualized using SuperSignal West Pico Chemiluminescent Substrate (Pierce) and images acquired using a GeneGnome imager (Syngene). Quantification was performed using GeneTools analysis software (Syngene).
Statistical analysis
LRRK2 membrane localization (Figs. 5B and C; n = 4) and quantitative YTH assays (Fig. 6E) were analyzed by paired Student's t-test with two-tailed distribution. Statistical significance of luciferase assays was performed by unpaired t-test with two-tailed distribution. In all cases, combined data from a minimum of three independent experiments was used. In all cases, P-values are indicated, with statistically significant values (P < 0.05) highlighted in bold.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by The Wellcome Trust (WT088145AIA, WT095010MA to K.H.), The Michael J. Fox Foundation (to D.C.B. and K.H.), a Vera Down British Medical Association Research Grant (to K.H.) and an Ibercaja Obra Social award (to Marian Blanca Ramirez). Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
ACKNOWLEDGEMENTS
K.H. and D.C.B. thank Marian Blanca Ramirez for technical assistance; Professor Robert Harvey for useful discussions and critical reading of this manuscript and Dr Ana Antunes-Martins for critical reading.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Gasser T. Mendelian forms of Parkinson's disease. Biochim. Biophys. Acta. 2009;1792:587–596. doi: 10.1016/j.bbadis.2008.12.007. doi:10.1016/j.bbadis.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 2.Kumari U., Tan E.K. LRRK2 in Parkinson's disease: genetic and clinical studies from patients. FEBS J. 2009;276:6455–6463. doi: 10.1111/j.1742-4658.2009.07344.x. doi:10.1111/j.1742-4658.2009.07344.x. [DOI] [PubMed] [Google Scholar]
- 3.Parkinson J. An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 2002;14:223–236. doi: 10.1176/jnp.14.2.223. discussion 222 doi:10.1176/appi.neuropsych.14.2.223. [DOI] [PubMed] [Google Scholar]
- 4.Berwick D.C., Harvey K. LRRK2 signaling pathways: the key to unlocking neurodegeneration. Trends Cell Biol. 2011;21:257–265. doi: 10.1016/j.tcb.2011.01.001. doi:10.1016/j.tcb.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Greggio E., Cookson M.R. Leucine-rich repeat kinase 2 mutations and Parkinson's disease: three questions. ASN Neuro. 2009;1:pii:e00002. doi: 10.1042/AN20090007. , [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maiese K., Li F., Chong Z.Z., Shang Y.C. The Wnt signaling pathway: aging gracefully as a protectionist? Pharmacol. Ther. 2008;118:58–81. doi: 10.1016/j.pharmthera.2008.01.004. doi:10.1016/j.pharmthera.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacDonald B.T., Tamai K., He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. doi:10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Amerongen R., Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–3214. doi: 10.1242/dev.033910. doi:10.1242/dev.033910. [DOI] [PubMed] [Google Scholar]
- 9.Taelman V.F., Dobrowolski R., Plouhinec J.L., Fuentealba L.C., Vorwald P.P., Gumper I., Sabatini D.D., De Robertis E.M. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143:1136–1148. doi: 10.1016/j.cell.2010.11.034. doi:10.1016/j.cell.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Avila M.E., Sepúlveda F.J., Burgos C.F., Moraga-Cid G., Parodi J., Moon R.T., Aguayo L.G., Opazo C., De Ferrari G.V. Canonical Wnt3a modulates intracellular calcium and enhances excitatory neurotransmission in hippocampal neurons. J. Biol. Chem. 2010;285:18939–18947. doi: 10.1074/jbc.M110.103028. doi:10.1074/jbc.M110.103028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cerpa W., Godoy J.A., Alfaro I., Farías G.G., Metcalfe M.J., Fuentealba R., Bonansco C., Inestrosa N.C. Wnt-7a modulates the synaptic vesicle cycle and synaptic transmission in hippocampal neurons. J. Biol. Chem. 2008;283:5918–5927. doi: 10.1074/jbc.M705943200. doi:10.1074/jbc.M705943200. [DOI] [PubMed] [Google Scholar]
- 12.Cerpa W., Gambrill A., Inestrosa N.C., Barria A. Regulation of NMDA-receptor synaptic transmission by Wnt signaling. J. Neurosci. 2011;31:9466–9471. doi: 10.1523/JNEUROSCI.6311-10.2011. doi:10.1523/JNEUROSCI.6311-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ciani L., Boyle K.A., Dickins E., Sahores M., Anane D., Lopes D.M., Gibb A.J., Salinas P.C. Wnt7a signaling promotes dendritic spine growth and synaptic strength through Ca2+/calmodulin-dependent protein kinase II. Proc. Natl Acad. Sci. USA. 2011;108:10732–10737. doi: 10.1073/pnas.1018132108. doi:10.1073/pnas.1018132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall A.C., Lucas F.R., Salinas P.C. Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT-7a signaling. Cell. 2000;100:525–535. doi: 10.1016/s0092-8674(00)80689-3. doi:10.1016/S0092-8674(00)80689-3. [DOI] [PubMed] [Google Scholar]
- 15.Inestrosa N.C., Arenas E. Emerging roles of Wnts in the adult nervous system. Nat. Rev. Neurosci. 2010;11:77–86. doi: 10.1038/nrn2755. doi:10.1038/nrn2755. [DOI] [PubMed] [Google Scholar]
- 16.Varela-Nallar L., Alfaro I.E., Serrano F.G., Parodi J., Inestrosa N.C. Wingless-type family member 5A (Wnt-5a) stimulates synaptic differentiation and function of glutamatergic synapses. Proc. Natl Acad. Sci. USA. 2010;107:21164–21169. doi: 10.1073/pnas.1010011107. doi:10.1073/pnas.1010011107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wisniewska M.B., Misztal K., Michowski W., Szczot M., Purta E., Lesniak W., Klejman M.E., Dabrowski M., Filipkowski R.K., Nagalski A., et al. LEF1/beta-catenin complex regulates transcription of the Cav3.1 calcium channel gene (Cacna1g) in thalamic neurons of the adult brain. J. Neurosci. 2010;30:4957–4969. doi: 10.1523/JNEUROSCI.1425-09.2010. doi:10.1523/JNEUROSCI.1425-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abe K., Takeichi M. NMDA-receptor activation induces calpain-mediated beta-catenin cleavages for triggering gene expression. Neuron. 2007;53:387–397. doi: 10.1016/j.neuron.2007.01.016. doi:10.1016/j.neuron.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 19.Hooper C., Killick R., Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J. Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. doi:10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rawal N., Corti O., Sacchetti P., Ardilla-Osorio H., Sehat B., Brice A., Arenas E. Parkin protects dopaminergic neurons from excessive Wnt/beta-catenin signaling. Biochem. Biophys. Res. Commun. 2009;388:473–478. doi: 10.1016/j.bbrc.2009.07.014. doi:10.1016/j.bbrc.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 21.Sancho R.M., Law B.M., Harvey K. Mutations in the LRRK2 Roc-COR tandem domain link Parkinson's disease to Wnt signalling pathways. Hum. Mol. Genet. 2009;18:3955–3968. doi: 10.1093/hmg/ddp337. doi:10.1093/hmg/ddp337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin C.H., Tsai P.I., Wu R.M., Chien C.T. LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3β. J. Neurosci. 2010;30:13138–13149. doi: 10.1523/JNEUROSCI.1737-10.2010. doi:10.1523/JNEUROSCI.1737-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bryja V., Gradl D., Schambony A., Arenas E., Schulte G. Beta-arrestin is a necessary component of Wnt/beta-catenin signaling in vitro and in vivo. Proc. Natl Acad. Sci. USA. 2007;104:6690–6695. doi: 10.1073/pnas.0611356104. doi:10.1073/pnas.0611356104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor J.P., Hulihan M.M., Kachergus J.M., Melrose H.L., Lincoln S.J., Hinkle K.M., Stone J.T., Ross O.A., Hauser R., Aasly J., Gasser T., Payami H., Wszolek Z.K., Farrer M.J. Leucine-rich repeat kinase 1: a paralog of LRRK2 and a candidate gene for Parkinson's disease. Neurogenetics. 2007;8:95–102. doi: 10.1007/s10048-006-0075-8. doi:10.1007/s10048-006-0075-8. [DOI] [PubMed] [Google Scholar]
- 25.Haugarvoll K., Toft M., Ross O.A., White L.R., Aasly J.O., Farrer M.J. Variants in the LRRK1 gene and susceptibility to Parkinson's disease in Norway. Neurosci. Lett. 2007;18:299–301. doi: 10.1016/j.neulet.2007.02.020. doi:10.1016/j.neulet.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 26.Greggio E., Lewis P.A., van der Brug M.P., Ahmad R., Kaganovich A., Ding J., Beilina A., Baker A.K., Cookson M.R. Mutations in LRRK2/dardarin associated with Parkinson disease are more toxic than equivalent mutations in the homologous kinase LRRK1. J. Neurochem. 2007;102:93–102. doi: 10.1111/j.1471-4159.2007.04523.x. doi:10.1111/j.1471-4159.2007.04523.x. [DOI] [PubMed] [Google Scholar]
- 27.Sutton L.P., Honardoust D., Mouyal J., Rajakumar N., Rushlow W.J. Activation of the canonical Wnt pathway by the antipsychotics haloperidol and clozapine involves dishevelled-3. J. Neurochem. 2007;102:153–169. doi: 10.1111/j.1471-4159.2007.04527.x. doi:10.1111/j.1471-4159.2007.04527.x. [DOI] [PubMed] [Google Scholar]
- 28.Veeman M.T., Slusarski D.C., Kaykas A., Louie S.H., Moon R.T. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr. Biol. 2003;13:680–685. doi: 10.1016/s0960-9822(03)00240-9. doi:10.1016/S0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- 29.Axelrod J.D., Miller J.R., Shulman J.M., Moon R.T., Perrimon N. Differential recruitment of Dishevelled provides signaling specificity in the planar cell polarity and Wingless signaling pathways. Genes Dev. 1998;12:2610–2622. doi: 10.1101/gad.12.16.2610. doi:10.1101/gad.12.16.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schultz A.M., Henderson L.E., Oroszlan S., Garber E.A., Hanafusa H. Amino terminal myristylation of the protein kinase p60src, a retroviral transforming protein. Science. 1985;227:427–429. doi: 10.1126/science.3917576. doi:10.1126/science.3917576. [DOI] [PubMed] [Google Scholar]
- 31.Kitten G.T., Nigg E.A. The CaaX motif is required for isoprenylation, carboxyl methylation, and nuclear membrane association of lamin B2. J. Cell Biol. 1991;113:13–23. doi: 10.1083/jcb.113.1.13. doi:10.1083/jcb.113.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng X., Dzamko N., Prescott A., Davies P., Liu Q., Yang Q., Lee J.D., Patricelli M.P., Nomanbhoy T.K., Alessi D.R., et al. Characterization of a selective inhibitor of the Parkinson s disease kinase LRRK2. Nat. Chem. Biol. 2011;7:203–205. doi: 10.1038/nchembio.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alegre-Abarrategui J., Christian H., Lufino M.M., Mutihac R., Venda L.L., Ansorge O., Wade-Martins R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009;18:4022–4034. doi: 10.1093/hmg/ddp346. doi:10.1093/hmg/ddp346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanafusa H., Ishikawa K., Kedashiro S., Saigo T., Iemura S., Natsume T., Komada M., Shibuya H., Nara A., Matsumoto K. Leucine-rich repeat kinase LRRK1 regulates endosomal trafficking of the EGF receptor. Nat. Commun. 2011;2:158. doi: 10.1038/ncomms1161. doi:10.1038/ncomms1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin N., Jeong H., Kwon J., Heo H.Y., Kwon J.J., Yun H.J., Kim C.H., Han B.S., Tong Y., Shen J., et al. LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 2008;314:2055–2065. doi: 10.1016/j.yexcr.2008.02.015. doi:10.1016/j.yexcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 36.Tong Y., Yamaguchi H., Giaime E., Boyle S., Kopan R., Kelleher R.J., Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl Acad. Sci. USA. 2010;107:9879–9884. doi: 10.1073/pnas.1004676107. doi:10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parr B.A., Shea M.J., Vassileva G., McMahon A.P. Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development. 1993;119:247–261. doi: 10.1242/dev.119.1.247. [DOI] [PubMed] [Google Scholar]
- 38.Oosterwegel M., van de Wetering M., Timmerman J., Kruisbeek A., Destree O., Meijlink F., Clevers H. Differential expression of the HMG box factors TCF-1 and LEF-1 during murine embryogenesis. Development. 1993;118:439–448. doi: 10.1242/dev.118.2.439. [DOI] [PubMed] [Google Scholar]
- 39.Patapoutian A., Reichardt L.F. Roles of Wnt proteins in neural development and maintenance. Curr. Opin. Neurobiol. 2000;10:392–399. doi: 10.1016/s0959-4388(00)00100-8. doi:10.1016/S0959-4388(00)00100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maretto S., Cordenonsi M., Dupont S., Braghetta P., Broccoli V., Hassan A.B., Volpin D., Bressan G.M., Piccolo S. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc. Natl Acad. Sci. USA. 2003;100:3299–3304. doi: 10.1073/pnas.0434590100. doi:10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen L., Zhou S., Glowacki J. Effects of age and gender on WNT gene expression in human bone marrow stromal cells. J. Cell Biochem. 2009;106:337–343. doi: 10.1002/jcb.22010. doi:10.1002/jcb.22010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castelo-Branco G., Wagner J., Rodriguez F.J., Kele J., Sousa K., Rawal N., Pasolli H.A., Fuchs E., Kitajewski J., Arenas E. Differential regulation of midbrain dopaminergic neuron development by Wnt-1, Wnt-3a, and Wnt-5a. Proc. Natl Acad. Sci. USA. 2003;100:12747–12752. doi: 10.1073/pnas.1534900100. doi:10.1073/pnas.1534900100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schulte G., Bryja V., Rawal N., Castelo-Branco G., Sousa K.M., Arenas E. Purified Wnt-5a increases differentiation of midbrain dopaminergic cells and dishevelled phosphorylation. J. Neurochem. 2005;92:1550–1553. doi: 10.1111/j.1471-4159.2004.03022.x. doi:10.1111/j.1471-4159.2004.03022.x. [DOI] [PubMed] [Google Scholar]
- 44.Rawal N., Castelo-Branco G., Sousa K.M., Kele J., Kobayashi K., Okano H., Arenas E. Dynamic temporal and cell type-specific expression of Wnt signaling components in the developing midbrain. Exp. Cell Res. 2006;312:1626–1636. doi: 10.1016/j.yexcr.2006.01.032. doi:10.1016/j.yexcr.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 45.Castelo-Branco G., Sousa K.M., Bryja V., Pinto L., Wagner J., Arenas E. Ventral midbrain glia express region-specific transcription factors and regulate dopaminergic neurogenesis through Wnt-5a secretion. Mol. Cell Neurosci. 2006;31:251–262. doi: 10.1016/j.mcn.2005.09.014. doi:10.1016/j.mcn.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 46.Thomas K.R., Capecchi M.R. Targeted disruption of the murine int-1 proto-oncogene resulting in severe abnormalities in midbrain and cerebellar development. Nature. 1990;346:847–850. doi: 10.1038/346847a0. doi:10.1038/346847a0. [DOI] [PubMed] [Google Scholar]
- 47.Castelo-Branco G., Andersson E.R., Minina E., Sousa K.M., Ribeiro D., Kokubu C., Imai K., Prakash N., Wurst W., Arenas E. Delayed dopaminergic neuron differentiation in Lrp6 mutant mice. Dev. Dyn. 2010;239:211–221. doi: 10.1002/dvdy.22094. [DOI] [PubMed] [Google Scholar]
- 48.Min C., Cho D.I., Kwon K.J., Kim K.S., Shin C.Y., Kim K.M. Novel regulatory mechanism of canonical Wnt signaling by dopamine D2 receptor through direct interaction with beta-catenin. Mol. Pharmacol. 2011;80:68–78. doi: 10.1124/mol.111.071340. doi:10.1124/mol.111.071340. [DOI] [PubMed] [Google Scholar]
- 49.Biskup S., Moore D.J., Celsi F., Higashi S., West A.B., Andrabi S.A., Kurkinen K., Yu S.W., Savitt J.M., Waldvogel H.J., et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. doi:10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- 50.Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., van der Brug M.P., Beilina A., Blackinton J., Thomas K.J., et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. doi:10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 51.Miklossy J., Arai T., Guo J.P., Klegeris A., Yu S., McGeer E.G., McGeer P.L. LRRK2 expression in normal and pathologic human brain and in human cell lines. J. Neuropathol. Exp. Neurol. 2006;65:953–963. doi: 10.1097/01.jnen.0000235121.98052.54. doi:10.1097/01.jnen.0000235121.98052.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Higashi S., Moore D.J., Colebrooke R.E., Biskup S., Dawson V.L., Arai H., Dawson T.M., Emson P.C. Expression and localization of Parkinson's disease-associated leucine-rich repeat kinase 2 in the mouse brain. J. Neurochem. 2007;100:368–381. doi: 10.1111/j.1471-4159.2006.04246.x. doi:10.1111/j.1471-4159.2006.04246.x. [DOI] [PubMed] [Google Scholar]
- 53.Melrose H.L., Kent C.B., Taylor J.P., Dachsel J.C., Hinkle K.M., Lincoln S.J., Mok S.S., Culvenor J.G., Masters C.L., Tyndall G.M., et al. A comparative analysis of leucine-rich repeat kinase 2 (Lrrk2) expression in mouse brain and Lewy body disease. Neuroscience. 2007;147:1047–1058. doi: 10.1016/j.neuroscience.2007.05.027. doi:10.1016/j.neuroscience.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 54.Milosevic J., Schwarz S.C., Ogunlade V., Meyer A.K., Storch A., Schwarz J. Emerging role of LRRK2 in human neural progenitor cell cycle progression, survival and differentiation. Mol. Neurodegener. 2009;4:25. doi: 10.1186/1750-1326-4-25. doi:10.1186/1750-1326-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.L'episcopo F., Serapide M.F., Tirolo C., Testa N., Caniglia S., Morale M.C., Pluchino S., Marchetti B. A Wnt1 regulated Frizzled-1/β-Catenin signaling pathway as a candidate regulatory circuit controlling mesencephalic dopaminergic neuron-astrocyte crosstalk: therapeutical relevance for neuron survival and neuroprotection. Mol. Neurodegener. 2011;6:49. doi: 10.1186/1750-1326-6-49. doi:10.1186/1750-1326-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.L'Episcopo F., Tirolo C., Testa N., Caniglia S., Morale M.C., Cossetti C., D'Adamo P., Zardini E., Andreoni L., Ihekwaba A.E., et al. Reactive astrocytes and Wnt/β-catenin signaling link nigrostriatal injury to repair in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Neurobiol. Dis. 2011;41:508–527. doi: 10.1016/j.nbd.2010.10.023. doi:10.1016/j.nbd.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.L'Episcopo F., Tirolo C., Testa N., Caniglia S., Morale M.C., Deleidi M., Serapide M.F., Pluchino S., Marchetti B. Plasticity of subventricular zone neuroprogenitors in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) mouse model of Parkinson's disease involves cross talk between inflammatory and Wnt/β-catenin signaling pathways: functional consequences for neuroprotection and repair. J. Neurosci. 2012;32:2062–2085. doi: 10.1523/JNEUROSCI.5259-11.2012. doi:10.1523/JNEUROSCI.5259-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rosi M.C., Luccarini I., Grossi C., Fiorentini A., Spillantini M.G., Prisco A., Scali C., Gianfriddo M., Caricasole A., Terstappen G.C., et al. Increased Dickkopf-1 expression in transgenic mouse models of neurodegenerative disease. J. Neurochem. 2010;112:1539–1551. doi: 10.1111/j.1471-4159.2009.06566.x. doi:10.1111/j.1471-4159.2009.06566.x. [DOI] [PubMed] [Google Scholar]
- 59.Brault V., Moore R., Kutsch S., Ishibashi M., Rowitch D.H., McMahon A.P., Sommer L., Boussadia O., Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 60.Carpenter A.C., Rao S., Wells J.M., Campbell K., Lang R.A. Generation of mice with a conditional null allele for Wntless. Genesis. 2010;48:554–558. doi: 10.1002/dvg.20651. doi:10.1002/dvg.20651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parish C.L., Castelo-Branco G., Rawal N., Tonnesen J., Sorensen A.T., Salto C., Kokaia M., Lindvall O., Arenas E. Wnt5a-treated midbrain neural stem cells improve dopamine cell replacement therapy in parkinsonian mice. J. Clin. Invest. 2008;118:149–160. doi: 10.1172/JCI32273. doi:10.1172/JCI32273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wiedau-Pazos M., Wong E., Solomon E., Alarcon M., Geschwind D.H. Wnt-pathway activation during the early stage of neurodegeneration in FTDP-17 mice. Neurobiol. Aging. 2009;30:14–21. doi: 10.1016/j.neurobiolaging.2007.05.015. doi:10.1016/j.neurobiolaging.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cerpa W., Dinamarca M.C., Inestrosa N.C. Structure-function implications in Alzheimer's disease: effect of Abeta oligomers at central synapses. Curr. Alzheimer Res. 2008;5:233–243. doi: 10.2174/156720508784533321. doi:10.2174/156720508784533321. [DOI] [PubMed] [Google Scholar]
- 64.Ciani L., Krylova O., Smalley M.J., Dale T.C., Salinas P.C. A divergent canonical WNT-signaling pathway regulates microtubule dynamics: dishevelled signals locally to stabilize microtubules. J. Cell Biol. 2004;164:243–253. doi: 10.1083/jcb.200309096. doi:10.1083/jcb.200309096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ciani L., Salinas P.C. c-Jun N-terminal kinase (JNK) cooperates with Gsk3beta to regulate dishevelled-mediated microtubule stability. BMC Cell Biol. 2007;8:27. doi: 10.1186/1471-2121-8-27. doi:10.1186/1471-2121-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Purro S.A., Ciani L., Hoyos-Flight M., Stamatakou E., Siomou E., Salinas P.C. Wnt regulates axon behavior through changes in microtubule growth directionality: a new role for adenomatous polyposis coli. J. Neurosci. 2008;28:8644–8654. doi: 10.1523/JNEUROSCI.2320-08.2008. doi:10.1523/JNEUROSCI.2320-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Votin V., Nelson W.J., Barth A.I. Neurite outgrowth involves adenomatous polyposis coli protein and beta-catenin. J. Cell Sci. 2005;118:5699–5708. doi: 10.1242/jcs.02679. doi:10.1242/jcs.02679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Melrose H.L., Dächsel J.C., Behrouz B., Lincoln S.J., Yue M., Hinkle K.M., Kent C.B., Korvatska E., Taylor J.P., Witten L., et al. Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol. Dis. 2010;40:503–517. doi: 10.1016/j.nbd.2010.07.010. doi:10.1016/j.nbd.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mandelkow E.M., Drewes G., Biernat J., Gustke N., Van Lint J., Vandenheede J.R., Mandelkow E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314:315–321. doi: 10.1016/0014-5793(92)81496-9. doi:10.1016/0014-5793(92)81496-9. [DOI] [PubMed] [Google Scholar]
- 70.Ishihara L., Warren L., Gibson R., Amouri R., Lesage S., Dürr A., Tazir M., Wszolek Z.K., Uitti R.J., Nichols W.C., et al. Clinical features of Parkinson disease patients with homozygous leucine-rich repeat kinase 2 G2019S mutations. Arch. Neurol. 2006;63:1250–1254. doi: 10.1001/archneur.63.9.1250. doi:10.1001/archneur.63.9.1250. [DOI] [PubMed] [Google Scholar]
- 71.Rajput A., Dickson D.W., Robinson C.A., Ross O.A., Dächsel J.C., Lincoln S.J., Cobb S.A., Rajput M.L., Farrer M.J. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67:1506–1508. doi: 10.1212/01.wnl.0000240220.33950.0c. doi:10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- 72.Hassin-Baer S., Laitman Y., Azizi E., Molchadski I., Galore-Haskel G., Barak F., Cohen O.S., Friedman E. The leucine rich repeat kinase 2 (LRRK2) G2019S substitution mutation. Association with Parkinson disease, malignant melanoma and prevalence in ethnic groups in Israel. J. Neurol. 2009;256:483–487. doi: 10.1007/s00415-009-0117-x. doi:10.1007/s00415-009-0117-x. [DOI] [PubMed] [Google Scholar]
- 73.Saunders-Pullman R., Barrett M.J., Stanley K.M., Luciano M.S., Shanker V., Severt L., Hunt A., Raymond D., Ozelius L.J., Bressman S.B. LRRK2 G2019S mutations are associated with an increased cancer risk in Parkinson disease. Mov. Disord. 2010;25:2536–2541. doi: 10.1002/mds.23314. doi:10.1002/mds.23314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Terabayashi T., Funato Y., Fukuda M., Miki H. A coated vesicle-associated kinase of 104 kDa (CVAK104) induces lysosomal degradation of frizzled 5 (Fzd5) J. Biol. Chem. 2009;284:26716–26724. doi: 10.1074/jbc.M109.039313. doi:10.1074/jbc.M109.039313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pan W., Choi S.C., Wang H., Qin Y., Volpicelli-Daley L., Swan L., Lucast L., Khoo C., Zhang X., Li L., et al. Wnt3a-mediated formation of phosphatidylinositol 4,5-bisphosphate regulates LRP6 phosphorylation. Science. 2008;321:1350–1353. doi: 10.1126/science.1160741. doi:10.1126/science.1160741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Korr D., Toschi L., Donner P., Pohlenz H.D., Kreft B., Weiss B. LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell Signal. 2006;18:910–920. doi: 10.1016/j.cellsig.2005.08.015. doi:10.1016/j.cellsig.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 77.Häbig K., Walter M., Poths S., Riess O., Bonin M. RNA interference of LRRK2-microarray expression analysis of a Parkinson's disease key player. Neurogenetics. 2008;9:83–94. doi: 10.1007/s10048-007-0114-0. doi:10.1007/s10048-007-0114-0. [DOI] [PubMed] [Google Scholar]
- 78.Holden P., Horton W.A. Crude subcellular fractionation of cultured mammalian cell lines. BMC Res. Notes. 2009;2:243. doi: 10.1186/1756-0500-2-243. doi:10.1186/1756-0500-2-243. [DOI] [PMC free article] [PubMed] [Google Scholar]