

Abstract
Sonic hedgehog (SHh) signaling is important in the pathogenesis of various human cancers, such as medulloblastomas, and it has been identified as a valid target for anti-cancer therapeutics. The SHh inhibitor cyclopamine induces apoptosis. The bioactive sphingolipid ceramide mediates cell death in response to various chemotherapeutic agents; however, ceramide’s roles/mechanisms in cyclopamine-induced apoptosis are unknown. Here, we report that cyclopamine mediates ceramide generation selectively via induction of N-SMase2 expression in Daoy human medulloblastoma cells. Importantly, siRNA-mediated knockdown of N-SMase2 prevented cyclopamine-induced ceramide generation, and protected Daoy cells from drug-induced apoptosis. Accordingly, ectopic expression of wild type N-SMase2 caused cell death, compared to controls, expressing the catalytically inactive N-SMase2 mutant. Interestingly, knockdown of smoothened (Smo), a target protein for cyclopamine, or Gli1, a down-stream signaling transcription factor of Smo, did not affect N-SMase2 expression, or apoptosis. Mechanistically, our data showed cyclopamine induced N-SMase2 mRNA and cell death selectively via increased nitric oxide (NO) generation by neuronal-nitric oxide synthase (n-NOS) induction, in Daoy medulloblastoma, and multiple other human cancer cell lines. Accordingly, N-SMase2 activity-deficient skin fibroblasts isolated from homozygous fro/fro (fragilitas ossium) mice exhibited resistance to NO-induced cell death. Thus, our data suggest a novel off-target function of cyclopamine in inducing apoptosis, at least in part, by Nnos/NO-dependent induction of N-SMase2 expression/ceramide axis, independent of Smo/Gli inhibition.
Keywords: sphingolipids, ceramide, sphingomyelin, sphingomyelinase, neutral sphingomyelinase
Introduction
The Sonic hedgehog (SHh) signaling pathway plays a critical role in normal cerebellar development and has been implicated in the pathogenesis of medulloblastoma and other cancers of the brain, prostate, lung, breast, and colon (1–3). SHh is regulated by the transmembrane receptor patched (Ptch), which when altered or mutated, results in SHh pathway activation and cell growth dysregulation (4–6). The SHh ligand binds Ptch, which then alleviates Ptch-mediated suppression of Smo, which activates Gli, a family of transcription factors involved in the regulation of numerous genes controlling cell division, growth, and/or apoptosis, leading to proliferation and/or inhibition of cell death (7–8). Thus, SHh is a novel therapeutic target for the treatment of cancers, including brain tumors (9–10). Cyclopamine (Fig. 1A, top) is a Smo antagonist, which inhibits growth, and induces apoptosis in various cancers cells including medulloblastoma (11–15). However, off-target functions of cyclopamine in inducing apoptosis, independent of Smo/SHh inhibition, have not been clearly defined previously.
Fig. 1. Cyclopamine induces cell death via caspase-3 activation and loss of mitochondrial membrane potential in Daoy cells.

(A) Effects of increasing concentrations of cyclopamine at 0, 2.5, 5, 10, 25, and 50μg/mL (24–48 hours) on cell viability were measured by MTT assay. Activation of caspase-3 (B) or loss of mitochondrial membrane potential (C) in response to cyclopamine (10μg/mL, 24 hours), as compared with controls, were measured. (D) Effects of cyclopamine on total ceramide accumulation were measured by LC/MS-MS, normalized to inorganic phosphate (Pi).
The bioactive ceramide is a precursor for the synthesis of more complex sphingolipids via multiple pathways (16–18). Stress-induced ceramide generation in response to various stimuli, such as anti-cancer agents, mediates cell cycle arrest, growth inhibition, and/or apoptosis (19). Ceramide is generated mainly via de novo synthesis by ceramide synthases 1-6 (CerS1-6) (20), or via hydrolysis of sphingomyelin (SM) by SMases with pH optima in acidic, neutral, or alkaline conditions (21). Activation of neutral (N)-SMases1-2 in response to chemotherapy has been reported to generate ceramide, thereby inducing cell death in various cancer cells and models (22–24). However, whether cyclopamine induces ceramide generation has not been described previously.
Therefore, our focus was to define roles and mechanisms of cyclopamine-induced apoptosis, and determine if this occurs, at least in part, via induction of ceramide generation by inhibition of SHh/Smo-dependent or SHh/Smo-independent manner in Daoy human medulloblastoma and UM-SCC-14 or UM-SCC-1 human head and neck squamous cell carcinoma (HNSCC) cells.
Materials and Methods
Cell culture
The Daoy medulloblastoma line (American Type Culture Collection) was grown in minimum essential medium with 10% FBS and 1% penicillin/streptomycin. WT, +/fro, and activity-deficient fro/fro skin fibroblasts isolated from new born mice (25) were cultured in Dulbecco’s Modified Eagle’s Medium with 10% FBS and 1% penicillin/streptomycin. UM-SCC-1 and UM-SCC-14A cells obtained from Dr. Thomas Carey (University of Michigan). Cell lines used in this study were not authenticated. Cells were treated at final concentrations of 5 to 20 μg/mL from cyclopamine stock solution (20 mg/mL; LC Laboratories) dissolved in 100% ethanol. Cyclopamine aliquots were dissolved at 55°C.
Measurement of ceramide by LC/MS
Endogenous ceramides were measured using liquid chromatography/tandem mass spectrometry (LC/MS-MS) as described (26).
Short interfering RNA and plasmids
Short interfering RNA’s (siRNA) for nSMase1 and nSMase2 were obtained from Ambion (Applied Biosystems). Gli1 siRNA and SMO siRNAs were custom designed by Qiagen and Invitrogen, respectively. Non-targeting (scrambled, SCR) siRNA #2 was obtained from Dharmacon. Transfections (40nmol/L, 48 hours) were carried out with DharmaFECT as described by the manufacturer. Knockdown of inducible NO synthase (iNOS) and neuronal NO synthase (nNOS) was carried out with siRNAs obtained from Dharmacon. Plasmids for wild-type (wt) and mutant N-SMase2, containing V5 tags were used as described (27).
Detection of cell death
Cell death was measured by various techniques: (i) activation of caspase-3, measured by fluorometry using a caspase-3 activity assay kit (R&D Systems); (ii) loss of mitochondrial membrane potential, detected by flow cytometry using the JC-1 mitochondrial membrane potential detection kit (Cell Technology); (iii) depletion of cellular ATP, measured by the ATP colorimetric/fluorometric assay kit (Abcam); and (iv) detection of Annexin V-aminoactinomycin D (7-AAD) staining, measured by flow cytometry using the BD Pharmingen PE Annexin V Apoptosis Detection Kit, as described by the manufacturers. Growth Inhibition was measured using MTT and trypan blue assay.
Measurement of SMase activity
N-SMase and acid (A)-SMase activities were assayed in vitro as described previously, utilizing 14C-[methyl]-SM as substrate (27).
Detection of NO by flow cytometry
Treated and untreated cells were incubated with fluorescent dyes (1 hour). Cells were trypsinsized, washed with PBS, and stained with 7AAD, before flow cytometry analysis on BD FACScan (BD Biosciences). For hCatalase transduced cells, before addition of CD34 antibody, cells were blocked in 30% human AB-serum (Gemini) for 30 minutes, preventing background staining. Fluorochrome-labeled antibodies were added, and cells were stained with 7AAD, and flow cytometry analysis was conducted with the BD FACScan (BD Biosciences). Data for viable (7-AAD) cells were analyzed and visualized by the FlowJo software (Tree Star).
Statistical analysis
Data are represented as mean ± SEM, unless otherwise indicated. Data represent at least 2 independent trials carried out as duplicates. Error bars on graphs represent SDs. An unpaired Student t test was carried out with Prism/GraphPad software; P<0.05 was considered significant (28).
Details of chemicals, RNA isolation, Q-PCR, Western blotting, target sequences of N-SMase1 and N-SMase2 siRNAs, catalase expression and activity assays can be found in Supplementary Materials and Methods in Molecular Cancer Therapeutics on line.
Results
Cyclopamine induces cell death and increases ceramide generation/accumulation
Cyclopamine (Fig. 1A, top), has shown some efficacy against desmoplastic medulloblastomas in preclinical and clinical studies (29–31). To confirm cyclopamine induces cell death, we treated Daoy human desmoplastic cerebellar medulloblastoma cells with increasing concentrations of cyclopamine (0–50μg/ml), and examined its effects on cell growth and cell death; measuring survival, caspase-3 activity, and loss of mitochondrial membrane potential. Cyclopamine treatment inhibited growth in a dose-dependent manner (IC50 ~5μg/ml, 48 hours, and ~10μg/ml, 24 hours) compared with vehicle-treated controls (Fig. 1A, bottom). Accordingly, cyclopamine increased caspase-3 activity around 2-fold, which was consistent with a loss of mitochondrial membrane potential, as measured by increased accumulation of cytoplasmic JC-1 (~8-fold), compared with controls (Fig. 1B and C, respectively). Pre-treatment with z-VAD (10μg/ml) almost completely prevented caspase-3 activation and loss of mitochondrial membrane potential in response to cyclopamine treatment in these cells (Figs. 1B and C, respectively). Thus, these data are consistent with previous studies, demonstrating that cyclopamine induces caspase-3-dependent mitochondrial apoptosis.
Next, we investigated whether cyclopamine-induced apoptosis is mediated by induction of ceramide via Smo-signaling inhibition, or off-target functions of cyclopamine, independent of SHh/Smo inhibition. Cyclopamine (5 or 10μg/mL, 24 hours) increased total ceramide approximately 2.5- or 3-fold, respectively, increasing total ceramide from 20 (in controls) to 50 to 60pmol/nmol Pi cyclopamine-treated cells, respectively (Fig. 1D). There were no significant changes in sphingosine or S1P (and dihydro-sphingosine or dihydro-S1P) in response to cyclopamine (data not shown). Moreover, cyclopamine (10μg/ml, 24 hours) induced C14-, C16-, C18-, C20-, and C22-ceramide generation ~2.5-, 3.5-, 15-, 6-, or 6.5-fold, respectively, compared to vehicle-treated controls (Supplementary Fig. S1A). Comparable increases were also observed with dihydro-C14-C22-ceramides (Supplementary Fig. S1B). Similar data were also obtained when cells were treated with 5μg/mL cyclopamine (24 hours), which increased endogenous C14-C22-ceramide generation compared to controls (Supplementary Fig. S1A). Thus, these data suggest that cyclopamine induces endogenous ceramide generation, consistent with its proapoptotic effects in Daoy cells.
Cyclopamine-mediated ceramide generation is dependent on nSMase2 induction
To assess whether de novo generation of ceramide plays a role in cyclopamine-induced apoptosis, we pretreated cells fumonisin B1 (FB1) and myriocin (MYR) at 50μmol/L and 50nmol/L, respectively, and examined cyclopamine-induced caspase-3 activation in Daoy cells (10μg/ml, 24 hours). FB1 or MYR did not prevent cyclopamine-induced caspase-3 activation (Supplementary Fig. S2A), indicating de novo generation of ceramide might not be involved in cyclopamine-induced apoptosis. We also examined the effects of siRNA-mediated knockdown of CerS1 on growth inhibition in response to cyclopamine. CerS1 is known to mainly generate C18-ceramide, and cyclopamine induced C18-ceramide generation approximately 15-fold compared with controls in Daoy cells (see Supplementary Fig. S1A). Downregulation of CerS1 with siRNAs did not protect cells from cyclopamine-induced growth inhibition compared with controls, which were transfected with non-targeting scrambled (SCR) siRNAs (Supplementary Fig. S2B). Effectiveness of siRNAs for knockdown of CerS1 in Daoy cells was confirmed with qPCR. An approximately 40–60% decrease in CerS1 was observed compared to controls in the absence/presence of cyclopamine (Supplementary Fig. S2C). Similarly, ectopic expression of wt CerS1, or its catalytically inactive form with the H122A mutation (32), did not enhance or prevent the growth inhibitory effects of cyclopamine (Supplementary Fig. S2D). Expression of wt and mutant CerS1-FLAG proteins was confirmed by Western blotting using anti-FLAG antibody compared to vector-transfected controls (Supplementary Fig. S1E, lanes 2–3 and 1, respectively). Actin was used as a loading control (Supplementary Fig. S2E). Thus, these data suggest that cyclopamine-induced cell death is independent of CerS1 activation in these cells.
Because activation of neutral SMase2 (N-SMase2) is known to mediate caspase activation and apoptosis (33, 34), we determined whether cyclopamine affects nSMase2 with qPCR. Cyclopamine increased N-SMase2 approximately 3- and 6-fold (12–24 hours, respectively), whereas treatment at 6 hours had no effect (Fig. 2A). Cyclopamine treatment did not induce caspase-3 activity at 6 hours (Supplementary Fig. S3A), treatment at 12 hours slightly, but significantly, increased caspase-3 activity (~30%, P < 0.05). Knockdown of nSMase2, but not N-SMase1 (Supplementary Fig. S3B), (~75% compared to SCR controls, as determined by qPCR, Fig. 2B), prevented cyclopamine-induced caspase-3 activation (Fig. 2C). These data were also consistent with studies in which siRNA-mediated knockdown of nSMase2 abrogated cyclopamine-induced cell death, as measured by increased Annexin V staining or depletion of cellular ATP (Fig. 2D and 2E, respectively). Importantly, increased nSMase2 was also consistent with increased enzyme activity of N-SMase2 (~1.7-fold) in response to cyclopamine (10μg/mL, 12 hours), which was prevented by knockdown of nSMase2 using siRNAs (Fig. 2F). Cyclopamine had no effect on acid (A)-SMase activity in these cells (data not shown). Increase in nSMase2 by cyclopamine (10μg/mL) was also observed in UM-SCC-14A or UM-SCC-1 human HNSCC cells, in which it was increased approximately 4- and 8-fold, or 1.5-and 5-fold at 12 and 24h, respectively (Supplementary Figs. S3C and S3D, respectively).
Fig. 2. Cyclopamine induces nSMase2, and downregulation of nSMase2 protects cells from drug-induced apoptosis.

(A) Effects of cyclopamine on nSMase2 (0, 6, 12, and 24 hours) were determined in Daoy cells with qPCR. Ribosomal RNA was used as a control. (B) roles of siRNA-mediated knockdown of nSMase2 versus nSMase1 in cyclopamine-induced caspase 3 activity (12 hours) were assessed. Nontargeting SCR siRNA-treated cells were used as controls. (C) effectiveness of siRNAs for targeting/knockdown of nSMase2 in the absence/presence of cyclopamine (10ug/mL) compared with SCR-siRNA-transfected controls was confirmed with qPCR. D and E, effects of siRNA-mediated knockdown of nSMase2 on cyclopamine-induced cell death were measured by AnnexinV-7-AAD staining (D), or ATP depletion (E) in Daoy cells. (F) Effects of down-regulation of nSMase2 on cyclopamine-mediated increase in N-SMase enzyme activity (n=2) were determined.
Overall, these data suggest that cyclopamine induces nSMase2 in multiple human cancer cell lines, and knockdown of nSMase2 prevents cell death in response to cyclopamine, indicating an important role for nSMase2 in this process.
Down-regulation of nSMase-2 prevents cyclopamine-induced ceramide generation and cell death
To determine whether increased nSMase2 plays a role in cyclopamine-mediated ceramide generation (10μg/mL, 24 hours), we measured endogenous ceramide and SM by LC/MS-MS in the absence/presence of siRNAs against nSMase1 or nSMase2 in Daoy cells. Data revealed that cyclopamine increased total ceramide approximately 2- to 3.5-fold in the presence of SCR or N-SMase1 siRNAs, and knockdown of nSMase2 prevented cyclopamine-mediated ceramide generation (Fig. 3A). Consistent with these data, total SM was significantly decreased (~70%, P < 0.05) in response to cyclopamine in the absence/presence of SCR or nSMase1 siRNAs (Fig. 3B). Concentrations of ceramides and SM in response to cyclopamine in the absence/presence of nSMase1 or nSMase2 siRNAs were depicted in Supplementary Fig. S4A and S4B. As observed earlier, C18-ceramide was the main species generated by cyclopamine (~20-fold), and accordingly, a significant decrease in C18-SM was also observed (Supplementary Fig. S4C and S4D). Interestingly, although knockdown of nSMase2 prevented ceramide generation (Fig. 3A), it did not attenuate decrease in SM levels (Fig. 3B), suggesting the hydrolysis of SM without increased ceramide generation by an unknown mechanism, or inhibition of SM synthases. These data suggest that cyclopamine induces ceramide generation by induction of nSMase2.
Fig. 3. N-SMase2-generated ceramide is important in cyclopamine-induced cell death in Daoy cells.

(A-B) Ceramide (A) and SM (B) in response to cyclopamine (10μg/mL), in the absence/presence of siRNAs targeting nSMase1 or nSMase2, were measured in Daoy cells by LC/MS-MS. SCR-siRNA-treated cells were used as controls. (C) Effects of downregulation of nSMase2 versus nSMase1 with siRNAs on cell viability were determined with trypan blue exclusion assay. (D) Roles of nSMase2/ceramide in the induction of apoptosis were examined with trypan blue exclusion assay after transfection of Daoy cells with vectors containing cDNAs of the wt or catalytically inactive mutant of N-SMase2, compared with vector only transfected cells. Ectopic expression of wt or mutant N-SMase2-V5 was confirmed by measurement of SMase activity (E) and Western blotting (F) with anti-V5 antibody (as duplicates) in Daoy cells as compared with vector-transfected controls. Actin was used as a loading control in F (bottom).
We then examined whether downregulation of nSMase2, which prevented ceramide generation, also attenuated cyclopamine-mediated cell death (10μg/mL, 24 hours). Data showed that siRNA-mediated knockdown of nSMase2 significantly protected growth inhibition of Daoy cells (~50%, P < 0.05) compared with SCR-siRNA-transfected controls in response to cyclopamine (Fig. 3C), consistent with the protective effects of down-regulation of nSMase2 on cyclopamine-mediated apoptosis (Fig. 2C–E). Taken together, these data suggest increased ceramide generation by elevation of nSMase2 mRNA and activity is involved, at least in part, in caspase-3 activation, and cell death in response to cyclopamine.
Accordingly, wt-N-SMase2 expression significantly decreased cell growth, and increased apoptosis (~30%, P < 0.05) in Daoy cells compared with cells transfected with the catalytically inactive form of N-SMase2 (Fig. 3D), or vector-transfected controls (data not shown). Expression of wt- and mutant-N-SMase2-V5 (27) was confirmed by Western blotting using anti-V5 antibody, and activity of wt compared to catalytically inactive mutant-N-SMase2-V5 or vector-transfected controls were confirmed (Fig. 3E and 3F), supporting that the N-SMase2/ceramide axis plays an important role for inducing cell death in these cells.
Cyclopamine-induced nSMase-2 is independent of Smo inhibition
Because cyclopamine is an antagonist of Smo, it was important to determine whether cyclopamine-induced nSMase2 is dependent or independent of Smo inhibition. First, we examined the effects of downregulation of nSMase2 or nSMase1 using siRNAs on Smo and Gli mRNAs in the absence/presence of cyclopamine (10μg/ml, 24 hours) by qPCR in Daoy cells. Cyclopamine reduced Smo and Gli expression by approximately 60%, but downregulation of nSMase2 or nSMase1 had no effect on inhibition of the Smo/Gli axis by cyclopamine (Fig. 4A and 4B, respectively). Thus, these data confirmed that inhibition of the Smo/Gli axis by cyclopamine is regulated independently of N-SMase2 expression.
Fig. 4. Up-regulation of nSMase2 and ceramide generation in response to cyclopamine is independent of the inhibition of Smo/Gli of the SHh pathway in Daoy cells.
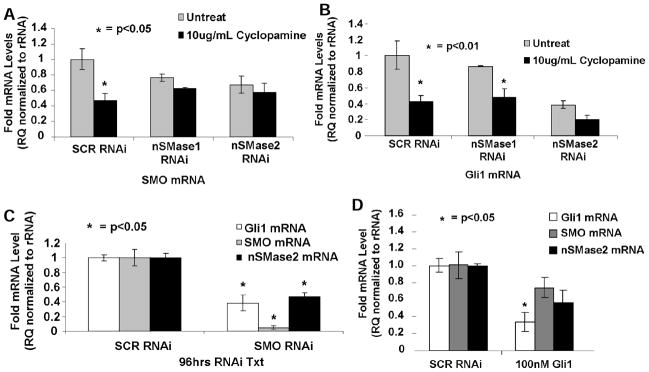
Effects of down-regulation of nSMase2 or nSMase1 using siRNAs on the inhibition of Smo (A) or Gli1 (B) expression in the absence/presence of cyclopamine (10μg/mL, 24 hours) were determined by qPCR. SCR-siRNA-transfected cells were used as controls. Potential roles of Smo or Gli1 inhibition on the regulation of nSMase2 were examined after siRNA-mediated knockdown of Smo (C) or Gli1 (D) using qPCR. Effectiveness of siRNAs on knockdown of Smo or Gli1 were also confirmed using qPCR (C and D, respectively). SCR-siRNA-transfected cells were used as controls.
Then, we examined the effects of down-regulation of Smo or Gli1 using siRNAs on nSMase2. We reasoned whether cyclopamine-mediated nSMase2 elevation is regulated downstream of Smo/Gli1 inhibition, knockdown of Smo or Gli1 should also increase N-SMase2. Interestingly, data showed while siRNAs successfully reduced Smo or Gli1 by approximately 90% or 80%, they did not increase N-SMase2 (Fig. 4C and 4D). Similarly, pharmacologic inhibitors of Gli or Smo, GANT-61 or SANT-1 (5–10μM, 24–48 hours) had no effect on nSMase2 (data not shown), despite successfully reducing expression of Gli1, and its downstream target Bcl2 (Supplementary Fig. S5A and S5B). Taken together, these data indicate that induction of nSMase2 in response to cyclopamine is independent of the Smo/Gli axis of SHh signaling in Daoy cells.
Cyclopamine-induced N-SMase-2 expression is regulated by oxidative stress
Because oxidative stress is known to induce N-SMase activity, and ceramide-mediated apoptosis (35–37), we then examined whether cyclopamine enhances N-SMase2 via induction of reactive oxygen species (ROS)/nitrogen species (RNS). First, we determined whether pretreatment with NAC (0.5 mmol/L), an anti-oxidant, altered nSMAse2 in the absence/presence of cyclopamine. Interestingly, while treatment with cyclopamine induced nSMase2 around 8-fold (10μg/mL, 24 hours), pre-treatment with NAC almost completely prevented cyclopamine-mediated nSMase2 in Daoy (Fig. 5A) and also in UM-SCC-14A cells (Fig. 5B), suggesting a role for cyclopamine-induced ROS/RNS in increased nSMase2, which was observed not only in Daoy cells, but was also observed in other human cancer cells, such as HNSCCs. Induction of ROS/RNS in response to cyclopamine (10μg/mL) at various time points was also confirmed by staining the cells with dichlorofluorescin diacetate (DCFDA) and flow cytometry analysis. Data showed cyclopamine treatment induced ROS/RNS generation within 3 to 6 hours of treatment, increasing DCFDA-fluorescence approximately 2- to 3-fold, respectively (Fig. 5C). Cyclopamine-mediated ROS/RNS generation was also detected by confocal microscopy, in which green fluorescence of DCFDA, a probe for ROS/RNS generation (38, 39), was visualized in Daoy cells compared with controls (Fig. 5D, bottom and top right, respectively). Interestingly, there was no significant colocalization of the green fluorescence of DCFDA with the red mitotracker in these cells in response to cyclopamine (Fig. 5D). Thus, these data suggest cyclopamine induces ROS/RNS generation, which is not selectively induced in mitochondria. Protective effects of NAC on cyclopamine-induced ROS/RNS generation were confirmed using DCFDA and flow cytometry. Pre-treatment with NAC prevented the generation of ROS/RNS, shifting the DCFDA fluorescence signal to the left when compared to cyclopamine-treated UM-SCC-14A or UM-SCC-1 cells, respectively (Fig. 5E and Supplementary Fig. S6A).
Fig. 5. Cyclopamine-mediated increased nSMase2 is regulated by ROS/RNS generation.

Effects of oxidative stress via ROS/RNS generation on cyclopamine-mediated overexpression of nSMase2 were examined in the absence/presence of antioxidant NAC (500 μmol/L) using qPCR in Daoy (A) and UM-SCC-14A (B) cells. (C) ROS/RNS induction in response to cyclopamine (10μg/mL) at 0, 3, 6, 9, 12, and 15 hours was measured by DCFDA by flow cytometry in Daoy cells. (D) Effects of cyclopamine (10μg/ml, 4 hours) on ROS/RNS generation (left) and potential mitochondrial colocalization (middle and right) in Daoy cells were visualized by confocal microscopy using DCFDA (green) and mitotracker (red) as compared to vehicle-treated controls (lower and upper, respectively). (E) Effects of NAC (500μmol/L) on prevention of ROS/RNS generation in response to cyclopamine (10μg/mL, 6 hours) in UM-SCC-14A cells were measured using DCFDA by flow cytometry.
Thus, these data suggest that cyclopamine induces early ROS/RNS generation (within 3–6 hours), which precedes increased nSMase2 detected at 12 hours, and that cyclopamine-mediated ROS/RNS is important in induction of nSMase2.
Cyclopamine increases nSMase-2 via induction of NO generation
We then determined the mechanism by which cyclopamine induces ROS/RNS generation, which subsequently leads to increased nSMAse2. First, to assess whether H2O2 generation was involved, we overexpressed human catalase, which increased its enzyme activity significantly (by >20-fold, P < 0.001) compared with controls (Supplementary Fig. S6B–S6D), and then determined its effects on nSMase2 and cell death in the absence/presence of cyclopamine (10μg/mL, 24 hours). Catalase overexpression did not prevent increased nSMase2 or Daoy cell death in response to cyclopamine (Supplementary Fig. S6B–D), suggesting they were independent of H2O2 generation. In addition, buthionine sulfoximine (2.5 mmol/L, 16 hours), a known inhibitor of glutathione (GSH) synthesis, inducing oxidative stress via accumulation of intracellular ROS, had no significant effect on nSMase2 or apoptosis (Supplementary Fig. S6E–S6F).
We next examined whether RNS generation plays a role in this process. To determine RNS generation, effects of cyclopamine on DAF fluorescence, a probe for peroxynitrite [ONOO(−)] and NO generation, was examined in Daoy cells using flow cytometry. As shown in Fig. 6A, cyclopamine (10μg/mL, 4 hours) enhanced DAF and DCFDA, but not DHE (which detects superoxide anion), fluorescence compared with controls, suggesting cyclopamine induces RNS, but not ROS, via generation of peroxynitrite (ONOO(−)) and/or NO. Pre-treatment with MnTBAP, a ONOO(−) scavenger, did not prevent nSMase2 upregulation (Supplementary Fig. S7A), whereas L-NAME pre-treatment, an inhibitor of NOS, almost completely blunted cyclopamine-induced nSMase2 (Fig. 6B).
Fig. 6. Cyclopamine-mediated induction of nSMase2 is regulated selectively by NO generation in Daoy cells.

(A) Generation of NO/ONOO(−) in response to cyclopamine (10μg/mL for 4 hours) was measured by DAF, DCFDA, or DHE by flow cytometry in Daoy cells. Effects of L-NAME (50μmol/L; B) versus DETA (3mmol/L, 12 hours; C) on nSMase2 in the absence/presence of cyclopamine (10μg/mL, 24 hours) were measured by qPCR. Effects of siRNA-mediated knockdown of n-NOS on nSMase2 (D), or cell death in Daoy (E) and UM-SCC-14A (F) cells were measured by qPCR, or trypan blue exclusion assay, respectively. (G) Cells isolated from wt or fro/fro mice were treated with increasing concentrations of cyclopamine, and effects on cell growth were measured by trypan blue exclusion assay. Vehicle-treated cells were used as controls.
In reciprocal experiments, DETA, a well-known NO donor, significantly increased nSMase2 (~15-fold, P < 0.05; Fig. 6C). Thus, these novel data indicate that cyclopamine-induced nSMase2 is selectively regulated via NO, but not via ONOO(−), generation in Daoy cells. To define the mechanism by which NO generation induces nSMase2 and cell death in response to cyclopamine, we examined the roles of i-NOS, endothelial NOS (e-NOS), or n-NOS. Cyclopamine (10μg/mL, 24 hours) increased (~2-fold, measured by qPCR) n-NOS (Supplementary Fig. S7B), but it had no significant effect on e-NOS or i-NOS (data not shown). Importantly, siRNA-mediated knockdown of n-NOS (~70%, measured by qPCR; Supplementary Fig. S7C) prevented nSMAse2 induction and cell death (Figs. 6D and 6E, respectively). Similar data were also obtained in UM-SCC-22A cells, in which knockdown of n-NOS using siRNAs significantly blocked cyclopamine-induced cell death, compared with SCR-siRNA transfected controls (Fig. 6F), suggesting a role for n-NOS in cyclopamine-mediated nSMase2 induction and cell death.
These results were also consistent when wt versus activity-deficient skin fibroblasts isolated from new born homozygous fro/fro (fragilitas ossium) mice (25) were treated with DETA. Treatment of wt cells with increasing concentrations of NO-donor DETA elevated N-SMase2 (data not shown), and inhibited cell viability (Fig. 6G), whereas fro/fro skin fibroblasts with inactive N-SMase2, exhibited resistance to DETA-mediated cell death (Fig. 6G), supporting the role of N-SMase2 in DETA/NO-mediated cell death. Interestingly, in contrast to human cancer cells, these noncancerous wt and fro/fro skin fibroblasts were equally sensitive to cyclopamine-mediated cell death, suggesting that SHh/Smo plays an important role for the growth and/or proliferation of these cells, regardless of their N-SMase activity (data not shown).
Overall, these data suggest that increased nSMase2/ceramide and cell death in response to cyclopamine is regulated, at least in part, by the n-NOS/NO axis, in Daoy medulloblastoma and HNSCC cells.
Discussion
Here, possible roles and mechanisms of interaction between the SHh inhibitor/anticancer drug cyclopamine, and the bioactive sphingolipid ceramide, in apoptosis of Daoy and HNSCC cells were examined. Unexpected and novel data revealed cyclopamine induces apoptosis in part via selective induction of nSMase2/increased ceramide. Mechanistically, induction of nSMAse2 was regulated selectively by n-NOS/NO in response to cyclopamine, which was independent of Smo/Gli1 inhibition.
Cyclopamine was to induce apoptosis (1,9,14). Another Smo antagonist GDC-0449 (40) is currently in clinical trials against medulloblastomas and various other cancers. In fact, amino acid substitution at a conserved aspartic acid residue of SMO, which interrupted GDC-0449 binding, was reported to cause drug resistance in the clinic (41). Ceramide mediates drug-induced apoptosis. However, any involvement of ceramide in cyclopamine-induced apoptosis has not previously been reported. Our novel data revealed that cyclopamine results in robust ceramide generation via induction of nSMase2, which is at least partly critical in cyclopamine-induced cell death in Daoy and HNSCC cells. These data are somewhat in agreement with a previous study, in which cyclopamine-induced apoptosis was partially rescued by Gli overexpression in Daoy cells, indicating an involvement of other mechanisms in this process (42). Unexpected data additionally suggested cyclopamine-induced N-SMase2/ceramide generation is independent of SHh/Smo/Gli inhibition, but is regulated by NO stress/signaling in these cells.
To our knowledge, no previous data involving cyclopamine-induced NO generation in cancer cells exists. Although ceramide was reported to play roles in sodium nitroprusside (an NO donor)-induced apoptosis (43), any role of NO in inducing nSMase2 has not been previously reported. However, many excellent previous reports exist (35–37), which showed the involvement of ROS/RNS in SMase regulation and cell death in various cell types. For example, H2O2 was shown to activate N-SMase2, which is prevented by glutathione (GSH) in human airway epithelial cells (HAE; ref. 44). In aging rat hepatocytes, however, decreased GSH induced N-SMase2, whereas in young hepatocytes, inhibition of GSH synthesis activated N-SMase (45), suggesting that ROS/N-SMase2 regulation might be context dependent. This was also consistent with the roles of RNS in SMase regulation. It was shown that ONOO(−) induced A-SMase, but not N-SMase, in HAE cells (35). However, our data indicate a role for n-NOS/NO in the activation of N-SMase but not A-SMase in Daoy and HNSCC cells. These data together suggest that SMase regulation by RNS is also context dependent, and it might be differentially regulated in non-cancerous HAE versus some cancer cell types. The cell type/context-dependent regulation of ceramide metabolism by NO is also consistent with earlier studies, in which NO donor induced apoptosis in cultured fibroblasts but not in keratinocytes (46). Moreover, L-NAME, inhibitor of NOS, increased ceramide formation and apoptosis in keratinocytes, but not in fibroblasts (46).
NO generation is regulated mainly by e-NOS, i-NOS, or n-NOS (47). Interestingly, our novel data suggest that cyclopamine increased n-NOS mRNA, and knockdown of n-NOS (48) prevented cyclopamine-induced N-SMase2 activation and cell death, indicating its involvement in this process. However, specific mechanisms involved in n-NOS/NO-generation in response to cyclopamine remain unknown.
Moreover, mechanisms by which cyclopamine-induced n-NOS/NO results in increased nSMase2 are unknown. It was reported previously that daunorubicin activated the nSMase2 promoter via Sp1/Sp3 transcription factors in MCF-7 human breast cancer cells, increasing ceramide accumulation and cell death (49). Recently, all-trans-retionic-acid was also shown to induce nSMase2, resulting in MCF-7 growth arrest (50). These studies are in agreement with our data, suggesting that various anti-cancer drugs, including cyclopamine, induce nSMase2, leading to increased ceramide generation and apoptosis.
In summary, our data show a novel off-target function of cyclopamine by inducing n-NOS/NO-dependent nSMase2 expression, and ceramide generation, which, in part, was necessary for drug-induced apoptosis. These data may have important implications for the Smo-independent apoptotic roles of cyclopamine in the treatment of various human cancers, in which the n-NOS/NO/N-SMase2/ceramide axis is intact, but SHh activation might be partly dispensable.
Supplementary Material
Acknowledgments
Grants/Funding Information: This work was supported in part by research funding obtained from the National Institutes of Health (CA088932, DE016572, CA097165), and from the Angel Walk, Rally Foundation for Childhood Cancer Research, Chase after a Cure, and Monica Kreber Golf Tournament. Lipid measurements and flow cytometry were conducted in facilities constructed with support from NIH (C06 RR015455).
We thank Dr. J. Schnellmann for her editorial review and Dr. J. Houghton and her group (Cleveland Clinic) for helpful discussions, and sharing SHh pathway molecular tools.
Footnotes
Financial Disclosure: The authors have no conflict of interest to disclose.
References
- 1.Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–354. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 2.Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8:97–106. doi: 10.1038/nrclinonc.2010.196. [DOI] [PubMed] [Google Scholar]
- 3.Ng JM, Curran T. The Hedgehog’s tale: developing strategies for targeting cancer. Nat Rev Cancer. 2011;11:493–501. doi: 10.1038/nrc3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mazumdar T, DeVecchio J, Shi T, Jones J, Agyeman A, Houghton JA. Hedgehog signaling drives cellular survival in human colon carcinoma cells. Cancer Res. 2011;71:1092–1102. doi: 10.1158/0008-5472.CAN-10-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Toole SA, Machalek DA, Shearer RF, Millar EK, Nair R, Schofield P, et al. Hedgehog Overexpression Is Associated with Stromal Interactions and Predicts for Poor Outcome in Breast Cancer. Cancer Res. 2011;71:4002–4014. doi: 10.1158/0008-5472.CAN-10-3738. [DOI] [PubMed] [Google Scholar]
- 6.Shi T, Mazumdar T, Devecchio J, Duan ZH, Agyeman A, Aziz M, et al. cDNA microarray gene expression profiling of hedgehog signaling pathway inhibition in human colon cancer cells. PLoS One. 2010;5:e13054. doi: 10.1371/journal.pone.0013054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bar EE, Chaudhry A, Farah MH, Eberhart CG. Hedgehog signaling promotes medulloblastoma survival via Bc/II. Am J Pathol. 2007;170:347–355. doi: 10.2353/ajpath.2007.060066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 9.Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297:1559–1561. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- 10.Sanchez P, Ruiz i Altaba A. In vivo inhibition of endogenous brain tumors through systemic interference of Hedgehog signaling in mice. Mech Dev. 2005;122:223–230. doi: 10.1016/j.mod.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 12.Kiselyov AS. Targeting the hedgehog signaling pathway with small molecules. Anticancer Agents Med Chem. 2006;6:445–449. doi: 10.2174/187152006778226495. [DOI] [PubMed] [Google Scholar]
- 13.Bar EE, Stearns D. New developments in medulloblastoma treatment: the potential of a cyclopamine-lovastatin combination. Expert Opin Investig Drugs. 2008;17:185–195. doi: 10.1517/13543784.17.2.185. [DOI] [PubMed] [Google Scholar]
- 14.Enguita-German M, Schiapparelli P, Rey JA, Castresana JS. CD133+ cells from medulloblastoma and PNET cell lines are more resistant to cyclopamine inhibition of the sonic hedgehog signaling pathway than CD133− cells. Tumour Biol. 2010;31:381–390. doi: 10.1007/s13277-010-0046-4. [DOI] [PubMed] [Google Scholar]
- 15.Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med. 2009;9:873–886. doi: 10.2174/156652409789105570. [DOI] [PubMed] [Google Scholar]
- 16.Pruett ST, Bushnev A, Hagedorn K, Adiga M, Haynes CA, Sullards MC, et al. Biodiversity of sphingoid bases (“sphingosines”) and related amino alcohols. J Lipid Res. 2008;49:1621–1639. doi: 10.1194/jlr.R800012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stiban J, Tidhar R, Futerman AH. Ceramide synthases: roles in cell physiology and signaling. Adv Exp Med Biol. 2010;688:60–71. doi: 10.1007/978-1-4419-6741-1_4. [DOI] [PubMed] [Google Scholar]
- 18.Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004;4:604–616. doi: 10.1038/nrc1411. [DOI] [PubMed] [Google Scholar]
- 19.Deng X, Yin X, Allan R, Lu DD, Maurer CW, Haimovitz-Friedman A, et al. Ceramide biogenesis is required for radiation-induced apoptosis in the germ line of C. elegans. Science. 2008;322:110–115. doi: 10.1126/science.1158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pewzner-Jung Y, Ben-Dor S, Futerman AH. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: Insights into the regulation of ceramide synthesis. J Biol Chem. 2006;281:25001–25005. doi: 10.1074/jbc.R600010200. [DOI] [PubMed] [Google Scholar]
- 21.Wu BX, Clarke CJ, Hannun YA. Mammalian neutral sphingomyelinases: regulation and roles in cell signaling responses. Neuromolecular Med. 2010;12:320–330. doi: 10.1007/s12017-010-8120-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edelmann B, Bertsch U, Tchikov V, Winoto-Morbach S, Perrotta C, Jakob M, et al. Caspase-8 and caspase-7 sequentially mediate proteolytic activation of acid sphingomyelinase in TNF-R1 receptosomes. Embo J. 2011;30:379–394. doi: 10.1038/emboj.2010.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Truman JP, Garcia-Barros M, Kaag M, Hambardzumyan D, Stancevic B, Chan M, et al. Endothelial membrane remodeling is obligate for anti-angiogenic radiosensitization during tumor radiosurgery. PLoS One. 2010;5:e12310. doi: 10.1371/journal.pone.0012310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filosto S, Castillo S, Danielson A, Franzi L, Khan E, Kenyon N, et al. Neutral sphingomyelinase 2: a novel target in cigarette smoke-induced apoptosis and lung injury. Am J Respir Cell Mol Biol. 2011;44:350–360. doi: 10.1165/rcmb.2009-0422OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aubin I, Adams CP, Opsahl S, Septier D, Bishop CE, Auge N, et al. A deletion in the gene encoding sphingomyelin phosphodiesterase 3 (Smpd3) results in osteogenesis and dentinogenesis imperfecta in the mouse. Nat Genet. 2005;37:803–805. doi: 10.1038/ng1603. [DOI] [PubMed] [Google Scholar]
- 26.Bielawski J, Pierce JS, Snider J, Rembiesa B, Szulc ZM, Bielawska A. Sphingolipid analysis by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) Adv Exp Med Biol. 2010;688:46–59. doi: 10.1007/978-1-4419-6741-1_3. [DOI] [PubMed] [Google Scholar]
- 27.Clarke CJ, Cloessner EA, Roddy PL, Hannun YA. Neutral Sphingomyelinase-2 (nSMase2) is the Primary Neutral Sphingomyelinase Isoform Activated by Tumor Necrosis Factor-alpha in MCF-7 cells. Biochem J. 2011;435:381–390. doi: 10.1042/BJ20101752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mukhopadhyay A, Saddoughi SA, Song P, Sultan I, Ponnusamy S, Senkal CE, et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. Faseb J. 2009;23:751–763. doi: 10.1096/fj.08-120550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coon V, Laukert T, Pedone CA, Laterra J, Kim KJ, Fults DW. Molecular therapy targeting Sonic hedgehog and hepatocyte growth factor signaling in a mouse model of medulloblastoma. Mol Cancer Ther. 2010;9:2627–2636. doi: 10.1158/1535-7163.MCT-10-0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishimaki H, Kasai K, Kozaki K, Takeo T, Ikeda H, Saga S, et al. A role of activated Sonic hedgehog signaling for the cellular proliferation of oral squamous cell carcinoma cell line. Biochem Biophys Res Commun. 2004;314:313–320. doi: 10.1016/j.bbrc.2003.12.097. [DOI] [PubMed] [Google Scholar]
- 31.Kelleher FC. Hedgehog signaling and therapeutics in pancreatic cancer. Carcinogenesis. 2011;32:445–451. doi: 10.1093/carcin/bgq280. [DOI] [PubMed] [Google Scholar]
- 32.Spassieva S, Seo JG, Jiang JC, Bielawski J, Alvarez-Vasquez F, Jazwinski SM, et al. Necessary role for the Lag1p motif in (dihydro)ceramide synthase activity. J Biol Chem. 2006;281:33931–33938. doi: 10.1074/jbc.M608092200. [DOI] [PubMed] [Google Scholar]
- 33.Canals D, Perry DM, Jenkins RW, Hannun YA. Drug targeting of sphingolipid metabolism: sphingomyelinases and ceramidases. Br J Pharmacol. 2011;163:694–712. doi: 10.1111/j.1476-5381.2011.01279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Devillard R, Galvani S, Thiers JC, Guenet JL, Hannun Y, Bielawski J, et al. Stress-induced sphingolipid signaling: role of type-2 neutral sphingomyelinase in murine cell apoptosis and proliferation. PLoS One. 2010;5:e9826. doi: 10.1371/journal.pone.0009826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castillo SS, Levy M, Thaikoottathil JV, Goldkorn T. Reactive nitrogen and oxygen species activate different sphingomyelinases to induce apoptosis in airway epithelial cells. Exp Cell Res. 2007;313:2680–2686. doi: 10.1016/j.yexcr.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Jana A, Pahan K. Oxidative stress kills human primary oligodendrocytes via neutral sphingomyelinase: implications for multiple sclerosis. J Neuroimmune Pharmacol. 2007;2:184–193. doi: 10.1007/s11481-007-9066-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ichi I, Kamikawa C, Nakagawa T, Kobayashi K, Kataoka R, Nagata E, et al. Neutral sphingomyelinase-induced ceramide accumulation by oxidative stress during carbon tetrachloride intoxication. Toxicology. 2009;261:33–40. doi: 10.1016/j.tox.2009.04.040. [DOI] [PubMed] [Google Scholar]
- 38.Ischiropoulos H, Gow A, Thom SR, Kooy NW, Royall JA, Crow JP. Detection of reactive nitrogen species using 2,7-dichlorodihydrofluorescein and dihydrorhodamine 123. Methods Enzymol. 1999;301:367–373. doi: 10.1016/s0076-6879(99)01100-3. [DOI] [PubMed] [Google Scholar]
- 39.Bilski P, Belanger AG, Chignell CF. Photosensitized oxidation of 2′,7′-dichlorofluorescin: singlet oxygen does not contribute to the formation of fluorescent oxidation product 2′,7′-dichlorofluorescein. Free Radic Biol Med. 2002;33:938–946. doi: 10.1016/s0891-5849(02)00982-6. [DOI] [PubMed] [Google Scholar]
- 40.Tremblay MR, Nesler M, Weatherhead R, Castro AC. Recent patents for Hedgehog pathway inhibitors for the treatment of malignancy. Expert Opin Ther Pat. 2009;19:1039–1056. doi: 10.1517/13543770903008551. [DOI] [PubMed] [Google Scholar]
- 41.Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–574. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bar EE, Chaudhry A, Farah MH, Eberhart CG. Hedgehog signaling promotes medulloblastoma survival via Bc/II. Am J Pathol. 2007;170:347–355. doi: 10.2353/ajpath.2007.060066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takeda Y, Tashima M, Takahashi A, Uchiyama T, Okazaki T. Ceramide generation in nitric oxide-induced apoptosis. Activation of magnesium-dependent neutral sphingomyelinase via caspase-3. J Biol Chem. 1999;274:10654–10660. doi: 10.1074/jbc.274.15.10654. [DOI] [PubMed] [Google Scholar]
- 44.Levy M, Castillo SS, Goldkorn T. nSMase2 activation and trafficking are modulated by oxidative stress to induce apoptosis. Biochem Biophys Res Commun. 2006;344:900–905. doi: 10.1016/j.bbrc.2006.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rutkute K, Asmis RH, Nikolova-Karakashian MN. Regulation of neutral sphingomyelinase-2 by GSH: a new insight to the role of oxidative stress in aging-associated inflammation. J Lipid Res. 2007;48:2443–2452. doi: 10.1194/jlr.M700227-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gallala H, Macheleidt O, Doering T, Schreiner V, Sandhoff K. Nitric oxide regulates synthesis of gene products involved in keratinocyte differentiation and ceramide metabolism. Eur J Cell Biol. 2004 Dec;83(11–12):667–79. doi: 10.1078/0171-9335-00425. [DOI] [PubMed] [Google Scholar]
- 47.Mocellin S. Nitric oxide: cancer target or anticancer agent? Curr Cancer Drug Targets. 2009 Mar;9(2):214–36. doi: 10.2174/156800909787581015. [DOI] [PubMed] [Google Scholar]
- 48.Hirst DG, Robson T. Nitric oxide physiology and pathology. Methods Mol Biol. 2011;704:1–13. doi: 10.1007/978-1-61737-964-2_1. [DOI] [PubMed] [Google Scholar]
- 49.Ito H, Murakami M, Furuhata A, Gao S, Yoshida K, Sobue S, et al. Transcriptional regulation of neutral sphingomyelinase 2 gene expression of a human breast cancer cell line, MCF-7, induced by the anti-cancer drug, daunorubicin. Biochim Biophys Acta. 2009;1789:681–690. doi: 10.1016/j.bbagrm.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 50.Clarke CJ, Mediwala K, Jenkins RW, Sutton CA, Tholanikunnel BG, Hannun YA. Neutral sphingomyelinase-2 mediates growth arrest by retinoic acid through modulation of ribosomal S6 kinase. J Biol Chem. 2011;286:21565–21576. doi: 10.1074/jbc.M110.193375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.