

Abstract
Many autoimmune chronic inflammatory diseases, including multiple sclerosis, are associated with the presence of Th1 and Th17 effector CD4 T cells. Paradoxically, the principal Th1 cytokine IFNγ does not appear necessary for disease, but the key Th1-associated transcription factor Tbet has been reported to be essential for disease development. This conundrum propelled us to investigate the regulation of this transcription factor during autoimmunity. Following the onset of experimental autoimmune encephalomyelitis (EAE), we observed a preferential upregulation of Tbet by CD4 T cells within the CNS, but not the secondary lymphoid organs. These Tbet-positive CD4 T cells were capable of producing the cytokine IFNγ, and a proportion of these cells produced both IFNγ and IL-17A. Interestingly, these Tbet-positive cells were present in high frequencies during disease in IFNγ-deficient mice. Moreover, we found that CD4 T cells from IFNγ-deficient/IFNγ reporter mice upregulated the Thy1.1 reporter, indicating the presence of Th1 or “Th1-like”, Tbet-positive CD4 T cells even in the absence of the cardinal Th1 cytokine, IFNγ. These IFNγ-deficient “Th1-like” cells not only maintain multiple Th1 properties but also exhibit increased expression of genes associated with the Th17 phenotype. We further examined the requirement of other Th1-associated molecules in controlling Tbet expression during EAE, and noted that STAT1, IL-12, and IFNγ were dispensable for the induction of Tbet in vivo. Hence, this study highlights the complex regulation of Tbet and the potential unrecognized role for Th1 cells during autoimmunity.
INTRODUCTION
Multiple sclerosis (MS) is a chronic autoimmune disease of the central nervous system (CNS) that results in immune destruction of the myelin sheath (1, 2). Experimental autoimmune encephalomyelitis (EAE) is a commonly employed mouse model for MS and has been proven invaluable in dissecting the cellular and molecular components that mediate disease pathogenesis. Immunization of mice with the myelin oligodendrocyte glycoprotein (MOG)35–55 peptide induces a chronic form of EAE in which effector CD4 T cells play a central role. In addition, the fundamental requirement for CD4 T cells in mediating disease development has been demonstrated using the proteolipid protein (PLP) relapsing-remitting model of EAE, as well as passive transfer of purified, myelin-specific CD4 T cells. Together, these data indicate that CD4 T cells are critical for the induction of EAE, however which effector functions of the CD4 T cells drive the autoimmune inflammation remain ill-defined.
CD4 T cells can be divided into distinct subsets based on their functional properties: T helper 1 (Th1) cells produce IFNγ, Th2 cells secrete IL-4, IL-5, and IL-13, Th17 cells make IL-17A, IL-17F, IL-21, and IL-22, and regulatory T (Treg) cells are Foxp3-positive and produce IL-10 (3–6). It was originally proposed that Th1 cells were the pathogenic cell population during MS and EAE, as high levels of IL-12 and IFNγ were found in the CNS, CD4 T cells producing IFNγ were prevalent during disease, and adoptive transfer of Th1 polarized CD4 T cells conferred disease (7–10). While these data suggested an important role for Th1 cells during disease, mice lacking IL-12 and IFNγ signaling (IL-12p35-, IL-12Rβ2-, IFNγ-, and IFNγR-deficient mice) remained susceptible to EAE (11–14), bringing into debate the function of these cells during EAE and, more importantly, MS. Moreover, in recent years an increasing amount of data has been published indicating a principle role for Th17 cells in mediating disease induction: IL-17-producing CD4 T cells are associated with the development of disease and mice lacking factors coupled with Th17 differentiation (IL-23p19, RORγt, or IL-6) are resistant to EAE (12, 15, 16). However, neutralization of IL-17A did not completely abrogate disease, suggesting that other molecules contribute to disease pathogenesis (17). One such cytokine may be GM-CSF, as recent studies have demonstrated that expression of this cytokine by CD4 T cells is essential for the development of EAE (18, 19).
Th1 effector CD4 T cells represent a paradox during EAE, in addition to other chronic autoimmune inflammatory disorders; the cardinal Th1 cytokine, IFNγ, is dispensable for disease, however the master Th1 transcription factor, Tbet, is required for the development of EAE (7, 14, 20–22). This highlights an unrecognized role for Tbet and potentially Th1 effector CD4 T cells during EAE and MS. Therefore it is critical to understand the factors that dictate Tbet expression during disease and which cells express Tbet, as well as determine which Tbet transcriptional targets are necessary for the development of disease. In this study we have probed the expression of Tbet among the effector CD4 T cells during active EAE and show that more than half of the CD4 T cells in the inflammatory sites are Tbet-positive, and that these cells are present during disease propagated by IFNγ-deficient CD4 T cells. We demonstrate that these IFNγ-deficient, Tbet-positive “Th1-like” cells retain expression of many Th1-signature genes, whereas a subset of these cells appears to acquire properties associated with the Th17 lineage. Furthermore, we establish that the marked upregulation of Tbet during EAE is independent of prototypic Th1-associated cytokine signaling. Collectively, our findings reveal a potentially pathogenic role of Tbet expressing Th1 cells during autoimmunity that is independent of IFNγ production.
MATERIALS AND METHODS
Mice
The following mice were purchased from the Jackson Laboratories and/or were bred at the University of Alabama at Birmingham: C57BL/6 (WT), B6.129S6-Tbx21tm1Glm/J (Tbet-deficient), B6.129S7-Ifngtm1Ts/J (IFNγ-deficient), B6.129S7-Ifngrtm1Agt/J (IFNγR-deficient), B6.129P2-Stat1tm1 (STAT1-deficient) and B6.129S1-Il12atm1Jm/J (IL-12p35-deficient) mice. Ifng/Thy1.1 Bac-In Tg (IFNγ BI) mice which were previously described (23) were bred with IFNγ-deficient mice in C57BL/6 background to generate IFNγ-deficient IFNγ BAC-In mice (IFNγ−/− × IFNγ BI). 129S6/SvEv-Stat1tm1Rds (STAT1-deficient) and 129SVE (WT) mice were purchased from the Taconic Farms. All animals were bred and maintained according to Institutional Animal Care and Use Committee regulations.
EAE induction, clinical scoring, and anti-IFNγ mAb treatment
EAE was induced in experimental mice between the age of 6–12 weeks by subcutaneous immunization with 200 μg myelin oligodendrocyte glycoprotein (MOG) 35–55 peptide (Biosynthesis) emulsified with CFA (final concentration of 300 μg Mycobacterium tuberculosis; Difco) and intraperitoneal pertussis toxin (500ng; List Biological Laboratories, Inc.) injections on day 0 and 2 (24). Disease scores were monitored daily with the following criteria: 0, no disease; 1, paralyzed tail; 2.0, hind limb paresis; 2.5, one hind limb paralyzed; 3.0, both hind limbs paralyzed; 4.0, forelimbs paralyzed; 5, moribund. Anti-IFNγ neutralizing mAb treatment was performed by intraperitoneal injection of 100μg anti-IFNγ mAb (clone XMG1.2; UAB hybridoma facility) to control C57BL/6 and IL-12p35-deficient mice every 3 days from day 0 to 9 (25).
Mononuclear cell isolation and activation
Experimental mice were anesthetized and perfused with sterile PBS between 12–20 days after immunization depending on the disease severity. Brains, spinal cords, spleens, and inguinal lymph nodes were removed and single-cell suspensions were prepared by mechanical disruption. Cells were prepared and cultured with RPMI1640 with 10% FCS, 2mM L-glutamine, 100 IU/ml of penicillin, 100μg/ml of streptomycin, 1 × non-essential amino acids, 1μM of sodium pyruvate and 2.5 μM β-mercaptoethanol (R10 media). Brain and spinal cord (CNS) mononuclear cells were purified by centrifugation over a 30/70% Percoll gradient and resuspended in R10 media. Mononuclear cells were left unstimulated, activated with 10 μM MOG35–55 peptide for 16 hrs, or restimulated with PMA (50ng/ml; Sigma) and ionomycin (750ng/ml; Calbiochem) for 4 hrs. Monensin was added for the final 3 hrs of culture according to the manufacturer’s instructions (BD Pharmingen).
Surface and intracellular staining
Standard cell surface staining was performed on single cell suspensions of spleen, lymph node, and CNS mononuclear cells with anti-CD4 PerCP-Cy5.5 (RM4–5; eBioscience) and anti-Thy1.1 FITC (OX-7; BD Pharmingen) (26). Cells were then either analyzed or fixed and permeabilized with permeabilization buffer (00-8333-56; eBioscience), and stained intracellularly with anti-IFNγ eFluor 450 (XMG1.2; eBioscience), anti-IL-17A PE (TC11-18H10; BD Pharmingen), anti-Tbet Alexa Fluor 647 (4B10; eBioscience), anti-IgG1κ Alexa Fluor 647 (eBioscience), anti-TNFα FITC (MP6-XT22; eBioscience), anti-IL-2 PE-Cy7 (JES6-5H4; eBioscience), or anti-GM-CSF PE (MP1-22E9; eBioscience) according to the manufacturer’s instructions. Samples were acquired using a LSRII flow cytometer (BD Biosciences) and data were analyzed using FlowJo software (Tree Star, Inc). For FACS sorting, pooled CNS mononuclear cells were stained with anti-CD4 eFluor 450 (RM4-5; eBioscience) and anti-Thy1.1 PE (OX-7; BD Pharmingen) and sorted using FACS-Aria and FACS-Aria II cell sorters (BD Biosciences) in the UAB CFAR and UAB CAMAC flow cytometry facilities, respectively.
RNA isolation, cDNA synthesis, and real-time PCR
RNA was extracted from the sorted CD4 T cells using the RNeasy Mini kit (QIAGEN), and cDNA synthesis was performed with the iScript cDNA Synthesis kit (Bio-Rad) according to the manufacturers’ instructions. Real-time PCR was performed using iQ SYBR Green Supermix (Bio-Rad) and the primer pairs listed in Supplemental Table 1, with β2-microglobulin as the housekeeping gene. Reactions were run in triplicate on the iQ5 Multicolor real-time PCR Detection System (Bio-Rad). Relative gene expression was calculated according to the ΔΔCt method in which ΔΔCt = (ΔCt of IFNγ deficient cells − ΔCt of WT (IFNγ sufficient) cells).
Statistics
Statistical significance was calculated by unpaired Student’s t-test using Prism software (GraphPad). P values ≤ 0.05 were considered significant, unless specifically indicated otherwise in the text.
RESULTS
The transcription factor Tbet is critical for IFNγ production by CD4 T cells, as well as for the generation of Th1 effector CD4 T cells (5, 27). It has been shown that Tbet is necessary for the development of EAE, and that CD4 T cell intrinsic expression of Tbet is necessary to confer disease upon adoptive transfer (7, 20–22). To investigate what proportion of the effector CD4 T cells during disease express Tbet and if these cells represent a functionally distinct population of pathogenic effector CD4 T cells, we have utilized the MOG35–55 peptide immunization model of EAE in C57BL/6 mice (Fig. 1). At the peak of the disease symptoms (day 20), cells were isolated from secondary lymphoid tissues (spleens and inguinal lymph nodes) and the sites of active inflammation (the brains and spinal cords), and direct ex vivo intracellular Tbet staining was performed without restimulation to determine the frequency of Tbet-positive CD4 T cells. Within the spleens and lymph nodes, only a minor fraction of the CD4 T cells expressed Tbet, however a significant proportion (> 75%) of the CNS-infiltrating CD4 T cells stained positive for Tbet (Fig. 1b). These data demonstrate that Tbet expressing effector CD4 T cells are present in the CNS during EAE.
Figure 1. Tbet is essential and highly expressed in infiltrating CD4 T cells in CNS during EAE.
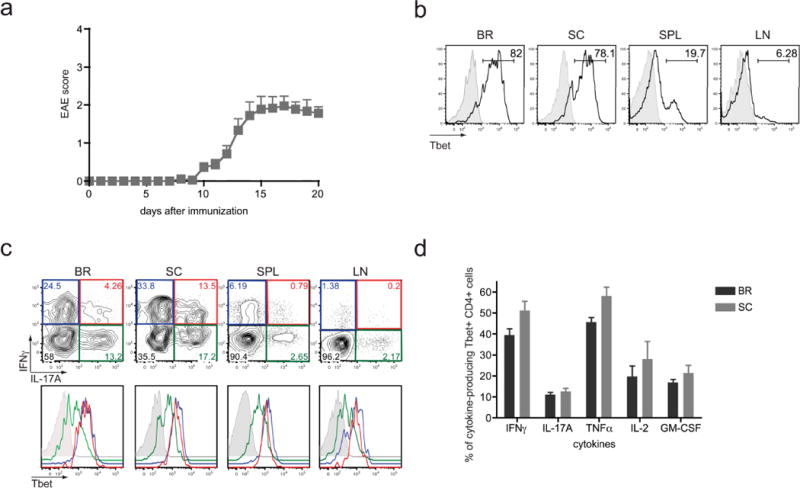
EAE was induced in WT C57BL/6 mice by MOG35–55 peptide immunization. (a) Disease severity in WT mice was monitored daily by the criteria described in Materials and Methods. Data represent two independent experiments with 5 mice in each group (means ± SEM). (b) Lymphocytes were purified from brain (BR), spinal cord (SC), spleen (SPL), and inguinal lymph nodes (LN) of WT mice 20 days after EAE induction. Samples were analyzed for Tbet expression ex vivo by intracellular staining with anti-Tbet (black histogram) and mouse IgG1κ isotype control (grey histogram) mAbs. Plots were gated on CD4 positive cells. (c) Single cell suspensions from the various tissues were restimulated with PMA/ionomycin for 4 hrs, and Tbet expression was evaluated by FACS in IFNγ single positive (blue line), IL-17A single positive (green line), and IFNγ/IL-17A double positive cells (red line) and shown by overlapping histograms. Tbet isotype control is shown in grey line. (d) Cells collected from BR and SC were restimulated with PMA/ionomycin for 4 hours and the production of IFNγ, IL-17A, TNFα, IL-2, and GM-CSF was analyzed by flow cytometry among CD4+ Tbet+ population. Data is representative of two experiments with a total of 8–9 mice.
The induction of EAE is associated with both IFNγ-positive Th1 cells and IL-17-positive Th17 cells (1, 28), yet Tbet expression is primarily linked with development of Th1 effector cells, not Th17 cells. Since we observed that the majority of the CD4 T cells in the CNS of diseased mice were Tbet-positive as well as produced IFNγ and IL-17A, we sought to determine if these cells were of the Th1 or Th17 phenotype. To visualize the cytokine production profile of the Tbet-positive CD4 T cells, intracellular staining for IFNγ, IL-17A, and Tbet was performed following a brief in vitro restimulation (Fig. 1c). As expected, three subsets of cytokine-producing CD4 T cells were detected: IFNγ single producing cells (IFNγ SP), IL-17A single producing cells (IL-17A SP), and IFNγ/IL-17A double producing cells (IFNγ/IL-17A DP). Analysis of the Tbet levels within these individual subsets revealed a strong correlation between Tbet expression and IFNγ production, as both the IFNγ SP and the IFNγ/IL-17A DP cells were Tbet-positive, while the IL-17A SP CD4 T cells demonstrated lower levels of Tbet staining. These data are consistent with published reports in which Tbet is an essential transcriptional regulator of IFNγ production in CD4 T cells and that Tbet expression is not required for IL-17A production, however this molecule can be expressed by Th17 cells under certain conditions (4, 6, 29).
In addition to the cytokines IFNγ and IL-17A, we also examined the ability of the Tbet-expressing CD4 T cells in the CNS to produce other effector cytokines. We observed that following polyclonal stimulation with PMA/ionomycin, between 25–50% of the Tbet-positive cells were capable of producing the pro-inflammatory cytokine TNFα, and a fraction of these cells made GM-CSF and/or IL-2 (Fig 1d). Similar trends were detected when cells were stimulated with the MOG35–55 peptide, however the frequencies of cytokine-producing cells were reduced, particularly for IL-2 (data not shown). Taken together, these data indicate that Tbet expression is not only associated with IFNγ production by CD4 T cells during EAE, but also additional pro-inflammatory cytokines that may contribute to disease pathogenesis.
The requirement for Tbet during EAE implies an important role for Th1 effector CD4 T cells in disease pathogenesis, nevertheless the signature Th1 cytokine, IFNγ, is not necessary for the development of the autoimmune state (7, 14, 20–22). Two potential explanations can reconcile these results: the induction of EAE in the absence of IFNγ is mediated by a Tbet-independent mechanism, or Tbet-positive CD4 T cells promote disease even in the absence of IFNγ, via an IFNγ-independent mechanism. To discriminate between these two possibilities, we examined the levels of Tbet within the effector CD4 T cells during EAE in IFNγ-deficient and IFNγR-deficient mice, both of which are devoid of IFNγ signaling. Interestingly, a significant fraction (between 40–80%) of the CNS-infiltrating CD4 T cells in the IFNγ-deficient and IFNγR-deficient mice expressed Tbet, and the frequencies and numbers of these Tbet-positive cells were similar to those observed in the WT control mice (Fig. 2b, c, e, f, and Supplemental Fig. 1). We also analyzed the effector cytokine profile of the CD4 T cells in the various cohorts of diseased mice. In keeping with previous findings that indicate IFNγ can suppress the differentiation of Th17 cells (4, 6), we detected elevated frequencies of IL-17A-producing CD4 T cells in the CNS-associated tissues of the IFNγ- and IFNγR-deficient mice (Fig. 3). Taken together, there is a correlation between the presence of Tbet-positive effector CD4 T cells in the CNS and disease pathogenesis, even when the cardinal Th1 cytokine IFNγ is not present.
Figure 2. IFNγ signaling is not required for Tbet expression in pathogenic CD4 T cells in CNS.
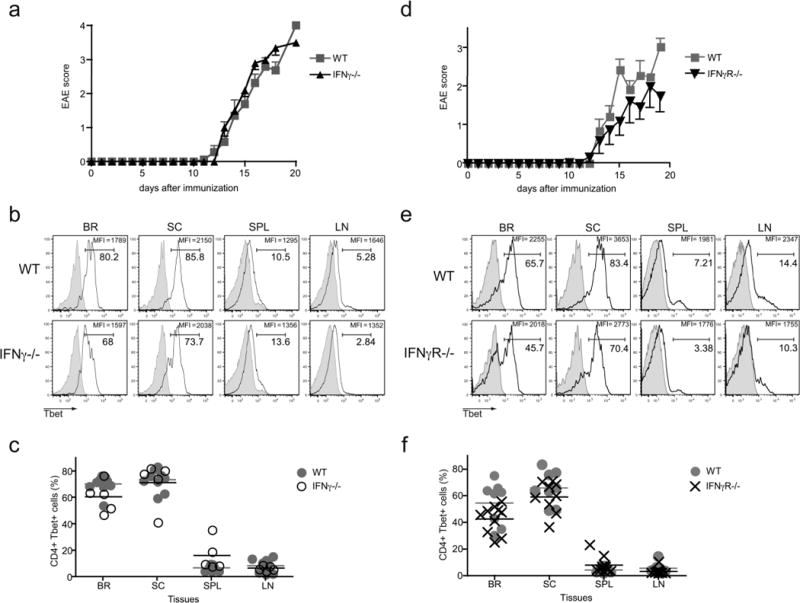
EAE was induced in WT, IFNγ-deficient, and IFNγR-deficient mice by MOG35–55 peptide immunization. (a) Disease severity in WT and IFNγ-deficient mice after EAE induction. Data is cumulative of two experiments with 5–9 mice total (means ± SEM). (b, c) Tbet expression was examined directly ex vivo in WT and IFNγ-deficient lymphocytes. (b) Representative histograms gated on CD4 positive cells are shown and percentage of Tbet-positive CD4 T cells, as well as the MFI of the Tbet staining is noted. Tbet isotype control staining is shown by the grey histograms. (c) Graphs indicate the percentages of Tbet-positive CD4 T cells in CNS from the WT and IFNγ-deficient mice. (d) Disease severity in WT and IFNγR-deficient mice after EAE induction. Results are pooled; 8–10 mice in two independent experiments (means ± SEM). (e, f) Tbet expression was evaluated in WT and IFNγR-deficient lymphocytes at 19–21 days after EAE induction. (e) Representative histograms display the Tbet staining in gated CD4 T cells. The percentage of Tbet-positive cells is noted, as well as the MFI of the Tbet staining. Isotype control staining is represented by the grey histograms. (f) Graphs denote the percentage of Tbet-positive cells in WT and IFNγR-deficient mice. Data is combined from 2 independent experiments.
Figure 3. IFNγ signaling inhibits IL-17A production in CNS infiltrating CD4 T cells during EAE.
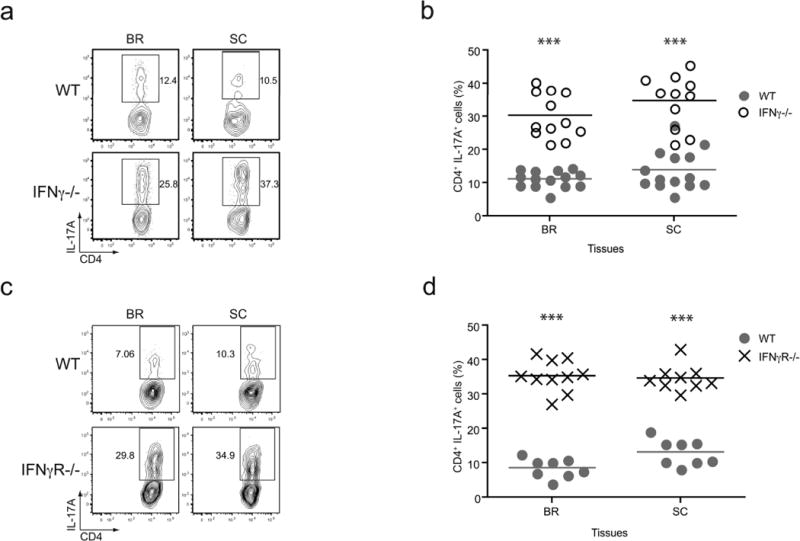
(a, b) Lymphocytes isolated from WT and IFNγ-deficient mice with EAE were restimulated with PMA/ionomycin, and subsequently stained for IL-17A production. (a) Representative plots are gated on CD4 positive T cells and the values indicate the percent of cells staining IL-17A-positive. (b) Pooled data of the frequency of IL-17A-positive CD4 T cells from multiple experiments are shown (n = 12–14, *** p < 0.005). (c, d) IL-17A production was analyzed in WT and IFNγR-deficient CD4 T cells following restimulation with PMA/ionomycin. (c) Representative plots show gated CD4 positive T cells and the percent IL-17A-positive cells is noted. (d) Collective data for the percent of IL-17A-positive CD4 T cells from multiple experiments are shown (n = 10, *** p < 0.005).
While the above data fit the explanation that Tbet-positive Th1 CD4 T cells function to promote autoimmune inflammation independent of IFNγ secretion, it is unclear if Th1 effector CD4 T cells are present in the IFNγ-deficient mice. Consistent with this concept, we noted that IFNγ-producing CD4 T cells were still present during EAE in IFNγR-deficient mice (Fig. 4), suggesting that these cells may function to drive disease separate from IFNγ secretion. To investigate the possibility that Th1 cells arise in IFNγ-deficient hosts, we have employed a unique IFNγ reporter mouse model, the IFNγ BAC-In (BI) transgenic mouse, which has been previously used to study the fate of in vivo generated Th1 effector CD4 T cells (23). These transgenic mice contain the Thy1.1 reporter molecule driven by the IFNγ promoter on a bacterial artificial chromosome and mark IFNγ-positive T cells with high fidelity (all Thy1.1+ T cells are IFNγ+, whereas 33–50% of the IFNγ+ T cells are Thy1.1+). The IFNγ BI transgenic mice were bred onto the IFNγ-deficient background, providing us with a system to identify Th1 or “Th1-like” CD4 T cells which could not produce IFNγ. Following induction of EAE in IFNγ BI and IFNγ-deficient IFNγ BI mice, we observed similar disease susceptibility between both cohorts (data not shown). Significantly, we found comparable frequencies of Thy1.1-positive CD4 T cells in the inflammatory sites between both groups of mice (Fig. 5a, b), indicating the presence of “Th1-like” effector CD4 T cells during EAE in IFNγ-deficient mice.
Figure 4. Functional Th1 cells are present during EAE irrespective of IFNγ signaling.
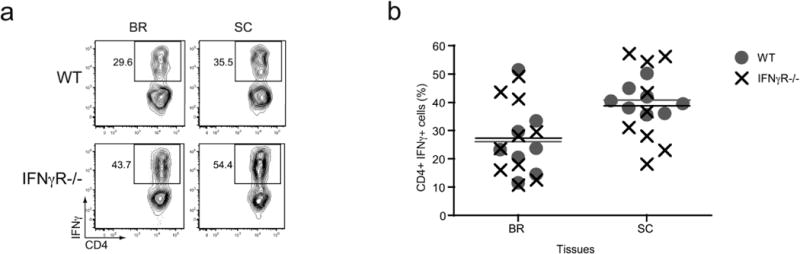
EAE was induced in WT and IFNγR-deficient mice and the CD4 T cells were analyzed for the ability to produce IFNγ after restimulation with PMA/ionomycin. (a) Representative plots show IFNγ staining of gated CD4 T cells. The frequency of IFNγ-producing CD4 T cells is noted. (b) Cumulative data of the percentage of IFNγ-positive CD4 T cells present in WT and IFNγR-deficient mice with EAE (n = 8–10).
Figure 5. The emergence of Th1 or “Th1-like” cells in during EAE.
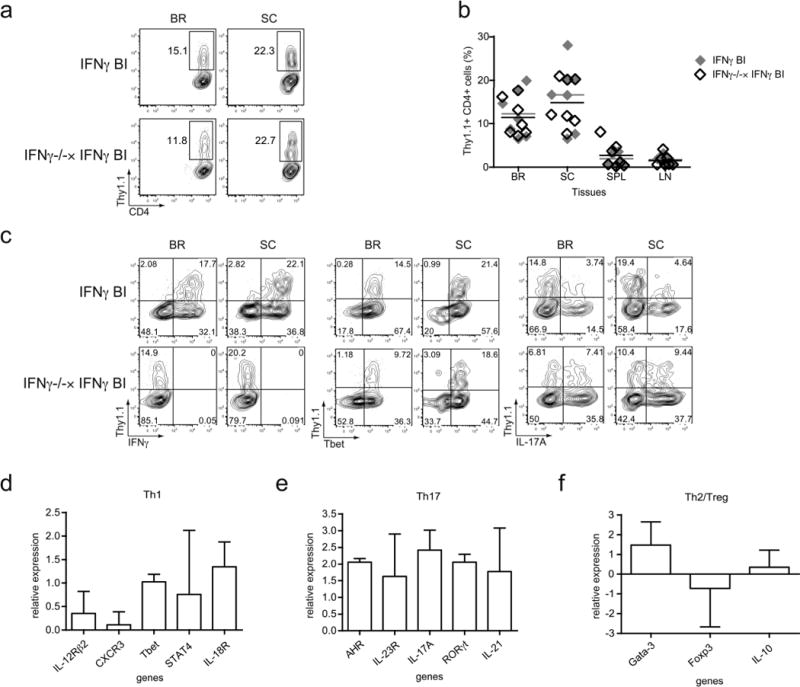
(a–c) EAE was induced in WT IFNγ BAC-In (IFNγ BI) and IFNγ-deficient IFNγ BAC-In (IFNγ−/− × IFNγ BI) transgenic mice and IFNγ reporter molecule (Thy1.1) expression was assessed 20–21 days later. (a) Representative contour plots are gated on CD4 T cells and the values indicate the percentage of Thy1.1-positive cells present in the gate. (b) Combined data from two separate experiments are shown (n = 7). (c) Cells from the BR and SC of WT and IFNγ-deficient IFNγ BI mice were stimulated for 4 hr with PMA/ionomycin and subsequently stained for CD4 and Thy1.1 in conjunction with IFNγ (left panel), Tbet (middle panel), or IL-17A (right panel). (d-f) Thy1.1-positive CD4 T cells were FACS-sorted from the CNS of WT and IFNγ-deficient IFNγ BI mice and real-time PCR was performed for genes related to the (d) Th1, (e) Th17, and (f) Th2/Treg lineages. Relative gene expression of IFNγ deficient compared to WT cells is shown as the ΔΔCt value, in which ΔΔCt = (ΔCt of the IFNγ-deficient cells – ΔCt of the WT cells). Data were combined from three independent experiments except the AHR data is from two experiments.
The observation that effector CD4 T cells from both WT and IFNγ-deficient mice express the IFNγ reporter molecule during EAE led us to examine the Th1 properties of these cells. By intracellular staining, we confirmed that the Thy1.1+ cells in the WT mice were IFNγ+, whereas the Thy1.1+ cells in the IFNγ-deficient hosts were negative for this cytokine (Fig. 5c). In keeping with our earlier data, regardless of their ability to produce the cytokine IFNγ, the Thy1.1+ CD4 T cells all expressed Tbet. In order to analyze additional genes associated with the Th1-lineage, we FACS-sorted Thy1.1+ CD4 T cells from the pooled brains and spinal cords of WT and IFNγ-deficient IFNγ BI mice and performed real-time PCR. Interestingly, the Thy1.1+ CD4 T cells from the IFNγ-deficient mice retained expression of multiple Th1-associated genes, including CXCR3, IL-12Rβ2, STAT4, and IL-18R (Fig. 5d). These data further support the concept that Tbet-positive, “Th1-like” effector CD4 T cells are present in IFNγ-deficient hosts and that these cells may contribute to disease pathogenesis independent of their ability to secrete the cardinal Th1 cytokine, IFNγ.
Our earlier data, as well as published reports, demonstrate the emergence of IFNγ+IL-17+ double-producing CD4 T cells during EAE, as well as elevated levels of Th17 cells when disease is induced in IFNγ-deficient hosts (Fig. 1c and 3). Therefore, we sought to determine if the IFNγ reporter-positive CD4 T cells that developed in the IFNγ-deficient mice exhibited properties of Th17 cells, as well as Th1 cells. Intracellular IL-17A staining was performed on CD4 T cells from the brains and spinal cords of WT and IFNγ-deficient IFNγ BI mice. In WT IFNγ BI mice a subset of Thy1.1+IL-17A+ cells was detectable which represented approximately 20% of the total Thy1.1+ cell population, a frequency that reflected the proportion of IFNγ/IL-17A DP cells out of the total IFNγ+ CD4 T cell population (Fig. 1c and 5c). However, when the IFNγ-deficient mice were analyzed we found a greater percentage of the Thy1.1+ CD4 T cells (about 50%) also expressed the cytokine IL-17A. Nevertheless, a subset of IFNγ reporter-positive cells did emerge in these hosts that was devoid of IL-17A expression. We extended this analysis by performing real-time PCR for several Th17-associated genes on FACS-sorted Thy1.1+ CD4 T cells from the CNS of WT and IFNγ-deficient mice. In keeping with the increased proportion of IL-17A+ cells, we also observed modest elevations in the expression of the Th17-associated transcription factors RORγt and AHR, as well as the cytokine IL-21 and IL-23R (Fig. 5e). Taken together, these data suggest that within the increased IL-17A population of cells seen in IFNγ-deficient hosts, a subset of these cells may display features that are characteristic of the IFNγ/IL-17A DP cells, not simply Th17 cells. Still, it does appear that a unique, “Th1-like” effector CD4 T cell population develops in the IFNγ-deficient hosts.
It has been previously published that induction of Tbet expression in CD4 T cells during Th1 differentiation is dependent on IFNγ and STAT1 (30, 31), however we found high levels of Tbet expression within CD4 T cells during EAE in the absence of IFNγ signaling (Fig 2). We did observe a slight reduction in the mean fluorescence intensity (MFI) of the Tbet staining, suggesting lower levels of Tbet protein on a per cell basis, but this did not reach statistical significance. Since IFNγ signaling via STAT1 has been shown to be important for Tbet upregulation in Th1 cells (31), we next analyzed Tbet levels in STAT1-deficient CD4 T cells during EAE in both B6 and 129S6 backgrounds. In the experiments utilizing mice on the B6 background, the percentage of Tbet-positive cells, as well as the disease intensity, was comparable between B6 WT and STAT1-deficient mice (Fig. 6a, b and data not shown). In 129S6 cohorts, even though the control mice exhibited mild disease and the Tbet expression was not as high as in B6 control mice, the STAT1-deficient mice did develop exacerbated EAE and high frequencies of Tbet-positive CD4 T cells were identified in the brains and spinal cords of diseased mice (Fig. 6b and Supplemental Table 2). A slight diminution in the MFI of the Tbet staining was detectable, suggesting that the IFNγ/STAT1 pathway is not necessary for Tbet gene expression but may function to augment Tbet levels. These findings demonstrate a potential alternative pathway regulates Tbet induction during EAE.
Figure 6. The development of Tbet-positive effector CD4 T cells during EAE is independent of STAT1, IL-12, and IFNγ signaling.
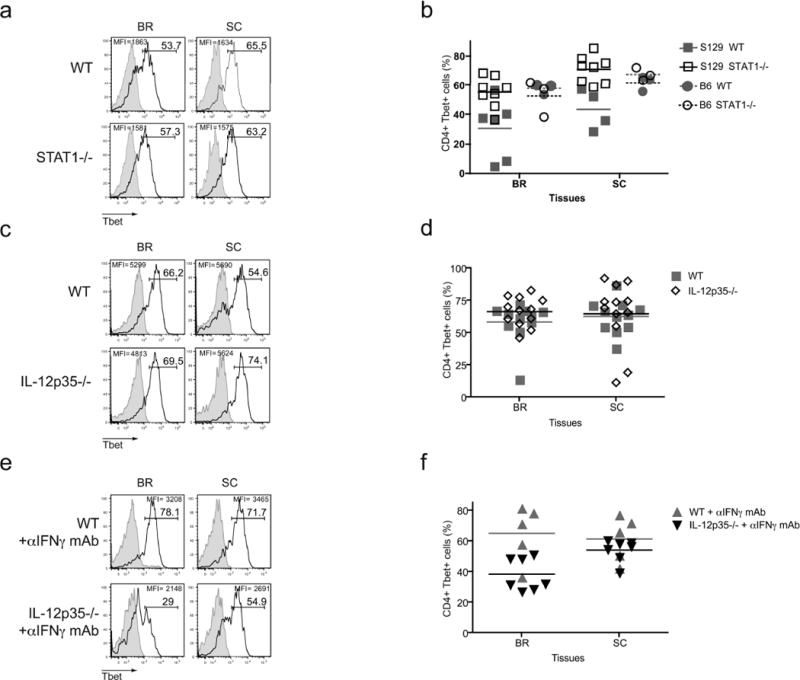
(a, b) WT and STAT1-deficient mice in either 129S6 or B6 background were actively induced with EAE. (a) The frequencies of Tbet-positive CD4 T cells in B6 cohorts were evaluated. (b) Combined data are shown for both 129S6 and B6 genetic backgrounds. (c, d) EAE was induced in WT and IL-12p35 deficient mice. CD4 T cells were examined by FACS for Tbet expression 20 days after disease induction. (e, f) WT and IL-12p35-deficient mice were immunized with MOG35–55 peptide and treated α-IFNγ mAb (100μg) every three days from day 0–9. Tbet expression was analyzed on day 13–21 after immunization. All histograms show gated CD4 T cells and isotype control staining is represented by the grey histograms. Experiments were performed 2–3 times, 5–15 mice total.
In vitro differentiation of Th1 cells is achieved by stimulating naïve CD4 T cells in the presence of IL-12, thus we wanted to test if the induction of Tbet expression during EAE was the result of IL-12 signaling (3, 32). To examine the role of IL-12 in Tbet regulation during EAE, we immunized WT and IL-12p35-deficient mice with MOG35–55 peptide and analyzed the CNS-infiltrating CD4 T cells at the peak of disease severity. These mice are known to develop EAE, as the IL-23 cytokine network is still intact (12). Staining for Tbet in the effector CD4 T cells revealed no differences in the percentages of Tbet-positive CD4 T cells, as well as the MFI of Tbet staining, between the control and IL-12p35-deficient mice, indicating IL-12 is not essential for Tbet expression during EAE (Fig. 6c, d). Still, it is possible that in the absence of IL-12, IFNγ signaling acts in a compensatory manner to promote Tbet induction and vice versa. Therefore, we investigated how the upregulation of Tbet in CD4 T cells was influenced when both IFNγ and IL-12 signaling were abrogated. Concurrent with EAE immunization, WT and IL-12p35-deficient mice were treated with anti-IFNγ neutralizing mAb to block IFNγ signaling. These treated mice developed EAE similar to control mice, as shown in Supplemental Table 2. CD4 T cells isolated from the brains and spinal cords were analyzed for Tbet expression at the peak of disease. We did observe an increase in the frequency of IL-17A-producing CD4 T cells in the IL-12p35-/-mice in which αIFNγ mAb was administered, indicating that the neutralization protocol did have an impact in vivo (Supplemental Fig. 2). Interestingly, we did note a decrease in the percentage of Tbet-positive CD4 T cells in the brains of mice lacking IL-12 and IFNγ signaling, however this difference in Tbet staining was not observed in the spinal cords of immunized mice (Fig. 6e, f). So, while the absence of IL-12 and IFNγ did have some impact on frequency of Tbet expressing CD4 T cells during EAE, it was not absolute. Therefore, other factors besides the traditionally Th1-associated cytokines must regulate Tbet induction in vivo.
DISCUSSION
During EAE, the major Th1 cytokine IFNγ is not necessary for disease induction, however the main Th1 associated transcription factor Tbet is, resulting in a conundrum regarding the role of Th1 cells during EAE (7, 14, 20–22). In this study we demonstrate that Tbet expressing CD4 T cells are present in the CNS in abundant frequencies during disease in WT, as well as IFNγ-deficient mice. These data, in conjunction with the upregulation of an IFNγ reporter transgene by CD4 T cells genetically deficient in IFNγ, suggest that Tbet-positive Th1 (or Th1-like) cells contribute to EAE independent of IFNγ expression. This is consistent with data published by Yang et al., who reported that IFNγ-deficient CD4 T cells from the CNS of mice with active EAE upregulate Tbet following an overnight stimulation with MOG35–55 peptide. Tbet is known to regulate the expression of many genes, both in a positive and negative manner, such as IL-12Rβ2, CXCR3, osteopontin, SOCS1, and SOCS3 (31, 33–35). Therefore Tbet may be important for the upregulation of pathogenic genes necessary for disease, or Tbet may be critical for the downmodulation of suppressive genes which prevent the onset of inflammation.
Induction of Tbet expression during in vitro Th1 differentiation has been shown to be IFNγ and STAT1 dependent (30, 31), however in this report we reveal that upregulation of Tbet within effector CD4 T cells during EAE is independent of both IFNγ and STAT1 signaling. Moreover, we show that Tbet expression during EAE does not require IL-12, a robust Th1 potentiating cytokine. TCR signaling and NFAT are known to be important for Tbet upregulation, however this pathway alone is unable to cause Tbet expression (WY and LEH, unpublished data). These data highlight the differences observed between the in vitro and in vivo studies regarding Tbet regulation and indicate that other factors besides the notable Th1-associated proinflammatory cytokines are likely to mediate Tbet expression during EAE. One possibility is that Tbet upregulation is actively suppressed, by molecules such as TGFβ, masking the positive impact of various molecules to turn on Tbet expression (29, 36).
In this study we demonstrate that Tbet was expressed by more than 60% (up to 80% in certain mice) of the CD4 T cells present in the inflammatory tissues during disease, as opposed to less than 20% of the CD4 T cells in the secondary lymphoid tissue. This differential expression of Tbet by the CNS-infiltrating CD4 T cells may reflect the preferential recruitment of Tbet-positive CD4 T cells to the inflammatory site or the upregulation of Tbet upon entry into the inflammatory area. Tbet was highly expressed in CD4 T cells which produced IFNγ, both the IFNγ SP and IFNγ/IL-17A DP cells, however lower levels of Tbet were detected in the IL-17A SP cells. Interestingly, within the IL-17A SP cells there are both Tbethi and Tbetlo populations, suggesting the Th17 cells in the CNS during EAE are heterogeneous. Tbet has been demonstrated to suppress IL-17A production in certain circumstances (4, 6, 29), hence the expression of Tbet by the IL-17A SP as well as IFNγ/IL-17A DP cells is quite interesting. Previously, it has been shown that in vitro polarized Th17 cells can transition into Th1 cells upon repeated stimulation and this evolution requires Tbet (37). It is tempting to speculate that these Tbet-positive, IL-17A SP or IFNγ/IL-17A DP cells found in the CNS during EAE are in the process of converting from a Th17 phenotype into a Th1 phenotype. Future studies will be necessary to decipher if this is the case and if this progression is essential for the onset of EAE. Recently, Herota et al., published a report supporting this hypothesis. This paper demonstrated that during EAE genetically marked IL-17A-positive CD4 T cells convert into IFNγ-producing cells overtime (38). Alternatively, another report has shown that Th17 cells polarized in the absence of TGFβ co-express Tbet and IL-17A, and MOG-specific TCR transgenic CD4 T cells activated under these conditions were able to confer EAE upon adoptive transfer into Rag-deficient hosts (29). It is possible that the Tbet-positive CD4 T cells we have identified in the CNS resemble these in vitro differentiated cells and have undergone a similar developmental program in vivo.
Our data in this report also raises interesting questions regarding the effector CD4 T cell populations that develop in IFNγ-deficient mice undergoing EAE. Using a novel strain of IFNγ reporter mice, the IFNγ-deficient IFNγ BI mice, we demonstrate that a subset of CD4 T cells turn on the IFNγ promoter in the CNS during EAE, irrespective of their ability to produce the actual cytokine. We have termed these cells “Th1-like” effector CD4 T cells because they appear to retain many of the characteristics of traditional Th1 cells. Interestingly, we noted two distinct subsets cells within this population of Thy1.1 reporter-positive CD4 T cells, those that express the Thy1.1 reporter alone and those that co-express the Thy1.1 reporter molecule in conjunction with IL-17A. We propose that the cells that co-express the Thy1.1 and IL-17A closely resemble the IFNγ/IL-17A DP cells seen in the CNS of WT mice, whereas the CD4 T cells which are Thy1.1-positive but do not express IL-17A are similar to the IFNγ SP cells. Hence the increased levels of Th17 cells observed in the IFNγ-deficient hosts are likely more heterogeneous in nature than previously believed. Future studies will need to be performed to determine how each of these cell populations contribute to the pathogenesis associated with EAE in the IFNγ-deficient hosts and if these cell populations are also present in other chronic inflammatory disorders.
Overall, in this report we demonstrate that the majority of effector CD4 T cells present in the CNS during EAE express Tbet, even under circumstances in which IFNγ-producing CD4 T cells are not present due to genetic deletion of the Ifng gene. This suggests a potential pathogenic function for Th1 CD4 T cells during EAE which is separate from the secretion of the cytokine IFNγ. Interestingly, the development of this autoreactive Tbet-positive CD4 T cell population is independent of the canonical Th1-inducing cytokines IFNγ and IL-12. Hence what molecules regulate Tbet expression in vivo and what genes downstream of Tbet drive EAE, represent prospective therapeutic targets for MS and other chronic inflammatory diseases.
Supplementary Material
Acknowledgments
We wish to thank the other members of the Harrington laboratory, as well as Eleonore Beurel and the Zajac laboratory, for helpful discussions and critical reading of this manuscript. We also thank Dr. Chander Raman for the generous gift of B6 STAT1-deficient mice, Dr. Xiangqin Cui for assistance with statistics of real-time PCR data, as well as Enid Keyser and Marion Spell for FACS sorting.
This study was supported by grants from the National Multiple Sclerosis Society (TA 3025-A-1) and NIH (DK084082) to LEH, and WY is supported by the National Multiple Sclerosis Society Award CA-1059-A-13 and the UAB Collaborative MS Research Center.
References
- 1.Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. 2009;9:393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 3.Zhu J, Paul W. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harrington L, Hatton R, Mangan P, Turner H, Murphy T, Murphy K, Weaver C. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 5.Szabo S, Kim S, Costa G, Zhang X, Fathman C, Glimcher L. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 6.Park H, Li Z, Yang X, Chang S, Nurieva R, Wang Y, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Y, Weiner J, Liu Y, Smith AJ, Huss DJ, Winger R, Peng H, Cravens PD, Racke MK, Lovett-Racke AE. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med. 2009;206:1549–1564. doi: 10.1084/jem.20082584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker D, O’Neill JK, Turk JL. Cytokines in the central nervous system of mice during chronic relapsing experimental allergic encephalomyelitis. Cell Immunol. 1991;134:505–510. doi: 10.1016/0008-8749(91)90321-2. [DOI] [PubMed] [Google Scholar]
- 9.Olsson T. Cytokines in neuroinflammatory disease: role of myelin autoreactive T cell production of interferon-gamma. J Neuroimmunol. 1992;40:211–218. doi: 10.1016/0165-5728(92)90135-8. [DOI] [PubMed] [Google Scholar]
- 10.Bright JJ, Musuro BF, Du C, Sriram S. Expression of IL-12 in CNS and lymphoid organs of mice with experimental allergic encephalitis. J Neuroimmunol. 1998;82:22–30. doi: 10.1016/S0165-5728(97)00184-7. [DOI] [PubMed] [Google Scholar]
- 11.Willenborg D, Fordham S, Staykova M, Ramshaw I, Cowden W. IFN-gamma is critical to the control of murine autoimmune encephalomyelitis and regulates both in the periphery and in the target tissue: a possible role for nitric oxide. J Immunol. 1999;163:5278–5286. [PubMed] [Google Scholar]
- 12.Cua D, Sherlock J, Chen Y, Murphy C, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira S, Gorman D, Kastelein R, Sedgwick J. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 13.Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, Kamoun M, Rostami A. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. 2003;170:2153–2160. doi: 10.4049/jimmunol.170.4.2153. [DOI] [PubMed] [Google Scholar]
- 14.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 15.Ivanov I, McKenzie B, Zhou L, Tadokoro C, Lepelley A, Lafaille J, Cua D, Littman D. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 16.Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom T, Oukka M, Kuchroo V. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haak S, Croxford A, Kreymborg K, Heppner F, Pouly S, Becher B, Waisman A. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. 2009;119:61–69. doi: 10.1172/JCI35997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 19.El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bettelli E, Sullivan B, Szabo S, Sobel R, Glimcher L, Kuchroo V. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nath N, Prasad R, Giri S, Singh A, Singh I. T-bet is essential for the progression of experimental autoimmune encephalomyelitis. Immunology. 2006;118:384–391. doi: 10.1111/j.1365-2567.2006.02385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gocke AR, Cravens PD, Ben LH, Hussain RZ, Northrop SC, Racke MK, Lovett-Racke AE. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol. 2007;178:1341–1348. doi: 10.4049/jimmunol.178.3.1341. [DOI] [PubMed] [Google Scholar]
- 23.Harrington L, Janowski K, Oliver J, Zajac A, Weaver C. Memory CD4 T cells emerge from effector T-cell progenitors. Nature. 2008;452:356–360. doi: 10.1038/nature06672. [DOI] [PubMed] [Google Scholar]
- 24.Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1:1810–1819. doi: 10.1038/nprot.2006.285. [DOI] [PubMed] [Google Scholar]
- 25.Yang Y, Xu J, Niu Y, Bromberg J, Ding Y. T-bet and eomesodermin play critical roles in directing T cell differentiation to Th1 versus Th17. J Immunol. 2008;181:8700–8710. doi: 10.4049/jimmunol.181.12.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, Weaver CT. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3-precursor cells in the absence of interleukin 10. Nat Immunol. 2007;8:931–941. doi: 10.1038/ni1504. [DOI] [PubMed] [Google Scholar]
- 27.Szabo S, Sullivan B, Stemmann C, Satoskar A, Sleckman B, Glimcher L. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 28.Axtell R, de Jong B, Boniface K, van der Voort L, Bhat R, De Sarno P, Naves R, Han M, Zhong F, Castellanos J, Mair R, Christakos A, Kolkowitz I, Katz L, Killestein J, Polman C, de Waal Malefyt R, Steinman L, Raman C. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med. 2010;16:406–412. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O’Shea JJ. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lighvani A, Frucht D, Jankovic D, Yamane H, Aliberti J, Hissong B, Nguyen B, Gadina M, Sher A, Paul W, O’Shea J. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci USA. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nature Immunology. 2002;3:549. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 32.Jacobson N, Szabo S, Weber-Nordt R, Zhong Z, Schreiber R, Darnell JJ, Murphy K. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J Exp Med. 1995;181:1755–1762. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dorfman DM, van den Elzen P, Weng AP, Shahsafaei A, Glimcher LH. Differential expression of T-bet, a T-box transcription factor required for Th1 T-cell development, in peripheral T-cell lymphomas. Am J Clin Pathol. 2003;120:866–873. doi: 10.1309/MLUF-X0HR-5B96-GVAX. [DOI] [PubMed] [Google Scholar]
- 34.Shinohara ML, Jansson M, Hwang ES, Werneck MB, Glimcher LH, Cantor H. T-bet-dependent expression of osteopontin contributes to T cell polarization. Proc Natl Acad Sci USA. 2005;102:17101–17106. doi: 10.1073/pnas.0508666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oestreich KJ, Huang AC, Weinmann AS. The lineage-defining factors T-bet and Bcl-6 collaborate to regulate Th1 gene expression patterns. J Exp Med. 2011;208:1001–1013. doi: 10.1084/jem.20102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hermann-Kleiter N, Baier G. NFAT pulls the strings during CD4+ T helper cell effector functions. Blood. 2010;115:2989–2997. doi: 10.1182/blood-2009-10-233585. [DOI] [PubMed] [Google Scholar]
- 37.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. 2011;12:255–263. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.