

Abstract
Background:
Cardiac glycosides such as digitoxin have been shown to directly cause apoptotic death of cancer cells both in vitro, and in vivo. However, the mechanism connecting cardiac glycoside action to apoptosis is not known. It has been reported that compounds resembling digitoxin are able to reduce c-MYC expression. Furthermore, it has been previously shown that the transcription of c-MYC depends on nuclear factor of activated T-cells (NFAT) binding sites in the c-MYC promoter. We have therefore hypothesized that NFAT might mediate digitoxin effects on c-MYC mRNA message.
Materials and Methods:
We have chosen to study this process in HeLa cells where structurally intact c-MYC genes in 8q24 co-localize with human papilloma virus 18 at all integration sites.
Results:
Here we show that within the 1st h following treatment with digitoxin, a significant reduction in c-MYC mRNA occurs. This is followed by a precipitous loss of c-MYC protein, activation of caspase 3, and subsequent apoptotic cell death. To test the NFAT-dependence mechanism, we analyzed the effects of digitoxin on NFAT isoform-dependent auto-activation of a NFAT-luciferase expression system. Drug dependent effects on expression varied according to each of the four canonical NFAT isoforms (1, 2, 3 or 4). The most digitoxin-sensitive NFAT isoform was NFAT1. Using c-MYC chromatin immune precipitation, we find that digitoxin inhibits interaction of NFAT1 with the proximal c-MYC promoter.
Conclusions:
These results suggest that the carcinotoxic activity of digitoxin includes suppression of NFAT-driven c-MYC expression.
Keywords: Cardiac glycosides, c-MYC, nuclear factor of activated T-cells
INTRODUCTION
Cardiac glycosides such as digitoxin and the digitoxin-analogue oleandrin have been shown to directly cause apoptotic death of cancer cells, both in vitro,[1–5] and in vivo.[6,7] In addition, adjuvant effects at the cellular level have also been observed. For example, oleandrin can independently sensitize cancer cells to radiation,[8] or to pro-apoptotic ligands such as apo2 L/TRAIL (TNF alpha Related Apoptosis-Inducing Ligand).[9] In mice with intact immune systems, equivalent carcinotoxic effects have been observed for chemically induced skin papillomas and pulmonary tumors with either digitoxin[6] or oleandrin.[7] In immune-compromised nude mouse models, growth of implanted castration resistant prostate cancer cells,[10] or A549 non-small cell lung cancer cells[11] have been substantially suppressed by the digitoxin analogue UNBS1450. Digoxin, a less active digitoxin analogue, has also been shown to inhibit the growth of neuroblastoma tumor cells in SCID (Severe Combined Immuno-Deficiency) mouse models, and angiogenesis in the chick chorioalantoic membrane.[12] Finally, ouabain, also a less active digitoxin analog, has been shown to efficiently suppress growth of retinoblastoma cell in SCID mouse xenografts.[13] Thus digitoxin, and its related active analogues are able to suppress chemically induced tumors in normal mice, or the growth of variety of human tumor cells, either in culture, or when xeno grafted into SCID mice.
However, at the level of cancer therapy in humans, only a few clinical parallels with the animal data have been reported. Patients with breast cancer and concomitant digitalis treatment have been shown to have tumors with much less growth potential.[14] However, digitalis is a crude mixture of digitoxin and other cardiac glycosides, thus complicating a specific interpretation. However, cancer patients from the Norwegian database, treated specifically with digitoxin, reportedly have a lower incidence of lymphoma/leukemia and kidney/urinary organ cancers.[15] Individual cardiac glycosides such as digitoxin have been reported to have anti-inflammatory properties.[16–19] Nonetheless, the mechanism(s) by which cardiac glycosides such as digitoxin and related cardenolides actually kill cancer cells remains largely unknown, and use of digitoxin to treat human cancers has languished.
Recent reports suggest that cardiac glycosides resembling digitoxin may suppress c-MYC expression. For example, it has been reported that both c-MYC mRNA and protein decline quickly and profoundly when Castration Resistant Prostate Cancer PC-3 cells are treated, either in vitro or in vivo, with the digitoxin analogue UNBS1450.[10] Relevantly, loss of c-MYC is among the most rapidly acting of the known pro-apoptotic processes. Both c-MYC mRNA and c-MYC protein have short half-lives, of approximately 30 min, respectively.[20] Of the known transcriptional drivers for c-MYC, one of the most critical appears to be nuclear factor of activated T-cells (NFAT). There are 4 classical NFAT isoforms (viz., NFAT1-4), of which at least one isoform, NFAT2 (also known as NFATc1), has been shown to be a transcriptionally required driver of c-MYC in human pancreatic cancer cells.[21] This action occurs both in vitro and in vivo. Consistently, both siRNA (small inhibiting RNA) against NFAT1/NFATc2, as well as the Ca++/calcineurin inhibitors cyclosporin A (CSA) and FK506, block NFAT1/NFATc2-dependent c-MYC expression and kill pancreatic cancer cells.[21]
Based on preliminary data, our hypothesis is that the mechanism by which cardiac glycosides, such as oleandrin and digitoxin, are carcinotoxic is by reducing NFAT, thereby inhibiting c-MYC transcription. To test this hypothesis we conducted a systematic analysis of digitoxin-induced cell death in the HeLa cervical cancer cell system. The HeLa cell is optimal for this study because all c-MYC genes colocalize at all integration sites with human papilloma virus (HPV18), the carcinogenic driver for HeLa cells. As shown below, our data indicate that digitoxin and oleandrin suppress c-MYC expression by reducing NFAT binding to the proximal c-MYC promoter. Loss of c-MYC expression leads to apoptotic cell death. We also implicate drug-dependent activation of caspase 3 in a global loss of NFAT isoforms. We suggest that knowledge of these specific mechanisms of action may serve to emphasize the potential translational value of digitoxin for tumors that are typically characterized by high levels of NFAT-dependent-c-MYC expression.
MATERIALS AND METHODS
Cells and drugs
HeLa cells were obtained from the ATCC (American Type Culture Collection, Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's medium, supplemented with 10% fetal bovine serum, 2 mM glutamine, penicillin (100 U/ml), and streptomycin (100 mg/ml). Oleandrin (I) was obtained from Indofine Chemical (Hillsborough, NJ; >98% purity). Digitoxin (II), digoxin, ouabain were obtained from Sigma (>95% purity). Drugs were solubilized as stock solutions in 100% ethanol and diluted for experiments to a final ethanol concentration of 0.1%. The caspase inhibitors (DEVD, IETD, LEHD) were from BD Pharmingen.
Antibodies and sources
Native NFAT isoforms were detected by routine western blot analyses. The abbreviations and equivalent nomenclatures are as follows: (i) NFAT1 (=NFATc2); (ii) NFAT2 (=NFATc, NFATc1); (iii) NFAT3 (=NFATc4); (iv) NFAT4 (=NFATc3). Antibodies were as follows: (i) NFAT1 (NF-ATc2, NF-ATp: Mouse monoclonal antibody from BD Pharmingen); (ii) NFAT2 (NF-ATc1, NF-ATc: Mouse monoclonal antibody from BD Pharmingen); (iii) NFAT3 (NFATc4: Rabbit monoclonal antibody from Cell Signaling); (iv) NFAT4 (NFATc3, NFATx: Rabbit monoclonal antibody from Cell Signaling). The antibodies for caspase 3, caspase 8, caspase 9, PARP (poly ADP ribose polymerase) were from BD Pharmingen. The antibodies for c-MYC and β-actin were from Sigma.
LDH assays
Cytotoxicity Detection Kits were purchased from Roche (Mannheim, Germany). HeLa cells were treated for 24 h with different concentrations of cardiac glycosides. The assay for released LDH (Lactate Dehydrogenase) was performed by mixing 100 μl supernatant volumes with 100 μl substrate volumes, followed by incubation at room temperature for 10-30 min. The reaction was terminated by adding 50 μl 1N HCl, and the absorbance was measured at 492 nm. The supernatants from untreated normal cells and Triton X-100 treated cells were used as lower controls and higher controls, respectively for the cytotoxicity calculation.
Western blot analysis
HeLa cells were lysed in M2 buffer (20 mM pH 7.0 Tris, 0.5% NP-40, 250 mM NaCl, 3 mM EDTA (ethylene diamine tetracetic acid), 3 mM EGTA (ethylene glycol tetraacetic acid), 2 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 20 mM β-glycerol phosphate, 10 mM 4-Nitrophenyl phosphate disodium salt, 1 mM sodium vanadate, 1 mg/ml leupeptin). 20 μg of the cell lysate from each sample were fractionated by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and immunoblotted. The blots were visualized with chemiluminescent substrate (Pierce).
RNA purification and qPCR analysis of MYC transcript levels 1
The RNAqueous-4PCR kit from AMBION Inc. was used for RNA purification. Briefly, HeLa cells treated with cardiac glycosides were lysed in guanidinium lysis buffer. The lysates were then adjusted to 64% ethanol and applied to silica-based filters. The filters were then washed with the buffers supplied by the manufacturer to remove resident DNA, protein and other contaminants. The RNA was then eluted in the supplied elution buffer. The RNA samples were further treated with DNase 1 to remove trace amount of DNA. Total RNA concentration was determined by spectrophotometry. Reverse transcription of 1 μg total RNA was completed using the iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA). qPCR (quantitative polymerase chain reaction) was performed on a CFX384 Real-Time PCR Detection System (Bio-Rad) using SsoFast EvaGreen Supermix (Bio-Rad) with 400 nM primer concentration and ~ 5 ng cDNA input. Raw Ct values were calculated as an average of four technical replicates. Comparative gene LDHA (lactate dehydrogenase A) expression values were calculated after normalizing with values from the housekeeping gene, and are relative to duplicate control samples. Primers used are: MYC forward = 5’- TCC TTG CAG CTG CTT AGA CGC T-3’, MYC reverse = 5’- AGC TAA CGT TGA GGG GCA TCG T - 3’, HPRT1 forward = 5’- TGA CAC TGG CAA AAC AAT GCA - 3’, HPRT1 reverse = 5’- GGT CCT TTT CAC CAG CAA GCT - 3’, LDHA forward = 5’- CTG CCA CCT CTG ACG CAC CA-3’, LDHA reverse = 5’- AAA CAT CCA CCT GGC TCA AGG GG - 3’.
c-MYC chromatin immuno-precipitation
Cardiac glycoside treated HeLa cells were rinsed with PBS (phosphopate buffered saline) and cross-linked with 1% formaldehyde at room temperature for 10 min. Cross-linking was terminated with 0.125 M glycine and cell pellets were washed and lysed to isolate nuclear extracts in high salt lysis buffer with protease inhibitors. Chromatin was sonicated for 10 cycles of 30 s ON and 30 s OFF in an UCD-200 Bioruptor sonication device (Diagenode). For immune precipitation, 300 μg of protein was pre cleared using Protein G agarose (Roche) before incubation with either anti-NFAT1 (sc-7295X, Santa Cruz) or mouse IgG (Immunoglobulin G) (I5381, Sigma) overnight at 4°C. Protein G beads were added for 2 h before washing and recovery in elution buffer. Reverse cross-linking was performed at 67°C overnight and DNA was isolated using the PCR Purification Kit (Qiagen). For quantitative PCR analysis of recovered DNA, amplification of a MYC promoter NFAT1 binding site and a control region 3,000 bp upstream was performed using the SsoFast EvaGreen Supermix on a CFX384 Real-Time PCR Detection System (Bio-Rad). The primers for the MYC promoter region are as previously published.[29] The primers for control upstream region were: Forward = 5’- ACCTTCTGCTGTGCCTCCCCTT-3’, reverse = 5’- CCAAACAAGCTTTAGCCCCCACGT-3’. Data were normalized to values corresponding to 10% input DNA standard wells.
Reactive oxygen species detection
HeLa cells were cultured in 24-well black visiplates (Wallac) and treated with cardiac glycosides for 1, 3, and 6 h. After each time point, the medium was replaced with 10 μM carboxy-H2DCFDA (Molecular Probes) in PBS and incubated for 30 min. Thereafter, the loading buffer was removed and the cells were returned to the growth medium. After incubation for 30 min, the fluorescence of the oxidized form of carboxy-H2DCFDA was measured by fluorescence (excitation wavelength, 495 nm; emission wavelength, 520 nm).
Glutathione determination
Intracellular GSH (Glutathione) content was determined using a colorimetric assay kit (Oxis Research). Briefly, HeLa cells were treated with cardiac glycosides for 1, 3, 6, 9, and 16 h. Following scrape-harvesting, cell pellets were resuspended in 4 volumes of 5% meta-phosphoric acid (Sigma), thoroughly mixed, and centrifuged at 3000 g at 4°C for 10 min. Clear supernants were transferred into 96-well plate and 5 μl each of R1 and R2 solutions were added to each well. One hour later, GSH content was measured at a wavelength of 400 nm.
Reporter gene assays
HeLa cells were seeded in 6 well plates overnight, then cotransfected overnight (16 h) with NFAT-luc and lacZ plasmids and then with either NFAT1, NFAT2 (both a gift from Dr. Anjana Rao), NFAT3 or NFAT4 (both a gift from Dr. Aviva Symes), using the Fugene6 transfection reagent (Roche). The cells were then treated with oleandrin and digitoxin at the different concentrations for 10 h. Cells were harvested and lysed with 1× passive lysis buffer. Luciferase assays were performed with the Promega Luciferase Assay System. The luciferase values were normalized to β-galactosidase activity.
Statistics
All experiments were run independently at least three (3) times. Data are analyzed by analysis of variance (ANOVA). Error bars give Standard Error of the Mean. A significant difference (*) is taken to be P < 0.05.
RESULTS
Cardiac glycosides are cytotoxic to cancer cells
As shown in Figure 1, we incubated different concentrations of digitoxin, or an active analogue, oleandrin, with HeLa cells for 24 h. At the end of the incubation period, the culture supernatants were assayed for LDH release as a marker for cytotoxic loss of plasma membrane integrity. Figure 1a shows that oleandrin induces cytotoxic effects in a dose-dependent manner, with a threshold concentration of ca. 30 nM. Figure 1b shows that a dose-dependent effect can also be detected with digitoxin, with a threshold concentration of ca. 100 nM. Apparent saturation of the cytotoxic effect at the 24 h time point occurs at >ca. 100 nM for oleandrin and ca. 300nM for digitoxin. These data indicate that both oleandrin and digitoxin are cytotoxically active in the HeLa cell system, and that oleandrin is substantially more cytotoxic at 24 h than digitoxin. We also tested two other cardiac glycosides: digoxin [Figure 2a] and ouabain [Figure 2b]. Both were also cytotoxic, with threshold concentrations in the 50-100 nM concentration ranges.
Figure 1.
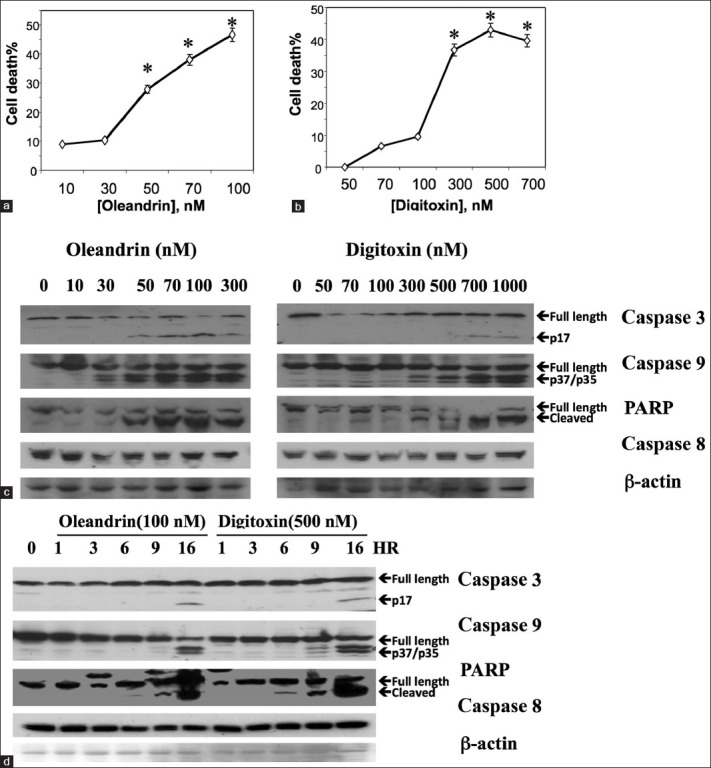
Cardiac glycosides induce cancer cell death by apoptosis. (a) Induction of cancer cell death by oleandrin. Cells were incubated in different concentrations of oleandrin for 24 h, and assayed for cell death by release of LDH. Assays were performed in triplicate (*P < 0.05, relative to 10 nM concentration point). Data are representative of three independent experiments. (b) Induction of cancer cell death by digitoxin. Cells were incubated in different concentrations of digitoxin for 24 h and assayed for cell death by release of LDH. Assays were performed in triplicate (*P < 0.05, relative to 50 nM concentration point). Data are representative of three independent experiments. (c) Drug concentration-dependent activation of caspases and PARP. Cells were incubated for 16 h in different concentrations of either oleandrin or digitoxin. Cells were then collected and subjected to Western blot analysis with antibodies against antigens as shown. (d) Time dependent changes in activation of caspases and PARP. Cells were incubated for different times in maximal concentrations of oleandrin (100 nM) or digitoxin (500 nM). Cells were then collected and subjected to western blot analysis with antibodies against antigens as shown
Figure 2.
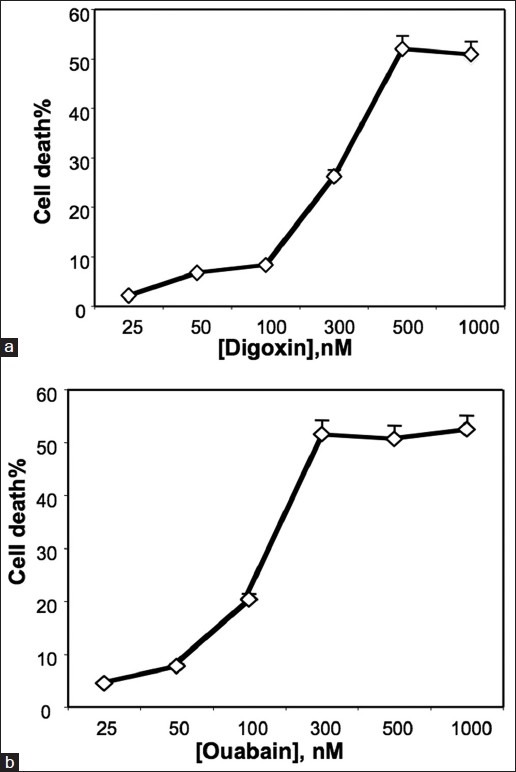
Influence of digoxin and ouabain on cell death. (a) Induction of cancer cell death by digoxin. Cells were incubated in different concentrations of digoxin for 24 h and assayed for cell death by release of LDH. (b) Induction of cancer cell death by ouabain. Cells were incubated in different concentrations of ouabain for 24 h and assayed for cell death by release of LDH
Cardiac glycosides cause apoptotic cell death
To test for the drug concentrations at which different classical molecular signs of apoptosis occurred, we studied changes in caspases 3, 8, 9, and PARP, occurring after 16 h of drug exposure. Figure 1c shows that in the case of oleandrin (left panel) caspase 9 starts to activate at ca. 30 nM oleandrin, while caspase 3 and PARP cleavage becomes evident at ca. 50 nM oleandrin. Changes in caspase 8, an obligatory partner for the death receptor pathway, are not yet detectable. In the case of digitoxin [Figure 1c, right panel], caspase 9 starts to activate at ca. 70 nM, while PARP and caspase 3 begin to become activated at 70 and 500 nM, respectively. As noted for oleandrin, digitoxin dependent changes in caspase 8 also cannot yet be detected. These data thus indicate that both cardiac glycosides appear to act initially through intrinsic apoptotic signaling processes, with oleandrin being somewhat more potent than digitoxin at the 16 h time point.
To test for how early in the cytotoxicity process these changes in apoptotic biomarkers occurred, we analyzed the time course of the process over the first 16 h after addition of 100 nM oleandrin or 500 nM digitoxin. Figure 1d shows that changes in PARP activation occur in digitoxin-treated cells by 6 h while PARP changes occur in oleandrin-treated cells between the 6th h and 9th h. Obvious activation of caspase 3 and caspase 9 occurs later at 16 h and 9 h, respectively, after addition of the drugs. Consistently, caspase 8 activation is still not detectable.
To test whether caspase activation might be a downstream effect of digitoxin action, we analyzed the effects of specific caspase blockers on both caspase activation and on cell death, occurring 24 h after addition of drugs. As shown in Figure 3, all three tested caspase inhibitors, DEVD (caspase 3), IETD (caspase 8) and LEHD (caspase 9), are able to suppress substantially, although not completely, both oleandrin [Figure 3a] and digitoxin [Figure 3b]-induced cell death. The fact that the specific caspase 8 inhibitor IETD also blocked cell death to some extent suggests that caspase 8 cleavage is contributing to the process, in spite of the relative insensitivity of the western blot analysis to detect caspase 8 cleavage. Figure 3c shows that the individual caspase inhibitors successfully blocked activation of their respective substrates by the 16-h time point.
Figure 3.
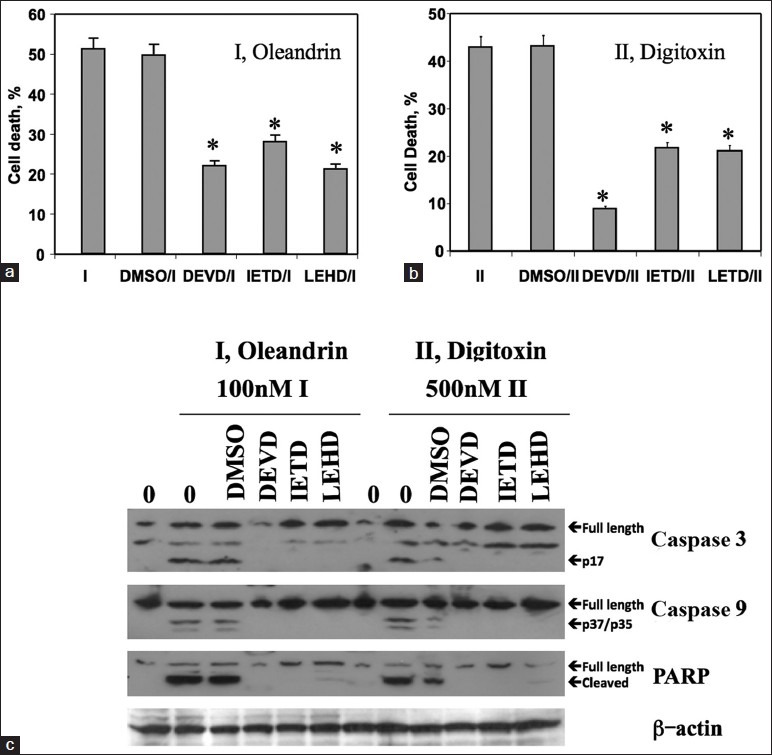
Protective effects of caspase-specific inhibitors on cardiac glycoside-induced cell death. (a) Influence of tetrapeptide inhibitors on oleandrin-induced cell death. Cells were pre - incubated in 10 μM inhibitors for 30 min and in oleandrin (I, 100 nM) for 24 h, and assayed for cell death by release of LDH. Inhibitors of caspase 3, 8, 9 blocked the cardiac glycoside-induced apoptosis pathway and cell death. (*: Significant at P < 0.05, relative to respective controls). (b) Cells were p re - incubated with inhibitors, and then with digitoxin (II, 500 nM) for 24 h and assayed for cell death by release of LDH. (*significant at P < 0.05, relative to respective controls). (c) Influence of tetrapeptide inhibitors on oleandrin (I) or digitoxin (II)-induced changes in caspase 3, caspase 9 and PARP. After 16 h incubation, cells were then collected and subjected to western blot analysis with antibodies against antigens as shown
Cardiac glycosides cause a reduction in c-MYC protein levels
Based on the recent result with the digitoxin analogue UNBS1450,[10] we turned our attention to the possibility that digitoxin itself might also induce loss of c-MYC. Figure 4a shows western blot analyses of c-MYC levels in cells which had been treated over a 16-h period with optimally cytotoxic concentrations of either oleandrin or digitoxin. Both the 64kD and 67kD splice variants of c-MYC are present, with the 64kD variant most prominent. By the respective 1 h time points, a clear difference in optical density is apparent. By 6 h, the c-MYC protein contents are reduced by more than 50%. Close scrutiny of the western blots indicates that the rate of loss of c-MYC in digitoxin may be slightly slower than the rate of loss in oleandrin. Figure 4b shows the same data graphed as a percentage of the optical density for the c-MYC band at zero time. From these data, we calculate that the cellular half-life of c-MYC is approximately 2-3 h in the presence of either drug. Since the half-life of c-MYC mRNA and c-MYC protein are both ca. thirty minutes under control conditions, it is possible that these cardiac glycosides have specifically inhibited production of c-MYC, either at the level of mRNA or at the level of protein synthesis.
Figure 4.
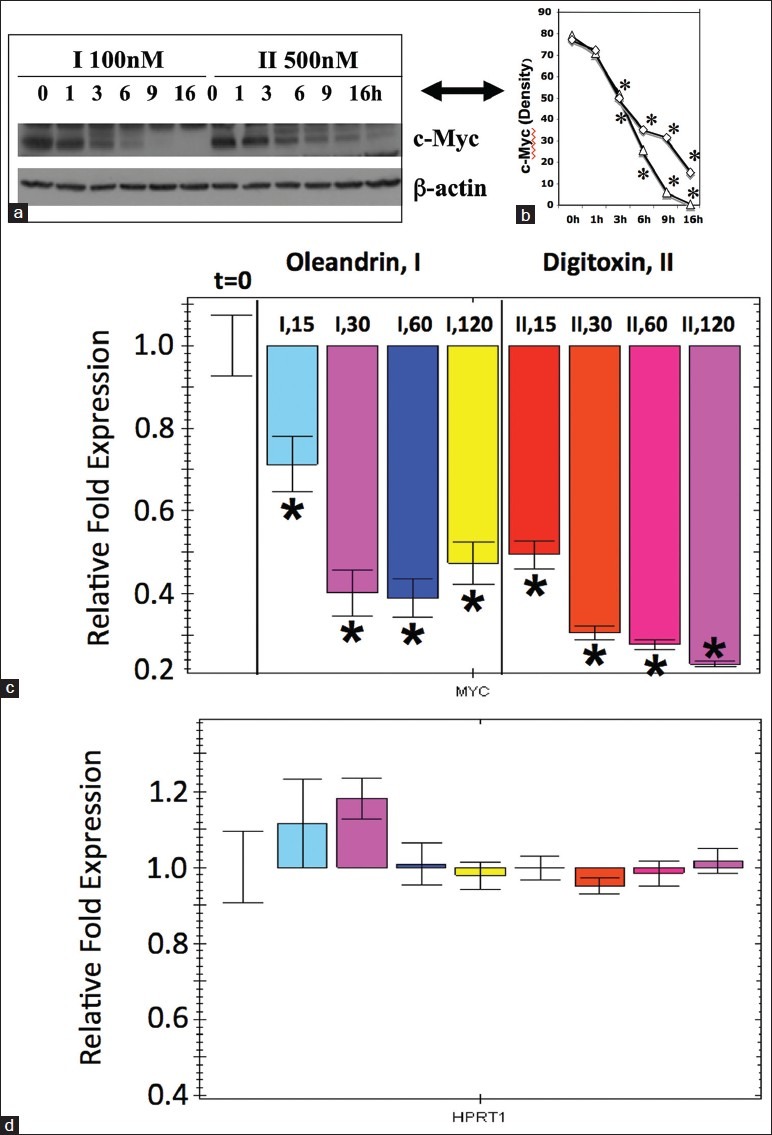
Cardiac glycosides induce rapid loss of c-MYC mRNA and protein. (a) Western blot analysis of c-MYC in cells treated with cardiac glycosides. Cells were treated with oleandrin (I, 100 nM) or digitoxin (II, 500 nM) for different times, at the concentrations shown. Western blot analysis was used to estimate levels of c-MYC remaining at each time point. The image is representative of three independent experiments.(b) Densitometric analysis of effect of cardiac glycosides on levels of immunoreactive c-MYC (Δ=I; ◊=II). Data are taken from the western blot data in a. Half-life of c-MYC in the presence of oleandrin or digitoxin is ca. 2-3 h. Differences are significant (*P < 0.05), relative to the 0-h time points. (c) Influence of cardiac glycosides on levels of c-MYC mRNA. Cells were treated with oleandrin (I, 100 nM) and digitoxin (II, 500 nM) for 15, 30, 60 and 120 min. C-MYC was quantitated by q-PCR (quantitative polymerase chain reaction) as described in the methods section. All data are statistically different from the 0-time condition on the extreme left hand side of the figure.(*P < 0.05). These data are representative of 3 independent experiments. (d) Influence of cardiac glycosides on levels of HPRT1 (hypoxanthine-guanine phosphoriribosyltransferase) in control mRNA. The same samples of RNA collected for the assay of c-MYC in c were also assayed by q-PCR for the HPRT1 mRNA. No statistically significant changes (*P < 0.05) were noted over the 2 h time period, in the presence of either oleandrin (I, 100 nM) or digitoxin (II, 500 nM). These data are representative of 3 independent experiments
Cardiac glycosides rapidly reduce cellular glutathione and increase reactive oxygen species
Cellular GSH levels are known to be tightly correlated with expression levels of c-MYC.[22–28] We therefore expected concomitantly prompt reductions in GSH following incubation with cardiac glycosides. As shown in Figure 5a by 1 h after exposure to cardiac glycosides, the initially high levels of GSH had begun to decline. By 6 h, GSH levels had fallen to ca. 50% of the initial values. Figure 5a also shows that the rate of GSH decrease in digitoxin is slightly slower than the rate in oleandrin. These slight but significant rate differences appears to parallel the drug-dependent differences in the rates of loss of c-MYC protein under the same conditions [Figure 4a and b].
Figure 5.
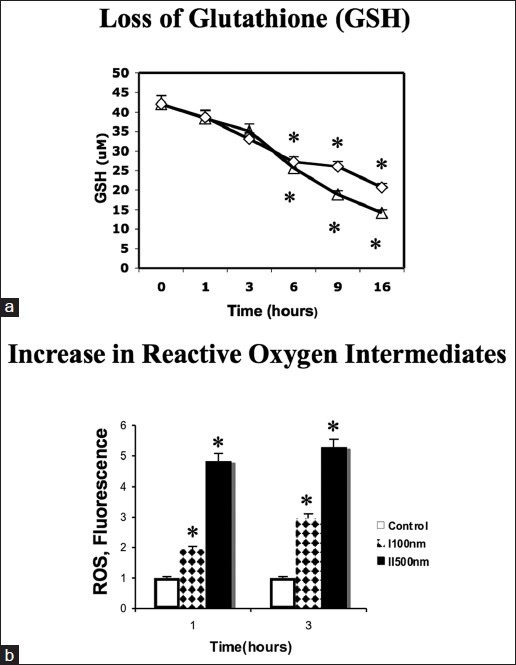
Cardiac glycosides induce rapid loss of glutathione, and elevation in reactive oxygen species. (a) Effect of oleandrin and digitoxin on levels of glutathione. Cells were treated with cardiac glycosides [Figure 3, part a] for different times. Cell pellets were suspended in 5% meta-phosphoric acid and vortexed thoroughly. Supernatant solutions were assayed for GSH. (Δ: Oleandrin (I, 100 nM); ◊: Digitoxin (II, 500 nM). Data are the average triplicate measurements and are representative of three independent experiments. Differences are significant at (*P < 0.05). (b) Effect of oleandrin and digitoxin on expression of reactive oxygen intermediates (ROS). HeLa cells were plated in black visiplates. Cells were then treated with either oleandrin (I, 100 nM) or digitoxin (II, 500 nM) for 1 and 3 h. Control and treated cells were then incubated 30 min with carboxy-H2DCFDA, and fluorescence read at Excitation 492 nm/ Emission 520 nm. Data are the average triplicate measurements and are representative of at least three independent experiments. Differences are significant at (*P < 0.05)
Levels of cellular ROS (Reactive Oxygen Species) are also known to be tightly correlated with expression levels of c-MYC.[23–28] As shown in Figure 5b, ROS levels also promptly rise following exposure to either oleandrin (I) or digitoxin (II). For example, with oleandrin treatment, ROS levels double within the 1st h of incubation, and then increase by 3 fold over the next 3 h. By contrast, with digitoxin, ROS levels increase five-fold in the 1st h, and then continue to increment slowly upwards over the 3 h time point. Thus, the drug-dependent increases in ROS levels are also approximately consistent with being downstream of the rapid time course of drug-dependent c-MYC protein loss. We therefore began a systematic analysis of factors that could mediate cardiac glycoside control of c-MYC protein expression, including changes in c-MYC mRNA and drug action at the level of the c-MYC promoter.
Cardiac glycosides cause rapid reduction of c-MYC mRNA
To determine whether the cardiac glycosides might be inhibiting c-MYC production at the level of mRNA, we isolated RNA from cells which had been treated with either oleandrin (I) or digitoxin (II). As shown in Figure 4c, within 15 min of the addition of oleandrin, the level of c-MYC mRNA drops by ca. 30% (see the I,15 bar). By 30 min, the level has dropped by ca. 60% (see the I, 30 bar). Figure 4c also shows equivalent data for digitoxin. In the latter case, levels of c-MYC have dropped by ca. 50% by 15 min after addition of drug (see the II, 15 min. bar), and by ca. 75% by 30 min after drug addition (see the II, 30 min. bar). Importantly, as shown in Figure 4d, neither oleandrin nor digitoxin caused losses in the control mRNA for HPRT1. These data thus clearly indicate that loss of c-MYC protein is kinetically heralded by profound and immediate loss of c-MYC mRNA. However, at this point it remained to be determined what mechanism might be responsible for the prompt, drug-dependent loss of c-MYC mRNA.
Cardiac glycosides suppress native and NFAT-driven luciferase expression
NFAT2 (same as NFATc1 in[21]), a member of the family of Ca2+ /calcineurin-responsive transcription factors, had been reported to upregulate c-MYC transcription in pancreatic cancer cells by binding within the proximal c-MYC promoter.[21,30] We therefore decided to investigate the possibility that digitoxin action on c-MYC might be mediated by suppression of NFAT. Our approach has been to transfect HeLa cells with a NFAT/luciferase fusion construct, and simultaneously with expression plasmids for NFAT1, 2, 3 or 4, respectively. Following a 24 h incubation period, we treated the cells with either oleandrin (I) or digitoxin (II) and assayed for luciferase activity 10 h later. As shown in Figure 6a, activities for all four NFAT isoforms were found to be reduced significantly, by ca. 70-80% compared to their respective controls. In all cases, as shown in Figure 6b, transfection of the NFAT (1-4) plasmids resulted in levels of recombinant NFAT (1-4) isoforms at substantially greater levels than those found for endogenous NFAT (1-4) isoforms.
Figure 6.
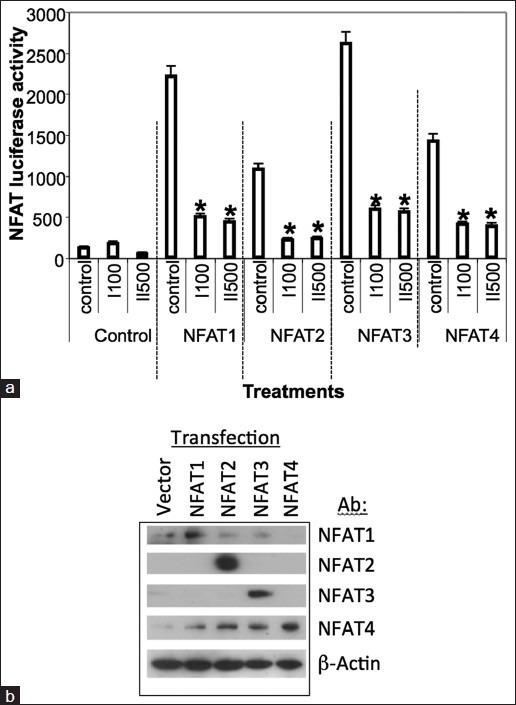
Influence of Oleandrin (I, 100nM) and digitoxin (II, 500 nM) on recombinant nuclear factor of activated T-cells 1-4 driven NFAT-luciferase - fusion expression. (a) Inhibition of nuclear factor of activated T-cells (NFAT)-driven luciferase expression by cardiac glycosides. Cells were treated with oleandrin (I, 100 nM) or digitoxin (II, 500 nM) in the presence of NFAT luciferase fusion construct and expression plasmids for NFAT1, 2, 3 or 4 for 24 h. Both drugs are effective inhibitors at these maximal concentrations. The luciferase fusion construct contains multimers of the NFAT binding site from IL-2. Data are the average of triplicate assays. Differences from respective controls are significant at (*P < 0.05). (b) Expression levels of recombinant nuclear factor of activated T-cells (NFAT) (1-4) following transfection. HeLa cells were transfected overnight with empty vector, or vector expressing NFAT1, NFAT2, NFAT3 or NFAT4. Cells were then harvested, and cultures individually analysed by western blot with antibodies against NFAT1, NFAT2, NFAT3 and NFAT4, respectively. Each cell culture listed under “Transfection” contains various amount of endogenous NFAT (1-4). Therefore, the different amounts of NFAT (1-4) detected in each culture, respectively, are indicated along the respective NFAT (1-4)-specific rows. Levels of recombinant NFAT (1-4) expression clearly depend on transfection efficiency, promoter efficiency and conditions of the cells. However, in all cases levels of recombinant protein are greater than endogenous levels
To investigate the mechanism of this process in more detail, we tested the concentration dependence of either oleandrin or digitoxin on the NFAT isoform-driven activity of the NFAT-luciferase fusion construct. For this experiment, the fusion plasmids, in combination with expression plasmids for NFAT1, NFAT2, NFAT3 or NFAT4, were transfected into HeLa cells overnight. The cells were then exposed for 10 h to different concentrations of either oleandrin or digitoxin. As shown in Figure 7a and b, both oleandrin (“I”) and digitoxin (“II”) significantly inhibit NFAT-driven luciferase activities induced by individual recombinant expressions of NFAT (1, 2, 3 or 4). The complete data sets are shown in Figure 8. For most isoforms, log/linear titrations of digitoxin and oleandrin show isoform-specific high and low affinity effects on NFAT (1, 2, 3 and 4)-driven luciferase expression. Figure 7c summarizes the highest affinity IC50s of cardiac glycosides for the inhibition of NFAT-luciferase activities, when activated individually by NFAT1, NFAT2, NFAT3, or NFAT4. These data show that NFAT1 and NFAT3 have the highest potencies for digitoxin (viz., 28 and 34 nM, respectively). NFAT2 and NFAT4 have the highest potencies for oleandrin (viz., 4.3 and 5.3 nM, respectively). Thus, each cardiac glycoside appears to exert unique suppressive effects on different NFAT isoforms as they drive NFAT dependent luciferase expression.
Figure 7.
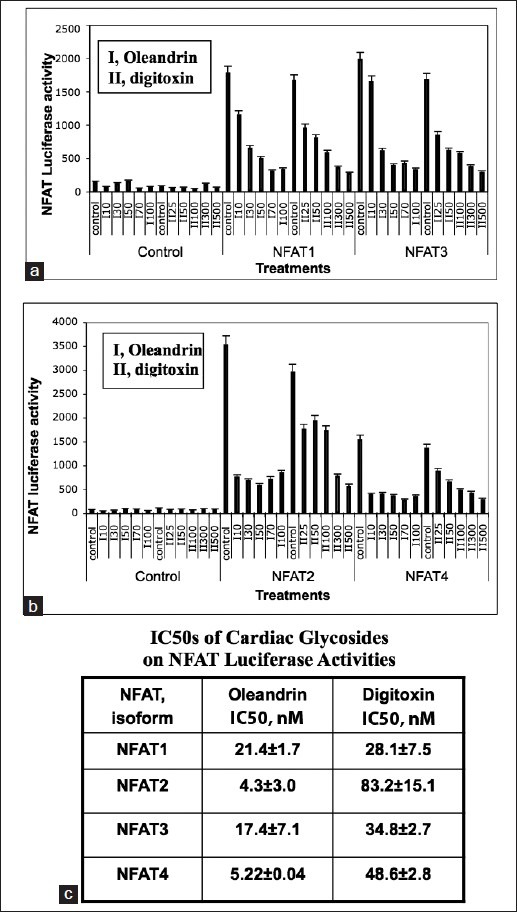
Titration of cardiac glycoside effects on nuclear factor of activated T-cells (NFAT)-driven luciferase expression, in the presence of recombinant NFAT isoforms (1-4). (a) Influence of cardiac glycosides on nuclear factor of activated T-cells (NFAT) 1 and NFAT3-driven transcription of NFAT-luciferase expression. Cells were treated for 10h with oleandrin (I: 10, 30, 50, 70, 100 nM) or digitoxin (II: 25, 50, 100, 300, 500 nM), in the presence of an NFAT luciferase fusion construct and expression plasmids for NFAT1 and NFAT3, for 24 h. The fusion construct contained multimers of the NFAT binding site from IL-2. Luciferase levels were measured and plotted. Data are the averages of triplicate assays. Differences from control for all doses of either oleandrin (I) or digitoxin (II) are significant at *P < 0.05. (b) Influence of cardiac glycosides on NFAT2 and NFAT4-driven transcription of NFAT-luciferase expression. Cells were treated for 10 h with oleandrin (I: 10, 30, 50, 70, 100 nM) or digitoxin (II: 25, 50, 100, 300, 500 nM), in the presence of an NFAT luciferase fusion construct, and expression plasmids for NFAT2 and NFAT4, for 24 h. Luciferase levels were measured and plotted. Data are the averages of triplicate assays. Differences from control for all doses of either oleandrin (I) or digitoxin (II) are significant at *P < 0.05. (c) IC50 values for high affinity cardiac glycoside inhibition of nuclear factor of activated T-cells NFAT (1-4) driven transcription of NFAT-luciferase expression. Summary data computed from Figure 8 are displayed
Figure 8.
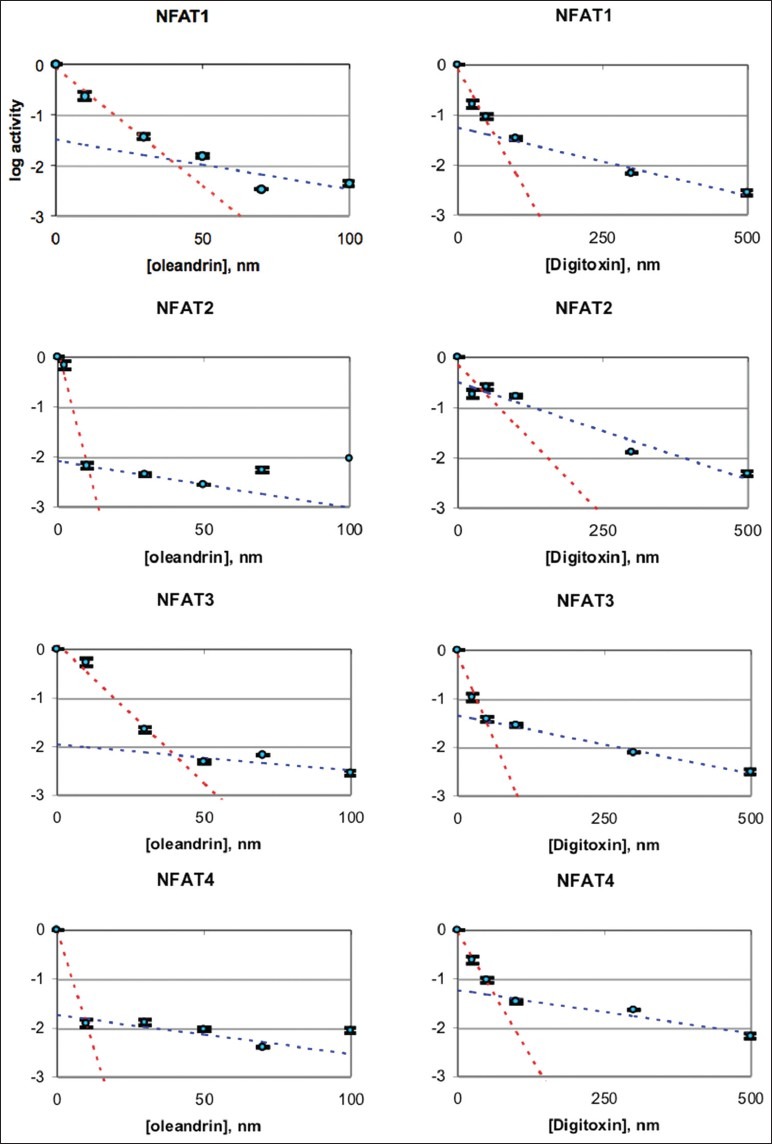
Nuclear factor of activated T-cells luciferase assays as a function of digitoxin and oleandrin concentrations, and expression of different isoforms of NFAT. The titrations of digitoxin and oleandrin occur as biphasic graphs, indicating high and low affinity effects. The values of IC50 for the high affinity effects are summarized in Figure 7c
Cardiac glycosides suppress native NFAT levels by activating caspase 3
NFAT isoforms have caspase 3 cleavage sites in their transactivation domains,[31] and we therefore tested whether digitoxin or oleandrin might suppress NFATs through activation of caspase 3. To test for cardiac glycoside effects on native NFAT isoforms, we therefore incubated cells with either oleandrin or digitoxin for 0, 6, 9 and 16 h. Figure 9a shows that the levels of native NFAT1, NFAT2, NFAT3 and NFAT4 are differentially, and significantly, reduced over this time period by treatment with either oleandrin (I) or digitoxin (II). To further test whether the reductions in native NFATs were due to caspase-3 action, we pretreated the cells for 30 min with the caspase 3 inhibitor DEVD, and then treated the cells with either oleandrin or digitoxin for 16 h. Western blot results [Figure 9b, left hand 3 lanes] show once again that the levels of native NFAT1, NFAT2, NFAT3 and NFAT4 are significantly reduced by treatment with either oleandrin (I) or digitoxin (II). However, in the DEVD pretreatment group, the levels of all NFATs are essentially at control levels. Thus, the caspase-3 inhibitor DEVD not only saves all four NFAT isoforms from cardiac glycoside-induced degradation, but also blocks cardiac glycoside-induced apoptosis and cell death [Figure 3].
Figure 9.
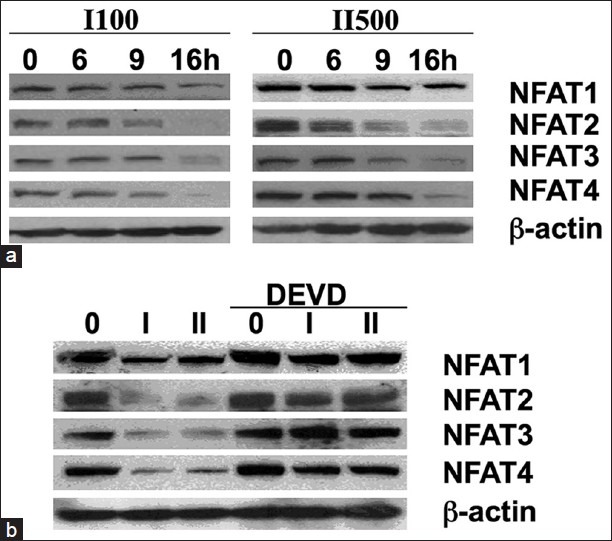
Influence of cardiac glycosides and caspase 3 inhibitor on levels of native nuclear factor of activated T-cells isoforms. (a) Time dependent changes in levels of nuclear factor of activated T-cells (NFAT) isoforms (1-4) in the presence of oleandrin (I) and digitoxin (II). Cells were treated with oleandrin (I, 100 nM) or digitoxin (II, 500 nM) for different times, at the concentrations shown. Western blot analysis was used to estimate levels of NFAT isoforms (1-4) remaining at each time point. A β-actin control is provided, which shows no significant change in β-actin relative to zero additions (*P < 0.05). (b) I nfluence o f the caspase 3-specific tetrapeptide inhibitor DEVD on native nuclear factor of activated T-cells (NFAT) isoform (1-4) levels in oleandrin-or digitoxin-treated cells. Cells were treated with oleandrin (I, 100 nM) or digitoxin (II, 500 nM) for 16 h, in the absence (left three lanes) or presence (right three lanes) of the caspase 3 inhibitor DEVD. DEVD preserves levels of native NFAT isoforms (1-4). A β-actin control is provided, which shows no significant change in β-actin relative to zero additions (*P < 0.05)
Cardiac glycosides decrease NFAT1 interaction with MYC promoter binding site
In order to determine whether decreased MYC expression after cardiac glycoside exposure is a result of decreased NFAT transactivation at MYC promoter binding sites, we tested for changes in direct interaction of NFAT1 with the proximal MYC promoter sequence. This exact sequence had been previously identified as NFAT-sensitive in pancreatic cancer cells by Buchholz et al., 2006.[21] In preparation for the experiment, we examined the sequence in silico, and verified the location and sequence of the NFAT binding domain using the Genomatix® program. To perform the experiment, we immunoprecipitated cardiac glycoside-treated HeLa cell chromatin with a NFAT1 antibody and tested for drug-dependent reductions in the c-MYC promoter. As shown in [Figure 10a], following exposure to digitoxin, we observed a 56% reduction of NFAT1 binding to the MYC promoter as compared to vehicle-only treated Hela cells. By contrast, following an oleandrin exposure we observed a 26% reduction of NFAT1 binding. Importantly, control experiments showed that cardiac glycoside exposure did not change chromatin binding for a control region upstream of the investigated promoter region, nor was chromatin binding to isotype control antibody altered during any experimental condition [Figure 10a]. A map of the locations of control and the TIE (TGFβ inhibitory element) domains is shown in Figure 10b. These results thus parallel the relative sensitivity of NFAT1 to the cardiac glycosides observed in luciferase reporter-based experiments. We suggest that these results also specifically support a rapid onset carcinotoxicity mechanism in which cardiac glycoside-dependent loss of NFAT1 suppresses c-MYC transcription, causing subsequent loss of c-MYC protein, and consequent apoptotic cell death.
Figure 10.
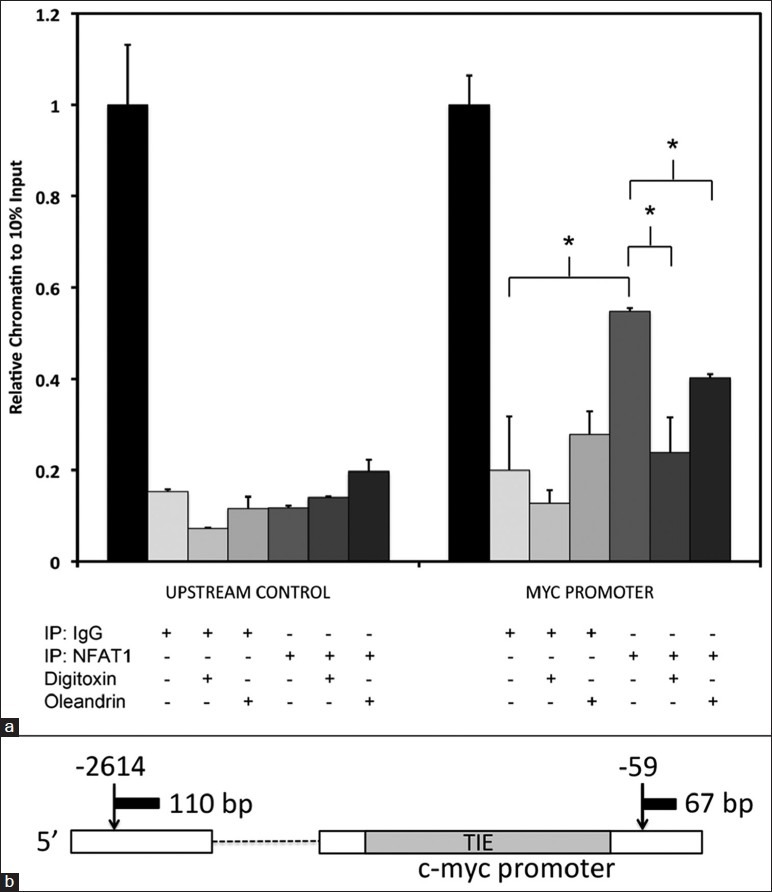
Inhibition of nuclear factor of activated T-cells 1-interaction with the c-MYC promoter by cardiac glycosides. (a) Cardiac glycoside effects on nuclear factor of activated T-cells (NFAT) 1 binding to the c-MYC promoter. Cells were treated with vehicle, digitoxin (500 nM), or oleandrin (100 nM) for 6 h. Chromatin immunoprecipitation (ChIP) was performed with either NFAT1 antibody or IgG (Immunoglobulin G) control. Quantitative PCR (polymerase chain reaction) analysis was used to estimate the level of MYC promoter or upstream control chromatin isolated during ChIP analysis. Values were normalized to those found during qPCR amplification of material from 10% of input sheared chromatin (*P < 0.05). (b) Schematic representation of qPCR amplicons for ChIP analysis of human c-Myc promoter element, including location of the TIE (TGFβ interacting element) for nuclear factor of activated T-cells (NFAT) binding. NFAT1 association was assayed by amplification of a nearby 67 bp product at position-59 from the transcription start site. The control ChIP background pull-down was assayed by amplification of a distant 110 bp product at position-2614 from the transcription start site
DISCUSSION
The data presented here support the hypothesis that digitoxin is carcinotoxic by virtue of its ability to rapidly inhibit expression of the pro-proliferative proto-oncogene c-MYC. The mechanism may depend in part on activating caspase 3, thereby proteolytically reducing the availability of NFAT to function as a transcriptional activator for the c-MYC promoter. Importantly, the digitoxin-dependent reduction in NFAT is independent of whether the stability experiment is performed on native or recombinant NFAT isoforms. This process of pro-proliferative regulation of c-MYC transcription by NFAT has been previously identified in pancreatic cancer cells,[21] and it is this specific process that digitoxin appears to also interfere with in our present HeLa cell system. Importantly, the rapid loss of c-MYC expression that occurs following treatment with digitoxin-like molecules has some precedent. Examples are PC-3 PC cells treated with the cardenolide digitoxin analogue UNBS1450,[10] and MDA breast cancer cells treated with the cardiac glycosides, peruvoside or ouabain.[32] However, our specific contribution is the demonstration that the mechanistic intermediary between digitoxin and c-MYC is caspase-3-dependent loss of NFAT isoforms 1-4, and the downstream loss of NFAT1-dependent transcriptional activation at the level of the c-MYC promoter. The relationship between digitoxin, caspase 3, and the NFAT/c-MYC signaling pathway are summarized in Figure 11.
Figure 11.
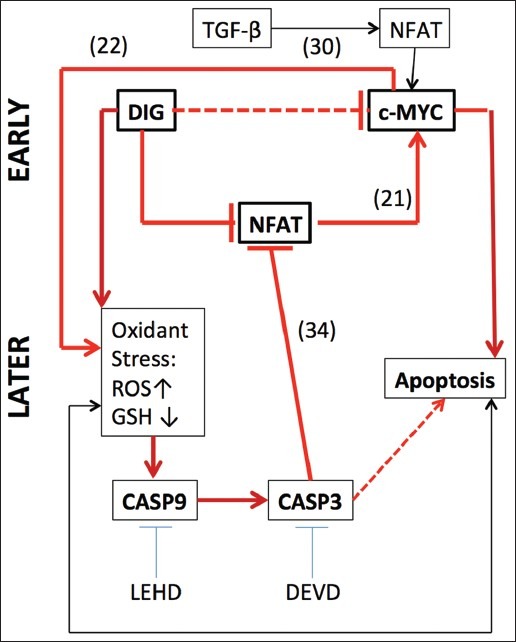
Mechanisms by which digitoxin kills tumor cells. (a) The principal finding is that at very early times after addition of digitoxin (DIG), or the analogue oleandrin, profound reductions are observed in the levels of c-MYC mRNA and protein. The delineation of EARLY and LATE time scales is more a function of how quickly endpoints for the different experiments can be reached rather than an absolute time scale. (b) The heavy horizontal dotted line indicates that multiple regulatory intermediates may be involved in the process. The mechanism (thick solid red line located in mid-Figure) depends on inhibition of nuclear factor of activated T-cells (NFAT) interaction with the proximal c-MYC promoter. This interpretation is based on chromat in immunoprecipitation studies. The interaction between specific NFAT isoforms and the promimal c-MYC promoter has a historical precedence.[21,30] (c) Markers of oxidative stress promptly appear as evidenced by elevated reactive oxygen intermediates (ROS) and loss of glutathione (GSH). Loss of c-MYC protein expression is known to be closely coupled to loss of GSH. Oxidative stress, mediated by digitoxin or loss of c-MYC, is coupled to caspase-9 (CASP9) activation, which leads to caspase-3 (CASP3)-driven proteolysis of NFAT itself.[31] However, the details of how digitoxin causes an increase in reactive oxygen species and subsequent CASP3 activation remain to be elucidated. (d) The prompt oxidant stress response may indicate that the drug action drives an intrinsic mitochondrial mechanism. The tetrapeptide inhibitors of CASP9 (LEHD) and of CASP3 (DEVD) both block digitoxin-activated apoptosis, thus providing support for the intrinsic mechanism. The apoptotic process itself also feeds back to drive the intrinsic mechanism and is marked by the lower thin black line with arrows at both ends
Mechanism for digitoxin-dependent reduction of c-MYC expression involves the destruction of NFATs
As further summarized in Figure 11, the unique contribution of this paper has been the discovery of how cardiac glycosides such as oleandrin and digitoxin might actually initiate reduction of c-MYC in treated cancer cells. Specifically, digitoxin reduces the interaction between the NFAT1 isoform and the NFAT binding site in the proximal c-MYC promoter sequence. The result is rapid suppression of c-MYC mRNA. C-MYC protein then also declines, albeit at a somewhat slower rate. This result is directly analogous to data described by Buchholz et al.,[21] who recently showed in pancreatic cancer cells that NFATc1 (NFAT2) was both constitutively overexpressed and constitutively activated. Furthermore, these investigators reported that of the 15 putative NFAT-specific sequences in the c-MYC promoter, at least one NFAT binding sequence in the proximal regulatory TIE (TGF-β inhibitory element) region, was critical for c-MYC transcriptional activity. It was therefore this region in which we chose to study digitoxin-suppression of the NFAT/c-MYC promoter interaction [Figure 10]. Although the TGF-β/SMAD (small mothers against dodecapentaplegic) pathway is inactivated in many cancers, the default process is for NFAT to activate c-MYC transcription by displacing the inhibitory SMAD transcription factor in the TIE.[30] Consistently, Buchholz et al.,[21] found that the proliferation of Panc-1 pancreatic cancer cells could be reduced by 80-90% by simply reducing NFATc1 (NFAT2) with specific siRNAs.
Nonetheless, using NFAT-luciferase expression plasmids for all four calcium-sensitive NFAT isoforms, we found that even at very low concentrations of cardiac glycosides, the luciferase fusion activities induced by all four NFAT isoforms could be significantly inhibited. The inhibition curves are not linear, thus suggesting multiple processes [viz., Figure 8, and calculated IC50 values for the respective high affinity binding sites in Figure 7c] Furthermore, the action of the caspase-3 inhibitor DEVD shows that all four NFATs are functional substrates of caspase 3. These data are therefore consistent with the demonstration by Wu et al.,[31] that at least NFATc1 (NFAT2) and NFATc2 (NFAT3) have functional caspase-3 cleavage sites in the N-terminal transactivation domain. Finally, we found that digitoxin and oleandrin have differential specificity for specific NFAT isoforms. NFAT1 and NFAT3 have the highest potencies for digitoxin (viz., 28 and 34 nM, respectively), while NFAT2 and NFAT4 have the highest potencies for oleandrin (viz., 4.3 and 5.3 nM, respectively). For this reason, we specifically picked the high potency NFAT1 for ChIP analysis of digitoxin-dependent interactions between NFAT and the proximal c-MYC promoter.
Cardiac glycosides may affect expression of other NFAT-dependent genes
Other cancer-related genes are also transcriptionally driven by NFAT, including NFAT itself,[33–36] as validated in the present paper [Figure 6, 7a and b], and HIF1 α. (hypoxia inducible factor alpha).[37] HIF1 α is a critical enabler of tumor growth in low oxygen tension environments and is sensitive to digitoxin and certain other cardiac glycosides.[38] In the case of HIF1 α,[38] digitoxin, and certain other cardiac glycosides, not only inhibit HIF1 α synthesis, but also block in vitro growth of PC PC3 cells, and liver hepatoma Hep3B-c1 cells. Furthermore, digoxin, a medicinal analogue of digitoxin Figure 2, blocks growth of P493-Myc and PC-3 prostate tumor xenografts in SCID mice.[38] Finally, independently of the cardiac glycoside narrative, Walczak-Drzeweicka et al.,[37] have demonstrated in mast cells that a well-defined relationship exists between NFAT and HIF1 α transcription. In these cells, the calcium-dependent activation of the Ca++/calcineurin signaling pathway results in translocation of cytosolic NFAT to the nucleus. There, it transcriptionally activates HIF1 α. Consistently, both activation of NFAT, and induced expression of HIF1 α are blocked by cyclosporine A (CSA) and FK506. Thus, by targeting NFAT expression and function, digitoxin, and similarly active homologues, can target multiple proliferative genes, which are independently associated with the neoplastic phenotype. In medicinal chemistry parlance, digitoxin may be a “shotgun drug,” engineered for polypharmacy by natural selection.
Ultimate cause of cancer cell death by cardiac glycosides may be cell-specific
The cytotoxic action of cardiac glycosides such as oleandrin and digitoxin have been studied in different types of cancer cells, and different candidate mechanisms of cell death have been suggested. Ouabain, digitoxin and oleandrin have been reported to induce apoptosis in androgen-independent PC cells, although the specific mechanism of drug action has not yet been identified.[33,39–46] Glioblastoma cells have been shown to be killed by an exotic cardiac glycoside-like cardenolide UNBS1450, which was tested on the basis of presumed specific activity against the NaKATPase.[44] However, the same compound was found to cause necrotic rather than apoptotic cell death of prostate cancer cells in vitro (100 nM for 24 h), and to delay cell death in mouse xenograft models.[10] These investigators also showed that within 2 h of addition of UNBS1450 to cultured prostate cancer PC-3 cells, c-MYC protein had declined to virtually zero, and stayed low for 24 h. Thus, this action on c-MYC in a prostate cancer cell line is consistent in time course with the current HeLa cell result. The difference is that a non-apoptotic process of cell death was identified for cell death due to UNBS1450. We interpret the latter data to suggest that our finding of a cardiac glycoside-dependent reduction in c-MYC in cancer cells is not without precedent, although the nature of the downstream cytotoxic consequences may be drug or cancer cell dependent. Importantly, the c-MYC oncogene is known to be activated in a wide variety of human tumors, including retinoblastoma,[13] melanoma,[45,46] pancreatic cancer,[21,29] hepatoma,[24] and others.[20,47] The HeLa cell is a particularly advantageous system for the analysis of c-MYC at the gene level since Human Papilloma Virus (HPV18) integrates at the 5’ -flanking sequence of c-MYC[48] on chromosome 8q24, without disrupting the structure of the c-MYC gene.[49] Thus it is possible that drugs such as digitoxin, which specifically target c-MYC, could have carcinolytic consequences for treatment of a broad range of malignancies. Digitoxin has been used for centuries as a chronic treatment for heart failure, and rapid translation of this insight to an off-label clinical application is therefore possible.
CONCLUSIONS
These results suggest that the carcinotoxic activity of digitoxin includes suppression of NFAT-driven c-MYC expression.
NOTE
While this article was in the PROOF stage, we became aware of an article describing a retrospective study of 145 French cancer patients, seen at the Institute Gustav Rousy, who had been coincidently treated with digoxin or digitoxin (Menger et al, 2012). These included a composite of patients with breast, colorectal, head and neck, hepatocellular, lung and prostate cancers, who, as a group, exhibited a substantial increase in survival, compared with 290 control patients, matched for disease, age and sex. (Reference: Menger L, Vacchelli E, Adjemian S, et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Science Translational Medicine Vol 4 Issue 143 143ra99, 2012)
AUTHOR's PROFILE
Qing Feng Yang, M.D., Ph.D., Department of Anatomy, Physiology and Genetics, Uniformed Services University School of Medicine, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, Bethesda, MD 20914, USA.
Clifton L. Dalgard, Ph.D., Department of Anatomy, Physiology and Genetics, Uniformed Services University School of Medicine, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, Bethesda, MD 20914, USA.
Ofer Eidelman, Ph.D., Department of Anatomy, Physiology and Genetics, Uniformed Services University School of Medicine, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, Bethesda, MD 20914, USA.
Catherine Jozwik, Ph.D., Department of Anatomy, Physiology and Genetics, Uniformed Services University School of Medicine, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, Bethesda, MD 20914, USA.
Bette S. Pollard, M.S. Office of Information Technology, Equal Employment Opportunity Commission, Washington, DC, 20500, USA.
Meera Srivastava, Ph.D., Department of Anatomy, Physiology and Genetics, Uniformed Services University School of Medicine, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, Bethesda, MD 20914, USA.
Harvey B. Pollard, M.D., Ph.D., Department of Anatomy, Physiology and Genetics, Uniformed Services University School of Medicine, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, Bethesda, MD 20914, USA.
ACKNOWLEDGMENT
The authors thank Dr. Aviva Symes for helpful discussions and for access to NFAT-related molecular biology materials.[50,51] These studies were supported by the National Institutes of Health: RO1-DK53051 (HBP) and NO1-HV-28187 (HBP).
Footnotes
Source of Support: National Institutes of Health: RO1-DK53051 (HBP) and NO1-HV-28187 (HBP)
Conflict of Interest: None declared.
REFERENCES
- 1.Haux J. Digitoxin is a potential anticancer agent for several types of cancer. Med Hypotheses. 1999;53:543–8. doi: 10.1054/mehy.1999.0985. [DOI] [PubMed] [Google Scholar]
- 2.Johansson S, Lindholm P, Gullbo J, Larsson R, Bohlin L, Claeson P. Cytotoxicity of digitoxin and related cardiac glycosides in human tumor cells. Anticancer Drugs. 2001;12:475–83. doi: 10.1097/00001813-200106000-00009. [DOI] [PubMed] [Google Scholar]
- 3.López-Lázaro M, Pastor N, Azrak SS, Ayuso MJ, Austin CA, Cortés F. Digitoxin inhibits the growth of cancer cell lines at concentrations commonly found in cardiac patients. J Nat Prod. 2005;68:1642–5. doi: 10.1021/np050226l. [DOI] [PubMed] [Google Scholar]
- 4.Ramirez-Ortega M, Maldonado-Lagunas V, Melendez-Zajgla J, Carrillo-Hernandez JF, Pastelín-Hernandez G, Picazo-Picazo O, et al. Proliferation and apoptosis of HeLa cells induced by in vitro stimulation with digitalis. Eur J Pharmacol. 2006;534:71–6. doi: 10.1016/j.ejphar.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 5.Sreenivasan Y, Raghavendra PB, Manna SK. Oleandrin-mediated expression of Fas potentiates apoptosis in tumor cells. J Clin Immunol. 2006;26:308–22. doi: 10.1007/s10875-006-9028-0. [DOI] [PubMed] [Google Scholar]
- 6.Inada A, Nakanishi T, Konoshima T, Kozuka M, Tokuda H, Nishino H, et al. Anti-tumor promoting activities of natural products. II Inhibitory effects of digitoxin on two-stage carcinogenesis of mouse skin tumors and mouse pulmonary tumors. Biol Pharm Bull. 1993;16:930–1. doi: 10.1248/bpb.16.930. [DOI] [PubMed] [Google Scholar]
- 7.Afaq F, Saleem M, Aziz MH, Mukhtar H. Inhibition of 12-O-tetradecanoyl phorbol-13-acetate-induced tumor promotion markers in CD-1 mouse skin by oleandrin. Toxicol Appl Pharmacol. 2004;195:361–9. doi: 10.1016/j.taap.2003.09.027. [DOI] [PubMed] [Google Scholar]
- 8.Nasu S, Milas L, Kawabe S, Raju U, Newman R. Enhancement of radiotherapy by oleandrin is a caspase-3 dependent process. Cancer Lett. 2002;185:145–51. doi: 10.1016/s0304-3835(02)00263-x. [DOI] [PubMed] [Google Scholar]
- 9.Frese S, Frese-Schaper M, Andres AC, Miescher D, Zumkehr B, Schmid RA. Cardiac glycosides initiate Apo2L/TRAIL-induced apoptosis in non-small cell lung cancer cells by up-regulation of death receptors 4 and 5. Cancer Res. 2006;66:5867–74. doi: 10.1158/0008-5472.CAN-05-3544. [DOI] [PubMed] [Google Scholar]
- 10.Mijatovic T, De Nève N, Gailly P, Mathieu V, Haibe-Kains B, Bontempi G, et al. Nucleolus and c-Myc: Potential targets of cardenolide-mediated antitumor activity. Mol Cancer Ther. 2008;7:1285–96. doi: 10.1158/1535-7163.MCT-07-2241. [DOI] [PubMed] [Google Scholar]
- 11.Mijatovic T, Op De Beeck A, Van Quaquebeke E, Dewelle J, Darro F, de Launoit Y, et al. The cardenolide UNBS1450 is able to deactivate nuclear factor kappaB-mediated cytoprotective effects in human non-small cell lung cancer cells. Mol Cancer Ther. 2006;5:391–9. doi: 10.1158/1535-7163.MCT-05-0367. [DOI] [PubMed] [Google Scholar]
- 12.Svensson A, Azarbayjani F, Bäckman U, Matsumoto T, Christofferson R. Digoxin inhibits neuroblastoma tumor growth in mice. Anticancer Res. 2005;25:207–12. [PubMed] [Google Scholar]
- 13.Antczak C, Kloepping C, Radu C, Genski T, Müller-Kuhrt L, Siems K, et al. Revisiting old drugs as novel agents for retinoblastoma: in vitro and in vivo antitumor activity of cardenolides. Invest Ophthalmol Vis Sci. 2009;50:3065–73. doi: 10.1167/iovs.08-3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stenkvist B, Pengtsson E, Dahlqvist B, Eriksson O, Jarkrans T, Nordin B. Cardiac glycosides and breast cancer, revisited. N Engl J Med. 1982;306:484. [PubMed] [Google Scholar]
- 15.Haux J, Klepp O, Spigset O, Tretli S. Digitoxin medication and cancer; case control and internal dose-response studies. BMC Cancer. 2001;1:11. doi: 10.1186/1471-2407-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Srivastava M, Eidelman O, Zhang J, Paweletz C, Caohuy H, Yang Q, et al. Digitoxin mimics gene therapy with CFTR and suppresses hypersecretion of IL-8 from cystic fibrosis lung epithelial cells. Proc Natl Acad Sci U S A. 2004;101:7693–8. doi: 10.1073/pnas.0402030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tabary O, Boncoeur E, de Martin R, Pepperkok R, Clément A, Schultz C, et al. Calcium-dependent regulation of NF-(kappa)B activation in cystic fibrosis airway epithelial cells. Cell Signal. 2006;18:652–60. doi: 10.1016/j.cellsig.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 18.Manna SK, Sah NK, Newman RA, Cisneros A, Aggarwal BB. Oleandrin suppresses activation of nuclear transcription factor-kappaB, activator protein-1, and c-Jun NH2-terminal kinase. Cancer Res. 2000;60:3838–47. [PubMed] [Google Scholar]
- 19.Yang Q, Huang W, Jozwik C, Lin Y, Glasman M, Caohuy H, et al. Cardiac glycosides inhibit TNF-alpha/NF-kappaB signaling by blocking recruitment of TNF receptor-associated death domain to the TNF receptor. Proc Natl Acad Sci U S A. 2005;102:9631–6. doi: 10.1073/pnas.0504097102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson EB. The many roles of c-Myc in apoptosis. Annu Rev Physiol. 1998;60:575–600. doi: 10.1146/annurev.physiol.60.1.575. [DOI] [PubMed] [Google Scholar]
- 21.Buchholz M, Schatz A, Wagner M, Michl P, Linhart T, Adler G, et al. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J. 2006;25:3714–24. doi: 10.1038/sj.emboj.7601246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biroccio A, Benassi B, Fiorentino F, Zupi G. Glutathione depletion induced by c-Myc downregulation triggers apoptosis on treatment with alkylating agents. Neoplasia. 2004;6:195–206. doi: 10.1593/neo.3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobson MD. Reactive oxygen species and programmed cell death. Trends Biochem Sci. 1996;21:83–6. [PubMed] [Google Scholar]
- 24.Xu Y, Nguyen Q, Lo DC, Czaja MJ. C-myc-Dependent hepatoma cell apoptosis results from oxidative stress and not a deficiency of growth factors. J Cell Physiol. 1997;170:192–9. doi: 10.1002/(SICI)1097-4652(199702)170:2<192::AID-JCP11>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 25.Benassi B, Fanciulli M, Fiorentino F, Porrello A, Chiorino G, Loda M, et al. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol Cell. 2006;21:509–19. doi: 10.1016/j.molcel.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: Nodes of coordination in immune signaling networks. Nat Immunol. 2009;10:348–55. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- 27.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 28.Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med. 2005;11:77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- 29.Köenig A, Linhart T, Schlengemann K, Reutlinger K, Wegele J, Adler G, et al. NFAT-induced histone acetylation relay switch promotes c-Myc-dependent growth in pancreatic cancer cells. Gastroenterology. 2010;138:1189–99. doi: 10.1053/j.gastro.2009.10.045. e1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh G, Singh SK, König A, Reutlinger K, Nye MD, Adhikary T, et al. Sequential activation of NFAT and c-Myc transcription factors mediates the TGF-beta switch from a suppressor to a promoter of cancer cell proliferation. J Biol Chem. 2010;285:27241–50. doi: 10.1074/jbc.M110.100438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu W, Misra RS, Russell JQ, Flavell RA, Rincón M, Budd RC. Proteolytic regulation of nuclear factor of activated T (NFAT) c2 cells and NFAT activity by caspase-3. J Biol Chem. 2006;281:10682–90. doi: 10.1074/jbc.M511759200. [DOI] [PubMed] [Google Scholar]
- 32.Prassas I, Paliouras M, Datti A, Diamandis EP. High-throughput screening identifies cardiac glycosides as potent inhibitors of human tissue kallikrein expression: Implications for cancer therapies. Clin Cancer Res. 2008;14:5778–84. doi: 10.1158/1078-0432.CCR-08-0706. [DOI] [PubMed] [Google Scholar]
- 33.Raghavendra PB, Sreenivasan Y, Ramesh GT, Manna SK. Cardiac glycoside induces cell death via FasL by activating calcineurin and NF-AT, but apoptosis initially proceeds through activation of caspases. Apoptosis. 2007;12:307–18. doi: 10.1007/s10495-006-0626-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Chuvpilo S, Jankevics E, Tyrsin D, Akimzhanov A, Moroz D, Jha MK, et al. Autoregulation of NFATc1/A expression facilitates effector T cells to escape from rapid apoptosis. Immunity. 2002;16:881–95. doi: 10.1016/s1074-7613(02)00329-1. [DOI] [PubMed] [Google Scholar]
- 35.Zhou B, Cron RQ, Wu B, Genin A, Wang Z, Liu S, et al. Regulation of the murine Nfatc1 gene by NFATc2. J Biol Chem. 2002;277:10704–11. doi: 10.1074/jbc.M107068200. [DOI] [PubMed] [Google Scholar]
- 36.Fretz JA, Shevde NK, Singh S, Darnay BG, Pike JW. Receptor activator of nuclear factor-kappaB ligand-induced nuclear factor of activated T cells (C1) autoregulates its own expression in osteoclasts and mediates the up-regulation of tartrate-resistant acid phosphatase. Mol Endocrinol. 2008;22:737–50. doi: 10.1210/me.2007-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walczak-Drzewiecka A, Ratajewski M, Wagner W, Dastych J. HIF-1alpha is up-regulated in activated mast cells by a process that involves calcineurin and NFAT. J Immunol. 2008;181:1665–72. doi: 10.4049/jimmunol.181.3.1665. [DOI] [PubMed] [Google Scholar]
- 38.Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105:19579–86. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arispe N, Diaz JC, Simakova O, Pollard HB. Heart failure drug digitoxin induces calcium uptake into cells by forming transmembrane calcium channels. Proc Natl Acad Sci U S A. 2008;105:2610–5. doi: 10.1073/pnas.0712270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McConkey DJ, Lin Y, Nutt LK, Ozel HZ, Newman RA. Cardiac glycosides stimulate Ca2+ increases and apoptosis in androgen-independent, metastatic human prostate adenocarcinoma cells. Cancer Res. 2000;60:3807–12. [PubMed] [Google Scholar]
- 41.Lin Y, Choksi S, Shen HM, Yang QF, Hur GM, Kim YS, et al. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem. 2004;279:10822–8. doi: 10.1074/jbc.M313141200. [DOI] [PubMed] [Google Scholar]
- 42.Yeh JY, Huang WJ, Kan SF, Wang PS. Inhibitory effects of digitalis on the proliferation of androgen dependent and independent prostate cancer cells. J Urol. 2001;166:1937–42. [PubMed] [Google Scholar]
- 43.Huang YT, Chueh SC, Teng CM, Guh JH. Investigation of ouabain-induced anticancer effect in human androgen-independent prostate cancer PC-3 cells. Biochem Pharmacol. 2004;67:727–33. doi: 10.1016/j.bcp.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 44.Lefranc F, Kiss R. The sodium pump alpha1 subunit as a potential target to combat apoptosis-resistant glioblastomas. Neoplasia. 2008;10:198–206. doi: 10.1593/neo.07928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biroccio A, Amodei S, Antonelli A, Benassi B, Zupi G. Inhibition of c-Myc oncoprotein limits the growth of human melanoma cells by inducing cellular crisis. J Biol Chem. 2003;278:35693–701. doi: 10.1074/jbc.M304597200. [DOI] [PubMed] [Google Scholar]
- 46.Newman RA, Yang P, Hittelman WN, Lu T, Ho DH, Ni D, et al. Oleandrin-mediated oxidative stress in human melanoma cells. J Exp Ther Oncol. 2006;5:167–81. [PubMed] [Google Scholar]
- 47.Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol. 2006;16:318–30. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 48.Dürst M, Croce CM, Gissmann L, Schwarz E, Huebner K. Papillomavirus sequences integrate near cellular oncogenes in some cervical carcinomas. Proc Natl Acad Sci U S A. 1987;84:10704. doi: 10.1073/pnas.84.4.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lazo PA, DiPaolo JA, Popescu NC. Amplification of the integrated viral transforming genes of human papillomavirus 18 and its 5’-flanking cellular sequence located near the myc protooncogene in HeLa cells. Cancer Res. 1989;49:4305–10. [PubMed] [Google Scholar]
- 50.Monticelli S, Rao A. NFAT1 and NFAT2 are positive regulators of IL-4 gene transcription. Eur J Immunol. 2002;32:2971–8. doi: 10.1002/1521-4141(2002010)32:10<2971::AID-IMMU2971>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 51.Jones EA, Sun D, Kobierski L, Symes AJ. NFAT4 is expressed in primary astrocytes and activated by glutamate. J Neurosci Res. 2003;72:191–7. doi: 10.1002/jnr.10584. [DOI] [PubMed] [Google Scholar]