

Abstract
Mildly oxidized low density lipoprotein (mLDL) acutely increases the permeability of the vascular endothelium to molecules that would not otherwise cross the barrier. We have shown that ascorbic acid tightens the permeability barrier in endothelial barrier in cells, so in this work we tested whether it might prevent the increase in endothelial permeability due to mLDL. Treatment of EA.hy926 endothelial cells with mLDL decreased intracellular GSH and activated the cells to further oxidize the mLDL. mLDL also increased endothelial permeability over 2 h to both inulin and ascorbate in cells cultured on semi-permeable filters. This effect was blocked by microtubule and microfilament inhibitors, but not by chelation of intracellular calcium. Intracellular ascorbate both prevented and reversed the mLDL-induced increase in endothelial permeability, an effect mimicked by other cell-penetrant antioxidants. These results suggest a role for endothelial cell ascorbate in ameliorating an important facet of endothelial dysfunction caused by mLDL.
Keywords: paracellular transport, oxidative stress, endothelial dysfunction
Introduction
Dysfunction of the vascular endothelium is an early stage of atherosclerosis that can be induced by oxidized low density lipoprotein [1,2]. One of the hallmarks of early endothelial dysfunction due to oxidized LDL is increased vascular permeability to serum proteins, which has been observed both in vivo [3,4] and in vitro [5,6]. Oxidized LDL increases endothelial permeability by causing a cytoskeleton-dependent contraction of the endothelial cells and opening of gaps between adjacent cells, which then allow large molecules such as albumin to diffuse across the endothelial barrier [5,7].
Given that at least part of the cytoxicity of oxidized LDL is due to oxidative damage to the endothelial cells, small molecule antioxidants such as vitamins C and E have been tested and found in most studies to protect endothelial cells in culture from oxidized LDL-induced oxidative damage and death [8,9]. This was especially evident for vitamin C, or ascorbic acid, which prevented both cytotoxicity to the cells and further oxidative modification of LDL [10,11]. Whether this might extend to protection against oxidized LDL-induced increases in endothelial barrier permeability is unknown. Ascorbate was shown to increase barrier function in cultured endothelial cells treated with daily additions of 10–100 μM ascorbate, although the effect was attributed for the most part to increased collagen deposition induced by ascorbate over 5 days in culture. We recently found, however, that intracellular ascorbate rapidly (over 15–60 min) increased the barrier function of both immortalized (EA.hy926) and human dermal capillary endothelial cells cultured on porous membrane supports [12]. Barrier function was measured as transfer of radiolabeled inulin (molecular weight 5000–5500) and of ascorbate itself across the cell layer and membrane filter. The effect was not due to collagen deposition and was prevented by the microtubule agent colchicine, suggesting that it involved acute cytoskeletal changes.
In the present work we tested whether intracellular ascorbate can prevent the acute increase in endothelial permeability induced by mildly oxidized LDL. Again, we studied EA.hy926 endothelial cells, which are a hybridoma line derived from human umbilical vein endothelial cells [13]. In addition to providing a permeability barrier to inulin and ascorbate when cultured on semi-porous membrane supports [12], they also manifest several other features of endothelial cells [13–15], including oxidative modification of LDL [14]. We found that minimally oxidized LDL increased permeability of the endothelial barrier to both inulin and ascorbate, an effect that was completely reversed by intracellular ascorbate, likely through an antioxidant mechanism.
Materials and Methods
Materials
Reagent chemicals, including ascorbate, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM), dehydroascorbic acid (DHA), and N-2-hydroxyethylpiperazine N'-2-ethanesulfonic acid (Hepes) were supplied by Sigma-Aldrich Chemical Co. (St. Louis, MO). BAPTA-AM was initially dissolved in a small amount of dimethylsulfoxide, and then diluted with culture medium such that the final dimethylsulfoxide concentration was 0.8%. Perkin-Elmer Life and Analytical Sciences, Inc. (Boston, MA) supplied the [carboxyl-14C]inulin (molecular weight range 5000–5500, 2 mCi/g, ~10 Ci/mol).
Cell Culture
EA.hy926 cells were originally provided by Dr. Dr. Cora Edgell (University of North Carolina, Chapel Hill, NC, USA). They were cultured to confluence at 37 °C in humidified air containing 5% CO2 in Dulbecco's minimal essential medium and 10% (v/v) heat-inactivated fetal bovine serum, which contained 20 mM D-glucose and HAT media supplement (Sigma-Aldrich Chemical Co., St. Louis, MO).
Preparation of minimally oxidized LDL
LDL-enriched lipoproteins were obtained from a volunteer with familial hypercholesterolemia at the time of plasma apheresis to lower LDL. In this procedure, apolipoprotin B-containing particles were removed during apheresis from heparinized plasma using a dextran sulfate-cellulose adsorbent column of a packed volume of 150 ml (LA-15, Liposorber, Kaneka Corp., Osaka, Japan). Apolipoprotein B-enriched lipoproteins were eluted from the column resin with 70 ml of 690 mM sodium chloride. The lipoprotein solution had a protein concentration of 2–4 mg/ml and was not concentrated before subsequent storage or oxidation. It was either stored at 3 °C or immediately oxidized by incubation of 2 mg/ml LDL with 5 μM copper sulfate at 23 °C for 24 h under nitrogen [15]. The reaction was terminated by addition of EDTA to 5 mM. This preparation was dialyzed under nitrogen for 24 hours at 3 °C against 3 changes of phosphate buffered saline (12.5 mM sodium phosphate, 140 mM sodium chloride, pH 7.4) to remove the copper and EDTA. It was stored up to 3 weeks before use under nitrogen in an opaque plastic tube at 3 °C. Gel electrophoresis showed that the oxidized LDL preparation consisted largely of lipoproteins migrating in the beta range, corresponding to LDL [16]. Copper oxidation did not affect electrophoretic migration, but doubled the thiobarbituric acid-reactive substances content of the preparation when measured by HPLC as malondialdehyde [16]. This suggests that although lipids underwent peroxidation, apolipoproteins were not damaged significantly, indicating that the oxidized LDL preparation was minimally oxidized LDL (mLDL). The amount of mLDL in experiments was determined as its protein concentration in aqueous solution [11] using the Bradford reagent.
Assay of trans-endothelial ascorbate and inulin transfer
EA.hy926 cells were cultured either on standard 6-well culture plates or on polyethylene terephthalate cell culture inserts (6-well , 0.4 micron pores at a density of 2 ± 0.2 × 106 pores per cm2, Falcon BD Biosciences, Franklin Lakes, NJ). Cells were cultured to confluence and thereafter for 6–8 days with 1.7 ml of medium in the upper well and 2.8 ml of medium in the lower well. Medium was changed every 3 days. After incubations as described, either ascorbate (0.3 mM) or [carboxyl-14C]inulin (10 μM), was added above the cells/filter and incubation was carried out at 37 °C for 1 h, except where noted. For assay of ascorbate transfer, medium below the cells/filter was sampled taken for ascorbate assay as described below.
Assay of [carboxyl-14C]inulin permeability was determined as previously described [17] with minor modifications [12]. The permeability coefficients for [carboxyl-14C]inulin were calculated, including adjustment for the rate of [carboxyl-14C]inulin transfer across filters after removal of cells [18]. This corrects in each well for any changes in permeability due to deposition of the matrix laid down by the cells during culture and the experiment. The permeability of inulin was calculated as the volume or space occupied by the inulin that was transferred from above the cells to below the cells over 1 h, divided by the surface area of the filter, giving units of cm/h.
Assay of ascorbate, GSH, and lipid peroxidation
For determination of ascorbate transferred across the cells and filter, ascorbate was measured in culture medium as follows. An aliquot of medium (0.1 ml) taken from below the cells was added to 0.1 ml of 25% metaphosphoric acid (w/v), mixed, neutralized with 0.35 ml of the above phosphate/EDTA buffer, and centrifuged to remove any precipitated solids before assay of ascorbate as described below.
For assay of intracellular ascorbate, after removal of the medium, cells on the 6-well plate or filter were rinsed 3 times with Krebs-Ringer Hepes buffer (KRH) that consisted of 20 mM Hepes, 128 mM NaCl, 5.2 mM KCl, 1 mM NaH2PO4, 1.4 mM MgSO4, and 1.4 mM CaCl2, pH 7.4. After removal of the last rinse, the cell monolayer was treated with 0.1 ml of 25 % (w/v) metaphosphoric acid, detached from the filter with a rubber spatula, and then treated with 0.35 ml of 0.1M Na2HPO4 and 0.05 mM EDTA, pH 8.0. The lysate was removed and centrifuged at 3 °C for 1 min at 13,000 × g and the supernatant was taken for assay of ascorbate. Assay of ascorbic acid was performed in duplicate by high performance liquid chromatography as previously described [19]. Intracellular GSH was measured in duplicate by the method of Hissin and Hilf [20] using fluorometric detection of a conjugate of o-phthalaldehyde with GSH on extract derived from the same samples as ascorbate. Intracellular concentrations of ascorbate and GSH were calculated based on the intracellular distribution space of 3-O-methylglucose in EA.hy926 cells, which was previously measured to be 3.6 ± 1.2 μl/mg protein [21].
Lipid hydroperoxides in mLDL present in the cell incubation medium was measured using the FOX-2 assay with tert-butyl hydroperoxide as a standard [22].
Data Analysis
Results are shown as mean + standard error. Statistical comparisons were made using SigmaStat 2.0 software (Jandel Scientific, San Rafael, CA). Differences between treatments were assessed by two-way analysis of variance with post-hoc testing using Tukey's test.
Results
mLDL generates acute oxidant stress in EA.hy926 cells
Although EA.hy926 cells do not contain appreciable ascorbate in culture, incubation of these cells with 0.5 mM DHA for 15 min raised the intracellular ascorbate concentration to almost 2 mM (Fig. 1A, time zero). This increase was due to uptake of DHA on glucose transporters and intracellular reduction to ascorbate [23]. Treatment of cells with 0.2 mg/ml mLDL caused a 50% depletion of ascorbate by about 90 min that persisted to 180 min of incubation at 37 °C (Fig. 1A). Intracellular GSH did not change significantly (Fig. 1B, circles), and lipid hydroperoxides in the mLDL increased by a small amount at the 120 and 180 min time points (Fig. 1C, circles). On the other hand, when cells were not loaded with ascorbate prior to incubation with the mLDL, intracellular GSH progressively decreased, with a significant fall by 90 min, reaching about 50% of the time zero value at 180 min (Fig. 1B, squares). Further, lipid hydroperoxides in the mLDL progressively increased in the absence of intracellular ascorbate (Fig. 1C, squares). Thus, although intracellular ascorbate decreased in response to treatment of the cells with mLDL, ascorbate spared intracellular GSH and largely prevented the rise in lipid hydroperoxides in the mLDL due to incubation with the cells. The same concentration of LDL that had not been oxidized had no effect on intracellular ascorbate or GSH over 180 min (results not shown).
Figure 1. Time dependence of oxidative stress due to mLDL in EA.hy926 cells.
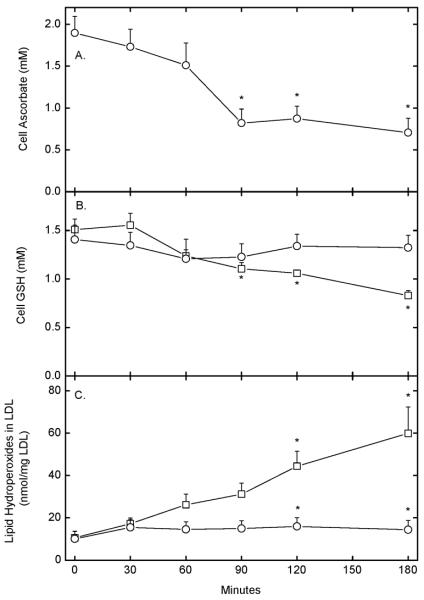
Cells were rinsed 3 times in KRH and incubated at 37 °C in KRH containing 5 mM D-glucose in the absence (squares) or presence (circles) of 0.3 mM DHA. After 15 min, 0.2 mg/ml mLDL was added and the incubation was continued for the times indicated, followed by removal of an aliquot of the medium for determination of lipid hydroperoxides (Panel C). The cells were rinsed 3 times in KRH and taken for determination of ascorbate (Panel A) and GSH (Panel B). Results are shown from 4–6 experiments for each assay, with an “*” indicating p < 0.05 compared to the zero time sample.
To assess the concentration dependence of mLDL-induced ascorbate depletion, and whether changes in extracellular ascorbate contributed to changes in intracellular ascorbate in response to mLDL, cells loaded with ascorbate by DHA treatment were incubated for 120 min at 37 °C with increasing amounts of mLDL and the ascorbate and GSH contents of the cells and the ascorbate concentration in the medium were measured. As shown in Fig. 2, extracellular ascorbate was about 4 μM and did not significantly change with increasing concentrations of mLDL. On the other hand, intracellular ascorbate decreased such that a significant difference from control was seen at mLDL concentrations of 0.1 mg/ml and 0.4 mg/ml decreased intracellular ascorbate to about 40% of normal. Intracellular GSH did not change with increasing mLDL (Fig. 2), in agreement with the time course experiment of Fig. 1B for cells loaded to about 2 mM ascorbate and treated with 0.2 mg/ml mLDL for 120 min. Thus, the mLDL-induced decrease in intracellular ascorbate depended on the concentration of mLDL and could not be accounted for by changes in extracellular ascorbate.
Figure 2. Effects of mLDL on extracellular ascorbate in DHA-loaded cells.
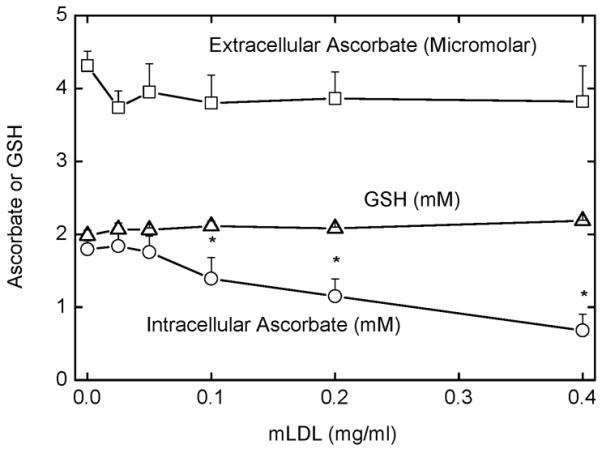
EA.hy926 cells were rinsed three times in KRH and incubated at 37 °C in KRH containing 5 mM D-glucose and 0.3 mM DHA. After 15 min, the indicated concentration of mLDL was added and the incubation was continued for an additional 2 h. Aliquots of the medium were sampled for ascorbate (squares), and intracellular ascorbate (circles) and GSH (triangles) were measured after removal of the cells. Results are shown from 4 experiments, with an “*” indicating p < 0.05 compared to the sample not exposed to mLDL.
mLDL increases endothelial barrier permeability
Incubation of EA.hy926 cells in culture on semi-porous membrane supports with 0.2 mg/ml mLDL gradually increased transfer of radiolabeled inulin across the cells and filter such that there was a significant difference by 45 min (Fig. 3). This is shown for each time point as inulin transfer. As depicted in the inset to Fig. 3,in a separate experiment, this increase persisted out to 140 min of incubation. Subsequent experiments were carried out after 1–2 h of incubation with mLDL. The increase in endothelial barrier permeability due to mLDL was concentration dependent, as shown for both inulin and ascorbate transfer in Fig. 4. This treatment did not cause morphologic changes evident on light microscopy (results not shown). Similar results have been found with mLDL prepared from different severely hypercholesterolemic donors (results not shown).
Figure 3. Time course of mLDL-induced increases in endothelial permeability.
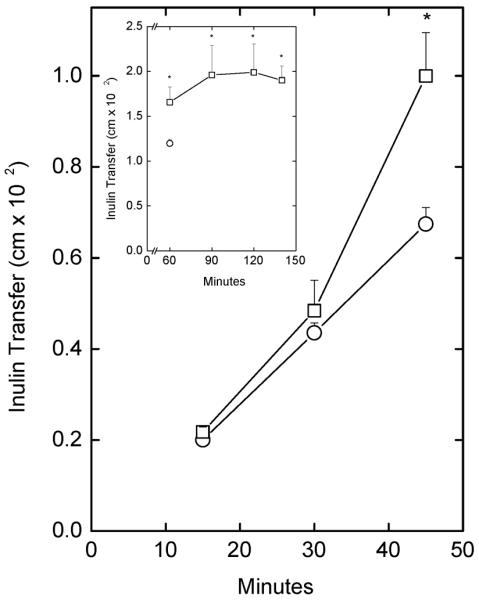
Cells cultured membrane filters in culture medium were treated without (circles) or with 0.2 mg/ml mLDL (squares) as well as 10 μM [carboxyl-14C]inulin at 37 °C. At the times indicated, the experiment was terminated and the extent of radiolabeled inulin transfer across the cells and filter was determined as described under Materials and Methods. Results are shown from 4 experiments, with an “*” indicating p < 0.05 compared to the sample at the same time point that did not receive mLDL. INSET: Longer time course of inulin transfer increase due to mLDL (squares) compared to the amount transferred at 60 min in the absence of mLDL treatment (circle). Results are shown from 6 experiments, with the asterisk indicating p < 0.05 compared to the 60 min sample without mLDL.
Figure 4. Concentration dependence of mLDL-induced increases in endothelial permeability.
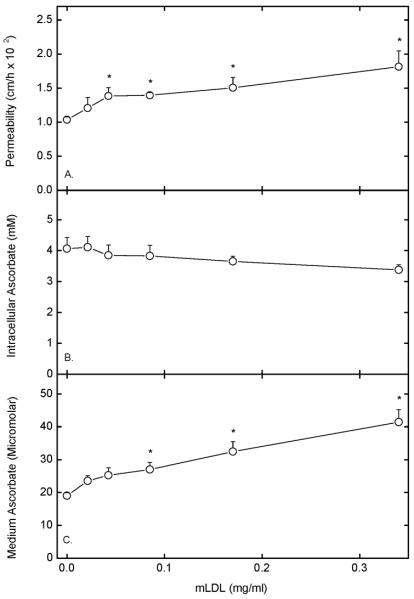
Cells cultured on filters were incubated in culture medium at 37 °C for 30 min with the indicated concentration of mLDL, followed by a 60 min assay of permeability of 10 μM [carboxyl-14C]inulin (Panel A) or of transfer of 0.3 mM ascorbate (Panel C). Intracellular ascorbate was also measured (Panel B). Results are shown from 7 experiments for inulin transfer and from 6 experiments for ascorbate transfer, with an “*” indicating p < 0.05 compared to the sample not treated with mLDL.
Permeability of the endothelial cell layer to radiolabeled inulin over a total of 90 min of incubation increased significantly at an mLDL concentration of 0.05 mg/ml and almost doubled at 0.35 mg/ml (Fig. 4A). Similar results were seen when ascorbate transfer was measured, determined as the concentration in the medium below the cells after 60 min of transfer of 0.3 mM ascorbate from above the cells (Fig. 4C). Cells accumulated ascorbate under these conditions to concentrations of about 4 mM, or twice that seen with DHA loading (Figs. 1 and 2). This uptake against a concentration gradient was greater than seen with DHA due to function of the SVCT2, which likely mediates energy- and sodium-dependent ascorbate transport in these cells [24]. Increasing mLDL caused a significant downward trend in intracellular ascorbate concentrations (p = 0.045 by linear regression analysis), although the decrease was less than that seen in DHA-loaded cells (Fig. 2), probably because of the availability of additional sources of reducing equivalents in culture medium compared to KRH containing 5 mM D-glucose alone.
To determine whether changes in the cytoskeleton were responsible for the increased permeability due to mLDL, EA.hy926 cells were incubated with 10 μM colchicine for 30 min followed by 0.2 mg/ml mLDL for 1 h, and then by the radiolabeled inulin transfer assay. As shown in Fig. 5, whereas colchicine alone had no effect on basal radiolabeled inulin transfer, it completely prevented the increase due to mLDL.
Figure 5. Prevention of mLDL-induced increases in endothelial permeability by colchicine.
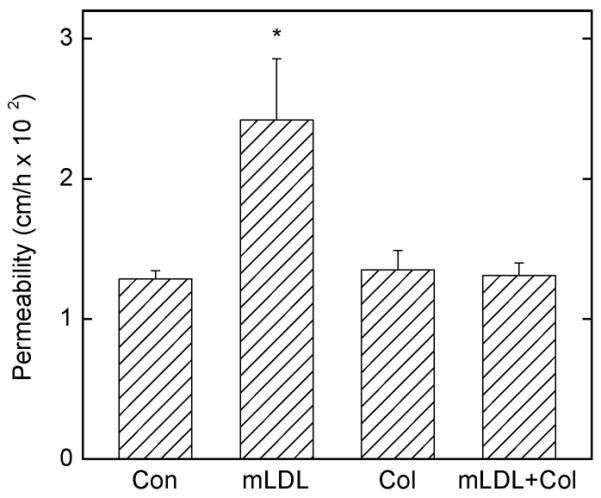
EA.hy926 cells cultured on filters were treated in culture medium at 37 °C for 30 min without (Con) or with 10 μM colchicine (Col) followed by 0.2 mg/ml mLDL as indicated for 2 h followed by the radiolabeled inulin transfer assay. Results are from 4 experiments, with an “*” indicating p < 0.05 compared to the other treatments.
To assess whether the increase in endothelial barrier permeability due to mLDL was affected by intracellular calcium, cells were incubated with increasing concentrations of the intracellular calcium chelator BAPTA-AM for 30 min, followed by 0.2 mg/ml mLDL for 60 min, and used for assays of permeability by both radiolabeled inulin and ascorbate. As shown in Fig. 6, although there was a non-significant trend downward in radiolabeled inulin transfer (Panel A), this was not seen for the mLDL-induced increase in transfer of ascorbate (Panel B). BAPTA-AM alone at concentrations up to 30 μM had no effect on transfer of either radiolabeled inulin or ascorbate (data not shown). These results show that the ability of mLDL to increase endothelial cell permeability was unaffected by chelation of intracellular calcium.
Figure 6. Failure of intracellular calcium chelation to affect the mLDL-induced increase in endothelial permeability to radiolabeled inulin or ascorbate.
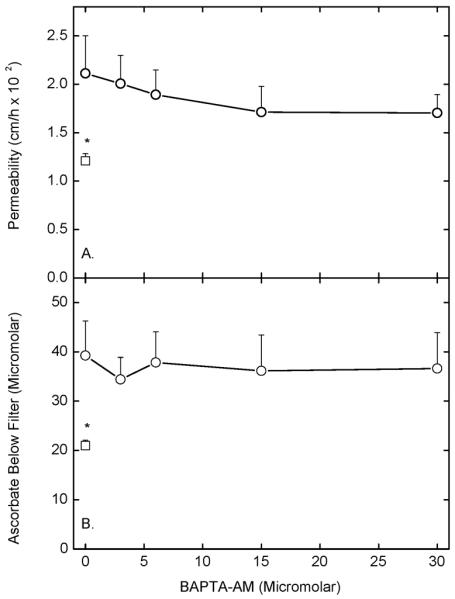
Cells cultured on filters were treated in culture medium at 37 °C for 30 min with the indicated concentration of BAPTA-AM followed by 0.2 mg/ml mLDL (circles). A control sample (squares) received neither agent. After 60 min, either 10 μM [carboxyl-14C]inulin (Panel A) or 0.3 mM ascorbate (Panel B) were added above the cells followed after 60 by assay of inulin permeability (Panel A) or measurement of the concentration of ascorbate in the well below the cells and filter (Panel B). Results are shown from 6 experiments for inulin transfer and 5 experiments for ascorbate transfer, with an asterisk indicating p < 0.05 compared to the sample that received mLDL alone.
Prevention of mLDL-induced increases in endothelial barrier permeability by ascorbate
To test whether ascorbate alone can prevent mLDL-induced increases in endothelial barrier permeability, EA.hy926 cells cultured on semi-porous filters were treated simultaneously with 0.2 mg/ml mLDL and varying concentrations of ascorbate for 60 min, followed by assay of inulin permeability. As shown in Fig. 7, 0.3 mM ascorbate alone had the expected effect to decrease inulin transfer, while mLDL alone doubled the rate of transfer. All three ascorbate concentrations tested completely prevented the mLDL-induced increase in transfer. In addition, transfer rates were suppressed to levels no different than either untreated cells or cells treated with ascorbate alone. Although not shown, the ascorbate concentration above the cells was unaffected by mLDL under these conditions (ascorbate alone: 0.32 ± 0.01 mM vs mLDL + ascorbate: 0.33 ± 0.01 mM, N = 2).
Figure 7. Inhibition of the mLDL-induced increase in endothelial permeability by ascorbate.
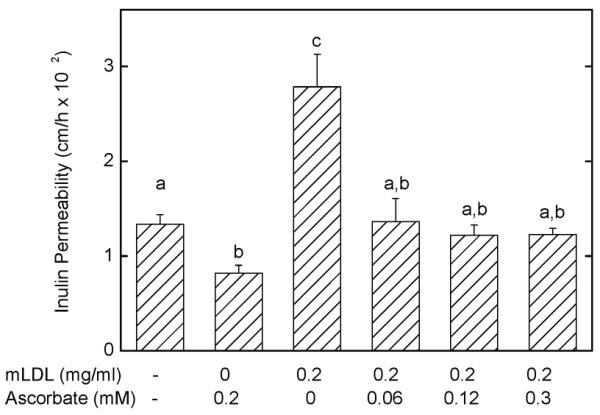
Ea.hy926 cells in culture on microporous filters were treated with mLDL or ascorbate above the cells as noted for 60 min at 37 °C, followed by addition of 10 μM [carboxyl-14C]inulin and determination of inulin permeability after an additional 60 min. Results are shown from 6 experiments with p < 0.05 differences for bars not sharing the same letters.
Intracellular ascorbate was shown in our earlier work to tighten the endothelial barrier to transfer of both radiolabeled inulin and ascorbate itself [12]. To assess whether intracellular ascorbate (as opposed to extracellular ascorbate) might affect the mLDL-induced increase in endothelial barrier permeability, cells were either loaded first with ascorbate by a 15 min incubation with DHA followed by addition of 0.2 mg/ml mLDL for 60 min (Fig. 8, open bars), or they were treated first for 60 min with 0.2 mg/ml mLDL followed by loading ascorbate with DHA for 60 min (Fig. 8, hatched bars). This different order of addition tested whether intracellular ascorbate could both prevent and reverse the increase in endothelial permeability due to mLDL. Transfer of both inulin (Fig. 8, Panel A) and ascorbate (Fig. 8, Panel B) were measured for each order of addition. As expected, intracellular ascorbate alone decreased transfer of both radiolabeled inulin (Panel A) and ascorbate (Panel B) by 30–50%. As also expected, mLDL alone increased the permeability of both inulin (Panel A) and ascorbate (Panel B). When DHA was added first, the mLDL-induced increase in permeability was decreased to control levels for both inulin and ascorbate transfer by initial DHA concentrations of 60 μM and greater (Fig. 8, last 3 open bars). When mLDL was added before DHA, a significant decrease in transfer of both agents required addition of a concentration of 120 mM DHA (Fig. 8, last 3 hatched bars). Thus, intracellular ascorbate can both prevent and reverse the increase in endothelial permeability due to mLDL, although not to levels seen with ascorbate loading alone.
Figure 8. Inhibition of the mLDL-induced increase in endothelial permeability by intracellular ascorbate.

Ea.hy926 cells in culture on filters were either treated first for 15 min with DHA followed by a 60 min treatment with mLDL (open bars), or were treated first with mLDL for 60 min followed by a 15 min treatment with DHA (hatched bars). This was followed by the [carboxyl-14C]inulin transfer assay (Panel A) or by measurement of transfer of 0.3 mM ascorbate from above to below the cells. Results are shown from 5–7 experiments, with p < 0.05 differences for bars of the same type not sharing the same letters in the same panels.
To assess whether the effect of ascorbate to prevent the mLDL-induced increase in endothelial membrane permeability might be due to an antioxidant effect, responses to several different antioxidants were examined. EA.hy926 endothelial cells that had been exposed to mLDL for 60 min were treated above the filters with the indicated concentrations DHA, Tempol, dithiothreitol, and lipoic acid. After another 30 min, the inulin transfer assay was carried out, with the results shown in Fig. 9. Of the agents tested, only DHA and dithiothreitol decreased basal endothelial permeability (Fig. 9A). On the other hand, when cells pre-treated with mLDL were then exposed to the antioxidants, each of them prevented the mLDL-induced increase in membrane permeability, although not below control rates (Fig. 9B). These results suggest that the effect of mLDL to increase endothelial permeability is due to increases in cellular oxidant stress.
Figure 9. Inhibition of mLDL-induced increases in endothelial permeability by antioxidants.

Panel A: EA.hy926 cells in culture on microporous filters were treated for 60 min above the cells with the agents noted, followed by the 60 min inulin transfer assay. Results are shown from 6 assays, with an “*” indicates p < 0.05 compared to samples receiving no additions. Panel B: Cells were treated for 60 min without or with the indicated concentration of mLDL as noted, followed by addition above the cells of the antioxidants noted. After 30 min of incubation, 10 μM [carboxyl-14C]inulin was added and the 60 min inulin transfer assay was performed. Results are shown from 5 assays, with an “*” indicating p < 0.05 compared to all other bars in Panel B.
Discussion
Treatment of endothelial cells for 90–180 min with minimally oxidized LDL caused an oxidative stress that depleted the intracellular antioxidants ascorbate and GSH, activated the cells to further oxidize the LDL, and increased permeability of the endothelial barrier to both inulin and ascorbate. When cells exposed to mLDL were loaded with ascorbate, GSH was preserved, there was minimal further oxidation of mLDL, and the increase in endothelial barrier permeability due to mLDL was both prevented and reversed.
The increase in endothelial permeability to large molecules caused by mLDL is due to cytoskeletal rearrangement with cellular contraction and opening of pores between the cells through which otherwise impermeant molecules can pass [25,26,5,7]. That this mechanism was operative in the present studies is supported by the finding that the mLDL-induced increase in endothelial permeability was prevented by colchicine, a microtubule inhibitor.
The cellular mechanism by which exposure to mLDL for minutes to several hours causes endothelial cell contraction and loss of the permeability barrier may be multifactorial. It is likely due initially to an increase in oxidative stress generated by further oxidation of mLDL in mitochondria [34] or lysosomes [27]. The effect has also been linked to mLDL-dependent increases in intracellular calcium in both cultured arterial and venous endothelial cells [28,26]. Similar acute increases in intracellular calcium following cell treatment with thrombin and histamine are known to increase endothelial barrier permeability [29]. Increased calcium in turn activates calmodulin-dependent myosin light chain kinase to phosphorylate myosin light chain, leading to cytoskeletal rearrangement and cell contraction [29–31]. Subsequent studies in cultured human umbilical vein endothelial cells failed to show an mLDL-induced increase in intracellular calcium; rather, the mLDL-induced increase in permeability was due to activation of Rho/Rho kinase causing phosphorylation and inhibition of myosin light chain phosphatase. The resulting increase in myosin light chain phosphorylation then caused increases in endothelial permeability [5]. The same group showed that a similar mechanism may also occur in response to thrombin stimulation [30]. Additional support for this mechanism derives from the observation that oxidized LDL activates the receptor for advanced glycation end products (RAGE) in vivo [31]. RAGE has recently been shown in endothelial cells to activate Rho/Rho kinase [32]. The results of the present study, that the mLDL-induced increase in endothelial barrier permeability was not reversed by chelation of intracellular calcium with BAPTA, also support the Rho/Rho kinase mechanism of activation of myosin light chain kinase with subsequent cytoskeletal rearrangement and cell contraction.
The major finding in the present study was that ascorbate prevented the mLDL-induced increase in endothelial barrier permeability. To assess whether this effect was due to intracellular ascorbate, cells were treated for 15 min or more with DHA, which is taken up on glucose transporters and rapidly reduced to ascorbate within the cells. Under these conditions, extracellular ascorbate concentrations were low (~4 μM) and stable for 2 h during mLDL treatment. Ascorbate loading of cells using this approach both prevented and reversed the mLDL-induced increase in endothelial barrier permeability. The effect of ascorbate was evident at extracellular ascorbate concentrations of 60–120 μM and at intracellular concentrations of 0.5–2.0 mM, both of which are likely to be in the physiologic range for endothelial cells in vivo [33]. These results also suggest that endothelial cell ascorbate is a factor in baseline endothelial barrier function and should be considered in studies of endothelial dysfunction due to mLDL.
The mechanism by which ascorbate alone increases endothelial barrier function is under study, but it appears that an antioxidant effect could mediate its ability to antagonize mLDL-induced increases in endothelial barrier permeability. In this scenario, oxidized LDL would generate radicals through mitochondrial [34] or lysosomal [27] activation that would in turn cause an oxidative stress in the cells [35]. That direct oxidative stress can increase endothelial barrier permeability has been demonstrated for H2O2 [36] and menadione [37] in brain microvascular endothelial cells. H2O2 directly induces oxidative stress, whereas menadione does so by redox cycling within cells [38]. Two results of this work support an antioxidant effect of ascorbate. First, it preserved intracellular GSH, either by directly reacting with intracellular radicals or by repairing protein radicals [39] before they could react with GSH. Second, several cell-penetrant antioxidants with different mechanisms of action prevented the effect of mLDL. These included Tempol, a radical species able to donate a single electron to scavenge other radicals, as well as two thiol reagents, dithiothreitol and lipoic acid. Dithiothreitol is small 4-carbon cell-penetrant molecule with 2 thiol groups that acts directly as a reducing agent. Lipoic acid, on the other hand, is an 8-carbon fatty acid with a disulfide link between sulfur atoms on carbons 6 and 8. It must first enter the cell and be reduced to dihydrolipoic acid [40], which has two thiol groups on each molecule and is a potent antioxidant. The thiols would be expected to compensate for the mLDL-induced decrease in GSH as well as directly scavenge radical species.
Although a decrease in oxidative stress due to ascorbate is likely involved in its prevention and reversal of the mLDL effect on endothelial permeability, it may not do so directly. Endothelial cell activation by mLDL has been shown to generate lysophosphatidic acid from the mLDL [7], which through binding to its cellular receptor caused endothelial stress fiber and gap formation in cultured human umbilical vein endothelial cells [7] and a decrease in trans-endothelial resistance in brain endothelial cells [41]. Our finding that intracellular ascorbate decreased endothelial cell-dependent oxidation of mLDL could result in less release of lysophosphatidic acid from the mLDL, thus decreasing the subsequent cellular response.
In conclusion, the finding that intracellular ascorbate both prevents and reverses the mLDL-induced impairment in endothelial barrier function suggest that ascorbate could help to maintain endothelial barrier permeability in vivo and thus prevent endothelial dysfunction in response to mLDL. Although the cellular and molecular mechanisms of the ascorbate effect remain to be determined, the present results support a mechanism involving decreased oxidative stress in cells containing ascorbate.
Acknowledgments
Grant support: This work was supported by NIH grant DK050435 and by the Cell Culture Core of the Vanderbilt Diabetes Research and Training Center (DK020593).
Abbreviations used
- BAPTA-AM
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
- DHA
dehydroascorbic acid
- Hepes
N-2-hydroxyethylpiperazine-NN-2-ethanesulfonic acid
- KRH
Krebs-Ringer Hepes
- mLDL
oxidized low density lipoprotein
Footnotes
Author Disclosure Statement The authors have no disclosures.
References
- 1.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 3.Liao L, Aw TY, Kvietys PR, Granger DN. Oxidized LDL-induced microvascular dysfunction. Dependence on oxidation procedure. Arterioscler Thromb Vasc Biol. 1995;15:2305–2311. doi: 10.1161/01.atv.15.12.2305. [DOI] [PubMed] [Google Scholar]
- 4.Rangaswamy S, Penn MS, Saidel GM, Chisolm GM. Exogenous oxidized low-density lipoprotein injures and alters the barrier function of endothelium in rats in vivo. Circ Res. 1997;80:37–44. doi: 10.1161/01.res.80.1.37. [DOI] [PubMed] [Google Scholar]
- 5.Essler M, Retzer M, Bauer M, Heemskerk JW, Aepfelbacher M, Siess W. Mildly oxidized low density lipoprotein induces contraction of human endothelial cells through activation of Rho/Rho kinase and inhibition of myosin light chain phosphatase. J Biol Chem. 1999;274:30361–30364. doi: 10.1074/jbc.274.43.30361. [DOI] [PubMed] [Google Scholar]
- 6.Orr AW, Stockton R, Simmers MB, Sanders JM, Sarembock IJ, Blackman BR, Schwartz MA. Matrix-specific p21-activated kinase activation regulates vascular permeability in atherogenesis. J Cell Biol. 2007;176:719–727. doi: 10.1083/jcb.200609008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siess W, Zangl KJ, Essler M, Bauer M, Brandl R, Corrinth C, Bittman R, Tigyi G, Aepfelbacher M. Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc Natl Acad Sci U S A. 1999;96:6931–6936. doi: 10.1073/pnas.96.12.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Negre-Salvayre A, Mabile L, Delchambre J, Salvayre R. α-Tocopherol, ascorbic acid, and rutin inhibit synergistically the copper-promoted LDL oxidation and the cytotoxicity of oxidized LDL to cultured endothelial cells. Biol Trace Elem Res. 1995;47:81–91. doi: 10.1007/BF02790104. [DOI] [PubMed] [Google Scholar]
- 9.Mabile L, Fitoussi G, Periquet B, Schmitt A, Salvayre R, Nègre-Salvayre A. α-tocopherol and trolox block the early intracellular events (TBARS and calcium rises) elicited by oxidized low density lipoproteins in cultured endothelial cells. Free Radic Biol Med. 1995;19:177–187. doi: 10.1016/0891-5849(95)00006-j. [DOI] [PubMed] [Google Scholar]
- 10.Martin A, Frei B. Both intracellular and extracellular vitamin C inhibit atherogenic modification of LDL by human vascular endothelial cells. Arterioscler Thromb Vasc Biol. 1997;17:1583–1590. doi: 10.1161/01.atv.17.8.1583. [DOI] [PubMed] [Google Scholar]
- 11.Sabharwal AK, May JM. alpha-Lipoic acid and ascorbate prevent LDL oxidation and oxidant stress in endothelial cells. Mol Cell Biochem. 2008;309:125–132. doi: 10.1007/s11010-007-9650-z. [DOI] [PubMed] [Google Scholar]
- 12.May JM, Qu ZC, Qiao H. Transfer of ascorbic acid across the vascular endothelium: mechanism and self-regulation. Am J Physiol Cell Physiol. 2009;297:C169–C178. doi: 10.1152/ajpcell.00674.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bauer J, Margolis M, Schreiner C, Edgell CJ, Azizkhan J, Lazarowski E, Juliano RL. In vitro model of angiogenesis using a human endothelium-derived permanent cell line: contributions of induced gene expression, G-proteins, and integrins. J Cell Physiol. 1992;153:437–449. doi: 10.1002/jcp.1041530302. [DOI] [PubMed] [Google Scholar]
- 14.Pech-Amsellem MA, Myara I, Pico I, Maziere C, Maziere JC, Moatti N. Oxidative modifications of low-density lipoproteins (LDL) by the human endothelial cell line EA.hy 926. Experientia. 1996;52:234–238. doi: 10.1007/BF01920713. [DOI] [PubMed] [Google Scholar]
- 15.Steinbrecher UP, Parthasarathy S, Leake DS, Witztum JL, Steinberg D. Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc Natl Acad Sci USA. 1984;81:3883–3887. doi: 10.1073/pnas.81.12.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chi X, May JM. Oxidized lipoprotein induces the macrophage ascorbate transporter (SVCT2): protection by intracellular ascorbate against oxidant stress and apoptosis. Arch Biochem Biophys. 2009;485:174–182. doi: 10.1016/j.abb.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siflinger-Birnboim A, del Vecchio PJ, Cooper JA, Blumenstock FA, Shepard JM, Malik AB. Molecular sieving characteristics of the cultured endothelial monolayer. J Cell Physiol. 1987;132:111–117. doi: 10.1002/jcp.1041320115. [DOI] [PubMed] [Google Scholar]
- 18.Utoguchi N, Ikeda K, Saeki K, Oka N, Mizuguchi H, Kubo K, Nakagawa S, Mayumi T. Ascorbic acid stimulates barrier function of cultured endothelial cell monolayer. J Cell Physiol. 1995;163:393–399. doi: 10.1002/jcp.1041630219. [DOI] [PubMed] [Google Scholar]
- 19.May JM, Qu Z-C, Mendiratta S. Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch Biochem Biophys. 1998;349:281–289. doi: 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- 20.Hissin PJ, Hilf R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem. 1976;74:214–226. doi: 10.1016/0003-2697(76)90326-2. [DOI] [PubMed] [Google Scholar]
- 21.Jones W, Li X, Perriott LM, Whitesell RR, May JM. Uptake, recycling, and antioxidant functions of α-lipoic acid in endothelial cells. Free Radic Biol Med. 2002;33:83–93. doi: 10.1016/s0891-5849(02)00862-6. [DOI] [PubMed] [Google Scholar]
- 22.Wolff SP. Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol. 1994;233:182–189. [Google Scholar]
- 23.May JM, Qu ZC, Li X. Requirement for GSH in recycling of ascorbic acid in endothelial cells. Biochem Pharmacol. 2001;62:873–881. doi: 10.1016/s0006-2952(01)00736-5. [DOI] [PubMed] [Google Scholar]
- 24.May JM, Qu ZC. Transport and intracellular accumulation of vitamin C in endothelial cells: relevance to collagen synthesis. Arch Biochem Biophys. 2005;434:178–186. doi: 10.1016/j.abb.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 25.Zhao B, Zhang Y, Liu B, Nawroth P, Dierichs R. Endothelial cells injured by oxidized low density lipoprotein. Am J Hematol. 1995;49:250–252. doi: 10.1002/ajh.2830490315. [DOI] [PubMed] [Google Scholar]
- 26.Zhao B, Ehringer WD, Dierichs R, Miller FN. Oxidized low-density lipoprotein increases endothelial intracellular calcium and alters cytoskeletal f-actin distribution. Eur J Clin Invest. 1997;27:48–54. doi: 10.1046/j.1365-2362.1997.750628.x. [DOI] [PubMed] [Google Scholar]
- 27.Wen Y, Leake DS. Low density lipoprotein undergoes oxidation within lysosomes in cells. Circ Res. 2007;100:1337–1343. doi: 10.1161/CIRCRESAHA.107.151704. [DOI] [PubMed] [Google Scholar]
- 28.Negre-Salvayre A, Fitoussi G, Reaud V, Pieraggi MT, Thiers JC, Salvayre R. A delayed and sustained rise of cytosolic calcium is elicited by oxidized LDL in cultured bovine aortic endothelial cells. FEBS Lett. 1992;299:60–65. doi: 10.1016/0014-5793(92)80101-l. [DOI] [PubMed] [Google Scholar]
- 29.Ehringer WD, Edwards MJ, Miller FN. Mechanisms of alpha-thrombin, histamine, and bradykinin induced endothelial permeability. J Cell Physiol. 1996;167:562–569. doi: 10.1002/(SICI)1097-4652(199606)167:3<562::AID-JCP20>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 30.Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M. Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J Biol Chem. 1998;273:21867–21874. doi: 10.1074/jbc.273.34.21867. [DOI] [PubMed] [Google Scholar]
- 31.Sun L, Ishida T, Yasuda T, Kojima Y, Honjo T, Yamamoto Y, Yamamoto H, Ishibashi S, Hirata K, Hayashi Y. RAGE mediates oxidized LDL-induced pro-inflammatory effects and atherosclerosis in non-diabetic LDL receptor-deficient mice. Cardiovasc Res. 2009;82:371–381. doi: 10.1093/cvr/cvp036. [DOI] [PubMed] [Google Scholar]
- 32.Hirose A, Tanikawa T, Mori H, Okada Y, Tanaka Y. Advanced glycation end products increase endothelial permeability through the RAGE/Rho signaling pathway. FEBS Lett. 2010;584:61–66. doi: 10.1016/j.febslet.2009.11.082. [DOI] [PubMed] [Google Scholar]
- 33.May JM, Qu ZC. Ascorbic acid efflux and re-uptake in endothelial cells: maintenance of intracellular ascorbate. Mol Cell Biochem. 2009;325:79–88. doi: 10.1007/s11010-008-0022-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mabile L, Meilhac O, Escargueil-Blanc I, Troly M, Pieraggi MT, Salvayre R, Negre-Salvayre A. Mitochondrial function is involved in LDL oxidation mediated by human cultured endothelial cells. Arterioscler Thromb Vasc Biol. 1997;17:1575–1582. doi: 10.1161/01.atv.17.8.1575. [DOI] [PubMed] [Google Scholar]
- 35.Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol. 2001;280:C719–C741. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- 36.Lee HS, Namkoong K, Kim DH, Kim KJ, Cheong YH, Kim SS, Lee WB, Kim KY. Hydrogen peroxide-induced alterations of tight junction proteins in bovine brain microvascular endothelial cells. Microvasc Res. 2004;68:231–238. doi: 10.1016/j.mvr.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 37.Lagrange P, Romero IA, Minn A, Revest PA. Transendothelial permeability changes induced by free radicals in an in vitro model of the blood-brain barrier. Free Radic Biol Med. 1999;27:667–672. doi: 10.1016/s0891-5849(99)00112-4. [DOI] [PubMed] [Google Scholar]
- 38.McCord JM, Fridovich I. The utility of superoxide dismutase in studying free radical reactions. II. The mechanism of the mediation of cytochrome c reduction by a variety of electron carriers. J Biol Chem. 1970;245:1374–1377. [PubMed] [Google Scholar]
- 39.Domazou AS, Koppenol WH, Gebicki JM. Efficient repair of protein radicals by ascorbate. Free Radic Biol Med. 2009;46:1049–1057. doi: 10.1016/j.freeradbiomed.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 40.Handelman GJ, Han D, Tritschler H, Packer L. α-lipoic acid reduction by mammalian cells to the dithiol form, and release into the culture medium. Biochem Pharmacol. 1994;47:1725–1730. doi: 10.1016/0006-2952(94)90298-4. [DOI] [PubMed] [Google Scholar]
- 41.Schulze C, Smales C, Rubin LL, Staddon JM. Lysophosphatidic acid increases tight junction permeability in cultured brain endothelial cells. J Neurochem. 1997;68:991–1000. doi: 10.1046/j.1471-4159.1997.68030991.x. [DOI] [PubMed] [Google Scholar]