

Abstract
Almost 30 years after its initial discovery, infection with the human immunodeficiency virus-1 (HIV-1) remains incurable and the virus persists due to reservoirs of latently infected CD4+ memory T-cells and sanctuary sites within the infected individual where drug penetration is poor. Reactivating latent viruses has been a key strategy to completely eliminate the virus from the host, but many difficulties and unanswered questions remain. In this review, the latest developments in HIV-persistence and latency research are presented.
Introduction
Before the introduction of highly active anti-retroviral therapy (HAART), a diagnosis of HIV/AIDS would have been a death sentence for most patients. However, modern anti-retroviral regimes are able to preserve the health of the patient and routinely reduce the plasma viral load to less than 50 copies of HIV-1 RNA ml−1 (Volberding & Deeks, 2010). Although HAART is very effective at blocking HIV-1 spread within the body, it is not a cure, as viral loads readily rebound when treatment is interrupted (Chun et al., 1999; Davey et al., 1999). Furthermore, ultrasensitive detection assays have shown that in most HAART-treated patients, a low-level viraemia of less than 5 copies ml−1 persists even after years of therapy (Chun et al., 2005; Palmer et al., 2008; Tobin et al., 2005). This low-level persistent viraemia is a major obstacle to the complete elimination of HIV-1 from the body.
HAART cannot fully restore the health of an infected individual. Long-term-treated HIV-1 patients have reduced lifespans and increased susceptibilities to non-AIDS related conditions such as cardiovascular disease, cancer, liver and kidney dysfunctions as well as neurological decline, which may be a consequence of the toxicity of the drugs or the chronic inflammation caused by HIV-1 infection (Deeks, 2011; d’Arminio et al., 2004). The financial cost of life-long treatment, especially in resource-poor settings, is prohibitive (Hecht et al., 2010). Until the discovery of an effective vaccine, or other interventions that can halt the continuing spread of HIV-1, it will become increasingly difficult for high disease burden countries in the developing world to control the epidemic using only current anti-retroviral regimes (Lewin et al., 2011). Thus, an effective cure of HIV/AIDS would not only alleviate the suffering of the millions of infected persons, it may be the only way to check the progress of the HIV-1 epidemic. In this article, we provide an overview of HIV-1 latency and address some of the major gaps in our understanding of the phenomenon. We examine recent advances in translational research aiming to find a sterilizing (complete eradication of the virus) or a functional (virus replication is on-going but does not lead to clinical problems) cure.
The question over the source of the persistent viraemia
The half-life of the HIV-1 virion in the plasma is very short (Ho et al., 1995; Ramratnam et al., 2000) and it is generally believed that the persistent viraemia is either the result of the reactivation of latently infected resting T-cells or on-going virus replication in ‘sanctuary’ sites within the body (Lewin et al., 2011; Palmer et al., 2011). The existence of a latently infected population of CD4+ T-cells was first indicated by the discovery that the number of cells expressing HIV-1 mRNA in vivo was lower than the number of cells carrying proviral DNA (Schnittman et al., 1989). Subsequent studies have demonstrated that a small number (approx. one million cells) of resting CD4+ T-cells in HAART-treated individuals harboured replication-competent latent viruses that could be reactivated by stimulation of the cells with mitogens (Chun et al., 1995, 1997; Finzi et al., 1997). While dormant, the virus is hidden from the host immune response and it has been shown that the decay rate of these latently infected resting CD4+ T-cells is very low, requiring an estimated period of 73.4 years for complete eradication using the current anti-retroviral regime (Siliciano et al., 2003). Alternatively, on-going low-level virus replication may be responsible for the persistent viraemia. Persistent virus production has been found within sanctuary sites such as the central nervous system (Canestri et al., 2010; Churchill et al., 2006; González-Scarano & Martín-García, 2005), the gastrointestinal tract (Chun et al., 2008) and the male and female genital tract (Halfon et al., 2010; Launay et al., 2011). Recent studies have indicated that anti-retroviral drug-penetration is site- and compound-specific, and drugs that penetrate poorly may allow virus replication at that site even when plasma viral load is below 50 copies ml−1 (Best et al., 2012; Di Mascio et al., 2009; Else et al., 2011; Halfon et al., 2010; Kwara et al., 2008; Launay et al., 2011). Since the rate of reactivation of latent viruses in resting T-cells is unknown in vivo (Siliciano & Siliciano, 2010), it is unclear whether it occurs frequently enough to maintain the low-level viraemia that is detected in patients. Thus, the most probable origin of the low-level viraemia may be the sanctuary sites where productive infection is expected to be occurring constantly.
In order to determine the contribution of each of these factors to the low-level viraemia in the body, phylogenetic studies were performed on the viral sequences isolated from the residual viraemia. The results were contradictory: while some studies showed a lack of evolution among the sequences found, suggesting that the progeny virions came from one stable reservoir among CD4+ T-cells (Bailey et al., 2006; Joos et al., 2008; Ruff et al., 2002), others found viral sequences that were not detected among the resting T-cell population, indicating another cellular source for the residual viraemia (Bailey et al., 2006; Brennan et al., 2009; Sahu et al., 2009). Another indication of on-going productive infection would be if treatment intensification (the addition of a fourth anti-retroviral to the standard three-drug regime) reduced the basal level of viraemia further. The majority of treatment intensification studies using the HIV-integrase inhibitor Raltegravir (RGV) showed no significant reduction of the residual plasma viraemia (Dinoso et al., 2009b; Gandhi et al., 2010; Hatano et al., 2011; McMahon et al., 2010). However, in one study RGV increased the number of ‘2-LTR (long terminal repeat) circles’ found in the PBMCs of 29 % of the treated subjects (Buzón et al., 2010). Since RGV blocks the integration of linear viral DNA from a productive infection and encourages the formation of 2-LTR circles (Middleton et al., 2004; Svarovskaia et al., 2004), these data indicate the presence of an on-going infection. In a separate study, intensification with RGV reduced unspliced HIV-1 RNA within the ileum, but caused no significant reduction in plasma viraemia (Yukl et al., 2010), illustrating the possibility of on-going infection occurring in a compartment other than the blood (Table 1).
Table 1. Summary of the evidence for and against the hypothesis that the persistent residual viraemia in HAART-treated patients originates from a single source.
| Persistent viraemia originated from one source (resting T-cells) | References |
| • Viral genomes recovered from persistent viraemia show little variation | Bailey et al. (2006); Joos et al. (2008); Ruff et al. (2002) |
| • HAART intensification does not reduce residual viraemia | Dinoso et al. (2009b); Gandhi et al. (2010); Hatano et al. (2011); McMahon et al. (2010) |
| Multiple sources contribute to persistent viraemia | |
| • Persistent HIV-1 infection has been found within different parts of the body | See text |
| • Viral sequences distinct from those isolated from resting T-cells are found | Bailey et al. (2006); Brennan et al. (2009); Sahu et al. (2009) |
| • HAART intensification increased 2-LTR circles from PBMCs | Buzón et al. (2010) |
| • HAART intensification reduced HIV-1 RNA within the ileum | Yukl et al. (2010) |
Most of the CD4+ T-cells in the body reside within the gastrointestinal tract and the lymphatic tissues rather than within peripheral blood (Mowat & Viney, 1997). In contrast, the majority of studies on HIV-1 replication dynamics and CD4+ T-cell depletion have been performed in peripheral blood because it is the easiest compartment to access. It has been shown that the gastrointestinal tract is the major site of HIV-1 replication and CD4+ T-cell depletion during all stages of HIV/AIDS (Brenchley et al., 2004a; Chun et al., 2008), and that the destruction of the CD4+ T-cell population within the gastrointestinal tract leads to the translocation of microbial products to the circulatory system and contributes to the chronic inflammation and immune exhaustion that are associated with HIV/AIDS (Douek et al., 2009). Thus, we may be overlooking vital pieces of the jigsaw if we focus solely on the peripheral blood compartment.
Apart from CD4+ T-cells, HIV-1 can also infect cells of the monocytic lineage (Coleman & Wu, 2009; Gartner et al., 1986; Le Douce et al., 2010). HIV-1 infection of macrophages tends to be less cytopathic than infection of activated T-cells (Ho et al., 1986, 1995; Nicholson et al., 1986). Also, infected monocytes can migrate to the central nervous system and the gastrointestinal tract before maturing into macrophages, potentially sheltering the virus from the full potency of HAART (Le Douce et al., 2010). The contribution of infected macrophages to HIV-related neurological decline is well documented (González-Scarano & Martín-García, 2005). It is also well known that dendritic cells can transport whole virions to lymph nodes where susceptible activated CD4+ T-cells reside. Moreover, dendritic cells themselves can become infected under certain circumstances (Coleman & Wu, 2009). However, it is not clear whether proviral clones or individual infected cells within the monocytic population can survive for long enough to function as long-lived latency reservoirs (Eisele & Siliciano, 2012). It is possible that within the safety of sanctuary sites and with continuous replenishment of susceptible cells, continuous productive infection may be maintained by macrophages and dendritic cells. In addition it has been proposed that infection of immature CD4+/CD8+ ‘double positive’ thymocytes during thymopoiesis may generate a population of latently infected naïve T-cells (Brooks et al., 2001). Despite the continuing debate over the true origin of the low-level viraemia, it can be agreed that a viable therapeutic intervention to cure HIV/AIDS should involve the elimination of all these proven and potential reservoirs.
Haematopoietic stem cells (HSCs) are a viral reservoir?
HSCs are a population of primitive, self-renewing precursor cells that reside in the bone marrow (Cabrita et al., 2003). HSCs can proliferate and differentiate into all the cell types found in peripheral blood. HSCs and other more mature precursor cell types such as the multipotent progenitor cells (MPPs) can express the HIV-1 receptors CD4, CCR5 and CXCR4; thus in theory these cells can be infected by HIV-1 (Alexaki & Wigdahl, 2008; McNamara & Collins, 2011). However, whether this is the case in vivo is controversial, with contradictory evidence emerging from different studies (Davis et al., 1991; Folks et al., 1988; Neal et al., 1995; Stanley et al., 1992).
If HSCs and other progenitor cells are proven to be latent reservoirs of HIV-1, it would make the difficult task of curing HIV/AIDS even more challenging as these cells are very long-lived, can self-propagate and the provirus in these cells may not be affected by HAART or any novel therapies that target latently infected CD4+ T-cells. Recently, it has been shown that CD34+ progenitor cells (that include HSCs and MPPs) are susceptible to latent infection ex vivo, and that integrated provirus was detected among CD34+ cells from HAART-treated patients (Carter et al., 2010). A follow-up study showed that only X4 or dual R5/X4-tropic viruses could efficiently infect these CD34+ progenitor cells (Carter et al., 2011). Furthermore, this study also showed that human HSCs infected with a GFP-reporter virus could be successfully engrafted into irradiated non-obese diabetic (NOD)/SCID IL-2Rγnull mice, leading to the detection of human leukocytes in the peripheral blood that were carrying the reporter virus 14–18 weeks post-engraftment (Carter et al., 2011). These results indicate that infected haematopoietic progenitor cells are a reservoir for HIV-1. However, other studies have failed to detect HIV-1 amongst the FACS-sorted CD34+ progenitor cells of HAART-treated patients (Durand et al., 2012; Josefsson et al., 2012) and it was suggested that the positive selection of CD34+ cells by magnetic beads as used in the studies by Carter et al. may be insufficient to remove all the contaminating CD4+ T-cells. Also, are the results from the engraftment experiment, which used a reporter virus and a highly artificial small animal model, relevant to the situation in the human body? The existence of an HSCs reservoir remains a controversy and requires further study.
Latent HIV-1 infection of resting CD4+ T-cells
Although HIV-1 can persistently replicate within sanctuary sites, improvements in drug penetration or HAART intensification may overcome this barrier to eradication in the future. However, enhancing the effectiveness of HAART will not affect the latent viruses hiding within the resting CD4+ T-cell populations of the body. Thus, the latent infection within resting T-cells remains the biggest proven obstacle to a sterilizing cure of HIV-1 infection. The majority of the circulating CD4+ T-cells in the body at any given time are in a resting state (Berard & Tough, 2002). These cells are typically defined by the lack of activation marker expression (CD25, CD69 and HLA-DR), as well as the maintenance of the cells in the G0 phase (Chun et al., 1997). They can be broadly divided into those that have not undergone antigen-stimulated expansion (naïve T-cells) and those that have remained behind after the end of an immune response (memory T-cells) (Berard & Tough, 2002). Among infected resting T-cells, HIV-1 gene expression is largely suppressed (Hermankova et al., 2003). However, some transcription of HIV mRNA can be detected within the resting T-cells of HAART-treated patients, although full virus production is inhibited by inefficiencies at various stages of the viral life cycle (Lassen et al., 2004, 2006; Vatakis et al., 2010) (Fig. 1). Since most of these latently infected resting CD4+ T-cells are CD45RO+ memory cells (Brenchley et al., 2004b; Chomont et al., 2009; Chun et al., 1997; Pierson et al., 2000), it is hypothesized that the majority of the latently infected T-cells come from activated CD4+ T-cells that were infected and then reverted back to a resting memory state before the start of virus replication (Han et al., 2007). Latent provirus can be maintained within the memory T-cell population by the homeostatic proliferation of the infected host cells, driven by IL-7 (Bosque et al., 2011; Chomont et al., 2009).
Fig. 1.
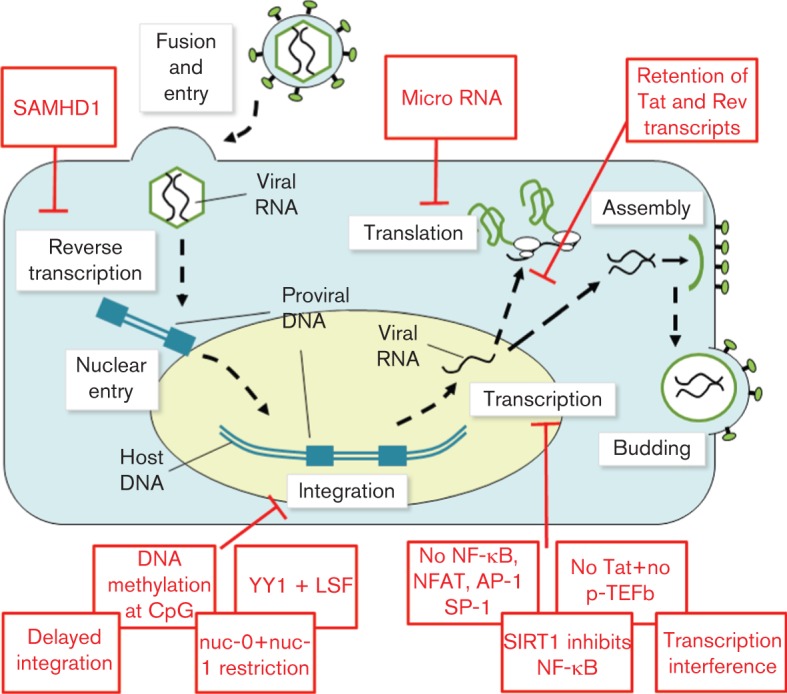
A summary of the multiple obstacles blocking productive HIV-1 infection of resting CD4+ T-cells. Inhibition of virus replication occurs at multiple steps during the viral life cycle. Transcription interference refers to promoter occlusion and the collision of RNA polymerases that hinder efficient viral gene expression.
Virus may infect resting T-cells directly and latent infection of naïve T-cells has been observed in patients, albeit at a lower frequency than memory T-cells (Chomont et al., 2009; Pierson et al., 2000). However, direct infection of resting T-cells is very inefficient (Pierson et al., 2000; Stevenson et al., 1990), with defects in reverse transcription and delays in integration in comparison with infection of activated CD4+ T-cells (Vatakis et al., 2007, 2009). Recently, two studies have implicated the innate restriction factor SAMHD1 in the inhibition of reverse transcription in resting CD4+ T-cells. Initially shown to be absent in transformed CD4+ T-cell lines, SAMHD1 was found to be expressed in both resting and activated primary CD4+ T-cells. In resting T-cells, SAMHD1 restricted reverse transcription by depleting the cellular pool of dNTPs (Baldauf et al., 2012; Descours et al., 2012). Nevertheless, integration of the viral genome can still occur in resting T-cells (Vatakis et al., 2009) and no method described to date has been able to distinguish between latently infected memory T-cells that were infected either during activation or during quiescence (Vatakis et al., 2010). Furthermore, a recent study showed that in patients receiving HAART treatment, the amount of HIV DNA in memory T-cells declined over time while the amount of HIV DNA in naïve cells remained constant, suggesting that direct infection of resting T-cells may be replenishing the latent viral reservoir as the disease progresses (Wightman et al., 2010). These observations are consistent with the finding that R5 tropic viruses [which are associated with acute infection (Choe et al., 1996; Feng et al., 1996; Zhu et al., 1993)] preferentially infect CCR5-expressing memory T-cells whereas X4 tropic viruses [which are associated with late disease progression (Connor et al., 1997)] exhibit a preference for CCR5- CXCR4+ naïve T-cells (Bleul et al., 1997; Ostrowski et al., 1999; Wu et al., 1997). The stimulatory effects of HIV-1 gp120 may enable the direct infection of resting T-cells by activating calcium flux and NFAT signalling down the CCR5 signalling pathway, as well as upregulating inositol triphosphate-mediated signalling and the expression of the IL-2 receptor (Cicala et al., 2006; Kornfeld et al., 1988; Weissman et al., 1997). Stimulation of CXCR4 signalling by HIV-1 gp120 induces cytoskeleton-remodelling activity in resting T-cells, increasing the efficiency of subsequent infection with HIV-1 (Yoder et al., 2008). Thus, it is possible that the direct infection of resting T-cells plays an increasingly important role in maintaining the viral reservoir as the disease progresses.
The molecular mechanisms of latency in HIV-1 infection have been reviewed extensively (Coiras et al., 2009; Colin & Van Lint, 2009; Marcello, 2006; Marsden & Zack, 2009; Richman et al., 2009; Siliciano & Greene, 2011). In general, HIV-1 latency may be divided into pre-integration or post-integration latency. Pre-integration latency refers to the partial or complete inhibition of the viral life cycle before the integration of the virus into the host genome (see above). Most of the HIV-1 DNA found in resting T-cells is unintegrated (Chun et al., 1997; Sloan & Wainberg, 2011). Although it has been shown that linear unintegrated viral DNA within resting CD4+ T-cells is able to complete integration after the activation of the cell (Bukrinsky et al., 1991), pre-integration latency is not thought to be relevant to the establishment of the reservoir of latently infected resting T-cells due to the labile nature of viral DNA in the cytoplasm of the cell (Pierson et al., 2002). Accordingly, the unintegrated viral DNA may no longer be replication competent after a protracted period inside the host cell (Han et al., 2007; Zhou et al., 2005). Post-integration latency is the failure of expression of the viral genome after it has been integrated into the host genome. While less than 0.05 %, or approximately 106 to 107 cells, carry integrated provirus, it is these integrated proviruses that are thought to constitute the latent viral reservoir (Chun et al., 1997).
Even after a successful integration event, there are still multiple barriers to productive HIV-1 replication within resting CD4+ T-cells (Fig. 1). Although HIV-1 genomes are generally integrated into genes that are actively expressed within resting T-cells (Han et al., 2004), viral gene expression may be downregulated by promoter occlusion (if the provirus is integrated in the same orientation as the host gene) (Greger et al., 1998) or by collisions between RNA Pol II molecules that are travelling in opposite directions (if the provirus is integrated in the opposite orientation as the host gene) (Han et al., 2008). Two nucleosomes, named nuc-0 and nuc-1, are frequently associated with the HIV-1 5′LTR and regulate the basal transcriptional activity of the viral genome by controlling the access of transcription factors to the LTR (Verdin et al., 1993). The remodelling of the nucleosomes is regulated by the acetylation status of their constituent histones, which is in turn controlled by enzymes such as histone acetyltransferases and histone deacetylases (Van Lint et al., 1996). This allows the manipulation of HIV-1 transcriptional activity by pharmacological means, potentially leading to a viable method to eliminate the virus reservoir from resting CD4+ T-cells (see Novel drug discovery). The presence of cellular transcriptional repressors, for example YY1 and LSF, as well as the methylation of the two CpG islands at the HIV-1 transcription start site, can recruit histone deacetylases to the HIV-1 LTR and reinforces latency (Blazkova et al., 2009; Coull et al., 2000; Kauder et al., 2009), while the binding of the transcription factor NF-κB stimulates proviral reactivation by recruiting histone acetyltransferases to the LTR and initiating early HIV-Tat production (Lusic et al., 2003; Williams et al., 2007). The lack of NF-κB, as well as transcription factors NFAT, SP-1 and AP-1 prevents the synthesis of Tat and the subsequent Tat-dependent, high level viral gene expression (Coiras et al., 2009; Mbonye & Karn, 2011; Williams & Greene, 2007). The transcriptional activity of Tat is highly dependent on interacting with the cellular factor P-TEFb, which triggers effective RNA Pol II elongation (Parada & Roeder, 1996), and the negative regulation of P-TEFb activity in resting T-cells further impairs the expression of HIV-1 genes (Ghose et al., 2001). Tat also interacts with several other cellular factors such as the histone acetyltransferases p300 and P/CAF to promote transactivation of viral genes (Benkirane et al., 1998). The acetylation of the RelA subunit of NF-κB by p300 increases its transcriptional activity (Chen et al., 2002) and this is countered by the cellular deacetylase SIRT1 (Yeung et al., 2004). SIRT1 activity is in turn blocked by HIV-1 Tat (Kwon et al., 2008). In addition, it has been demonstrated that HIV Tat and Rev transcripts are retained in the nuclei of resting CD4+ T-cells (Lassen et al., 2006) and that numerous host microRNAs can directly or indirectly downregulate HIV-1 gene expression, contributing to the maintenance of proviral latency (Chiang & Rice, 2012).
Due to the involvement of so many cellular factors, it has been proposed that there are different degrees of latency within the T-cell population, depending on the cell type and the extracellular environment (Pace et al., 2011). A recent in vitro study of HIV-1 latency using a central memory T-cell model system has shown that IL-7-driven homeostatic replication of infected cells can induce partial virus reactivation, while stimulation of the T-cell receptor signalling pathway with anti-CD3/anti-CD28 antibody induced full reactivation (Bosque et al., 2011). This supports the hypothesis of a dynamic reservoir of infected T-cells at various levels of cellular and viral activation.
An area of research which has, as yet, escaped the attention of the HIV-latency field is the molecular mechanism behind CD4+ T-cell quiescence. It has been known for some time that the quiescence state is actively maintained by factors such as LKLF, Tob, Foxo3a and Foxj1 (Coffer & Burgering, 2004; Tzachanis et al., 2004; Yusuf & Fruman, 2003). The role of these factors in HIV-latency has been explored by few laboratories so far (Haaland et al., 2005; van Grevenynghe et al., 2008) and further research may provide new insights into the mechanism of latency as well as potential therapeutic targets.
In vitro and in vivo models of latency
The latently infected CD4+ T-cell population within the patient is very small, thus making ex vivo experiments very difficult. The use of in vitro and in vivo models of latency has been and will continue to be vital to the understanding of HIV-1 latency and drug discovery. Early studies of lentiviral latency using cell lines such as ACH-2, U1 and J-Lat showed the involvement of host cytokine signalling pathways and chromatin reorganization in modulating latency (Folks et al., 1987, 1989; Jordan et al., 2003), but their transformed nature means their responses to treatments may not be physiologically relevant. For example, in the latently infected J-Lat cell line, HIV-1 preferentially integrates near the heterochromatin where transcriptional activity is low (Jordan et al., 2003). However, this preference is not observed within the latently infected resting CD4+ T-cells from HAART-treated patients, rather the provirus overwhelmingly favours integration into active transcriptional regions (Han et al., 2004; Schröder et al., 2002).
Most of the current in vitro models of HIV-1 latency involve the use of primary cells (Yang, 2011). These experiments are technically challenging, often taking weeks or months to complete in order to mimic the transition of activated T-cells to quiescent memory T-cells in vivo (Marini et al., 2008). Generating enough cells for experiments, especially in high-throughput screening of compounds, is another problem. Strategies such as the transduction of a survival gene into primary cells (Yang et al., 2009), using low levels of cytokines such as IL-2 or IL-7 (Bosque & Planelles, 2009; Marini et al., 2008) or co-culture with a feeder cell line (Sahu et al., 2006; Tyagi et al., 2010) have been described. Protocols to directly infect purified resting T-cells ex vivo have also been developed. To overcome the inefficient nature of infecting resting T-cells, methods such as spinoculation (O’Doherty et al., 2000) or stimulation with the chemokines CCL19 and CCL21 (Saleh et al., 2007, 2011) were used. The pros and cons of these in vitro model systems have been reviewed elsewhere (Pace et al., 2011; Wightman et al., 2012; Yang, 2011). Any model of latency would have to balance multiple conflicting demands such as maintaining the viability of the cells, while preserving a resting state and allowing viral integration without stimulating full-blown virus replication. A further complication is the fact that there are multiple types of cells that can be latently infected, such as central memory T-cells, transitional memory T-cells and naïve T-cells (Chomont et al., 2009; Wightman et al., 2010); any future treatments to reactivate the latent proviruses would have to be effective in all of these subsets of latently infected T-cells.
Non-human primates, in particular rhesus macaques infected with the simian immunodeficiency virus (SIV) or chimeric SIVs containing HIV-1 reverse transcriptase have been used to model HIV-1 latency in HAART-treated patients (Dinoso et al., 2009a; North et al., 2010; Shen et al., 2003). The major advantage of using non-human primates is that the locations of the persistent viral reservoirs mirror those in humans (North et al., 2010), which allows comparative in vivo studies. Also the progression of SIV in macaques resembles HIV-1 infection in humans, with distinctive acute and chronic phases of infection that may lead to immunodeficiency (Hirsch et al., 1996). However, there are significant differences between SIV infection of non-human primates and HIV-1 infection in humans. For example, the residual viraemia for SIV in rhesus macaques during chronic infection is higher than the levels seen in humans (Brenchley & Paiardini, 2011). The progression to AIDS appears to be more rapid in rhesus macaques than in humans (North et al., 2010). In African green monkeys or sooty mangabeys, although high levels of virus replication are observed during the chronic phase of infection, this is not accompanied by the destructive chronic immune activation seen in rhesus macaques or humans (Brenchley & Paiardini, 2011; Chahroudi et al., 2012). Also different strains of SIV can produce different pathologies in the same host (Hirsch et al., 2000).
The complexity of finding the correct host and SIV strain combination that mimics HIV-1 latent infection most closely, together with issues such as ethical concerns and high cost have led to the development of other, non-primate animal models for HIV-1 infection such as humanized SCID (SCID-hu) mouse models (Boberg et al., 2008; Brooks et al., 2001; Van Duyne et al., 2009). SCID-hu mice are created by transplanting SCID mice with human fetal thymus and liver tissues or peripheral blood lymphocytes to form SCID-hu Thy/Liv and SCID-hu PBL mice, respectively (Van Duyne et al., 2009). For example an in vitro model of latently infected immature CD4+/CD8+ thymocytes has been generated using SCID-hu Thy/Liv mice (Brooks et al., 2001; Burke et al., 2007). A major drawback of using SCID-mouse-based models is the failure to fully reconstitute the human immune system within the transplanted animals (Rossi et al., 2001; Van Duyne et al., 2009). Further improvement to efficiency of engraftment was achieved with the generation of the NOD/SCID mouse model (Hesselton et al., 1995) and later with the double knockout of the common cytokine receptor γC and the recombinase activating gene 2 (Rag2) (Goldman et al., 1998). The transplantation of human CD34+ stem cells into Rag2−/−γC−/− mice leads to the development of a functional model of the human immune system in the bodies of the mice (Traggiai et al., 2004) and forms the basis of a recent murine model of HIV-1 latency that contains infected resting T-cells in the peripheral blood and lymphoid tissues (Choudhary et al., 2009; 2012). Viable small animal models are vital in the preclinical evaluation of latency reversing therapies, especially if they can replicate latent infection compartments other than peripheral blood.
The feline model of HIV-1 latency
Feline immunodeficiency virus (FIV) was discovered in 1986 in California (Pedersen et al., 1987). Both HIV-1 and FIV target activated CD4+ T-cells (Yamamoto et al., 1988; Zagury et al., 1986), but whereas the primary receptor for HIV-1 is CD4 (Dalgleish et al., 1984), the primary receptor for FIV is CD134 (OX40) (Shimojima et al., 2004). HIV-1 utilizes CCR5 and CXCR4 as its secondary receptors (Choe et al., 1996; Deng et al., 1996; Feng et al., 1996), while FIV uses CXCR4 alone as its sole secondary receptor (Willett et al., 1997). FIV is transmitted mainly by biting (Yamamoto et al., 1989) and causes clinical signs in cats that are similar to AIDS in humans (Ackley et al., 1990; Barlough et al., 1991; Novotney et al., 1990).
Since FIV has a similar cell tropism to HIV-1, it is expected that the host response to FIV and its pathogenesis will be comparable to HIV-1. Cats mount both humoral and cytotoxic T-cell responses to FIV infection (Beatty et al., 1996; Egberink et al., 1992; Flynn et al., 2002). However, the hosts usually fail to clear the infection and may succumb to immunodeficiency. The mechanisms of pathogenesis of HIV-1 and FIV are remarkably similar. Both viruses cause massive depletion of the gastrointestinal tract CD4+ T-cell population (Brenchley et al., 2004a; Howard et al., 2010). The low fidelity of the HIV-1 and FIV reverse transcriptases results in the generation of a diverse pool of viral variants within the host, encouraging immune escape (Bebenek et al., 1993; Hosie et al., 2011; Kraase et al., 2010; Mansky & Temin, 1995). All of these factors promote chronic immune activation, eventually leading to the breakdown of the host immune system (Douek et al., 2009; Tompkins & Tompkins, 2008).
Previous studies of FIV in cats have shown that activated (CD4+ CD25+) and resting (CD4+ CD25–) CD4+ T-cells from peripheral blood can be latently infected ex vivo and that FIV replication can be reactivated by the application of ConA or IL-2 (Joshi et al., 2005, 2004), mirroring the crucial role of IL-2 in productive infection with HIV-1 (Oswald-Richter et al., 2004). In a separate study, cats challenged with a low-dose exposure to FIV-infected T-cells showed an aviraemic infection, and when cells from multiple tissues were stimulated by PMA, FIV gp120 production was detected (Assogba et al., 2007). More recently it has been shown that FIV establishes a latent infection within activated and resting T-cells of cats during the asymptomatic phase of infection, similar to the latent infection of the resting T-cell population by HIV-1 in humans (Murphy et al., 2012). These cells contained detectable FIV DNA but no FIV RNA. Furthermore, virus replication from these latently infected cells could be reactivated ex vivo by the mitogens PHA and PMA as well as the histone deacetylase inhibitor SAHA (McDonnel et al., 2012) (see Novel drug discovery). These findings support the proposal of using FIV-infected cats as an alternative small animal model for HIV-1 latency.
Stem cell transplantation and gene therapy approaches to curing HIV/AIDS
Recently, a HIV-1-positive patient who developed acute myeloid leukaemia was apparently cured of HIV-1 infection after receiving an HSC transplant from a donor who was homozygous for the CCR5 Δ32 allele (Hütter et al., 2009). The patient underwent intensive chemotherapy and radiotherapy to prepare for the transplant, which presumably also eliminated almost all the infected CD4+ T-cells within the body. In addition, the patient developed graft-versus-host disease after transplantation, indicating that the transplanted cells had replaced the host immune system. HAART treatment was then stopped and in the follow-up study the patient was shown to remain free from the virus (Allers et al., 2011). To subject otherwise healthy HAART-treated patients to this potentially lethal procedure is ethically questionable and practically not viable, especially in resource-poor settings. However, this unique case has raised an interesting question regarding the kind of intervention necessary to clear the body of HIV-1: was the cure achieved by the intensive chemotherapy and radiotherapy or by the transplant of the Δ32 HSCs, which gave rise to HIV-1 resistant CD4+ T-cells? Treatment of HIV-1-positive lymphoma patients with autologous stem cell transplants failed to eliminate the virus from the body (Cillo et al., 2012), which indicated the presence of residual virus or infected cells within the extracted autologous cell population or within the host. It also demonstrated the need to make the host CD4+ T-cells immune to HIV-1 infection before transplantation.
The CCR5 Δ32 mutation abrogates infection of CD4+ T-cells by R5 HIV-1 viruses (Dean et al., 1996), the strains most frequently associated with early stage infection and which are transmitted preferentially between individuals (Margolis & Shattock, 2006). Thus, the nascent CD4+ T-cells from the transplant would be resistant to new infection. Disruption of the CCR5 gene has no apparent undesirable effects on the normal functioning of HSCs (Bai et al., 2000; O’Brien & Moore, 2000). Various techniques have been developed to disrupt the CCR5 gene ex vivo, including the use of CCR5-specific siRNAs, ribozymes, intrabodies and zinc-finger nucleases (ZFNs) (Anderson et al., 2007; Bai et al., 2000; Holt et al., 2010; Kumar et al., 2008; Swan et al., 2006). Each of these treatments has been tested in mouse models and led to the production of modified HSCs which give rise to CD4+ T-cells that are resistant to R5 HIV-1 infection. ZFNs, which are engineered endonucleases containing zinc finger domains that recognize specific DNA sequences (Urnov et al., 2005), have also been used to disrupt the CCR5 gene in CD4+ T-cells in a mouse model of HIV-1 infection (Perez et al., 2008). More recently, ZFNs targeting CD4+ T-cells have been successfully tested in a phase I clinical trial, in which the treatment was well tolerated by patients, the modified CD4+ T-cells were able to persist in the body and there were improvements on the CD4+ T-cell count and CD4+:CD8+ T-cell ratio (June et al., 2012). ZFNs that can disrupt the CXCR4 gene in CD4+ T-cells have also been developed and it has been demonstrated that they confer resistance to cells against the X4-tropic HIV-1 strains associated with late-stage infection (Wilen et al., 2011; Yuan et al., 2012). Combining the disruption of CCR5 and CXCR4 may provide a viable gene therapy approach to a functional cure, in which the patient’s CD4+ T-cells are made resistant to HIV-1 ex vivo and are reintroduced back into the body. There may still be residual viraemia but the virus would not cause disease after the withdrawal of HAART. Potential problems with the use of the ZFNs include the possibility of non-specific cleavage of host DNA (Gabriel et al., 2011; Pattanayak et al., 2011) and the possibility of adverse effects from disrupting CXCR4, which has not been well studied at the time of writing.
Novel drug discovery
The main strategy that is currently being pursued by many laboratories to eradicate HIV-1 from the body is to reactivate the latent virus reservoir within resting CD4+ T-cells (Marsden & Zack, 2009; Richman et al., 2009). Early attempts at reactivation using powerful cytokines such as IL-2 and TNF-α stimulated virus production (Chun et al., 1998) but also caused dangerous side effects such as the non-specific, global activation of T-cells (Prins et al., 1999). In contrast, the cytokine IL-7 has also been shown to have potent anti-HIV latency effects without inducing T-cell activation (Levy et al., 2009; Scripture-Adams et al., 2002; Wang et al., 2005). IL-7 is well-tolerated in vivo (Levy et al., 2012) and it has been used in a clinical trial to reduce the latent reservoir size (ERAMUNE 01, due to finish in January 2013. http://clinicaltrials.gov). However, a potential problem with the use of IL-7 to deplete the latent reservoir is that at low concentrations, IL-7 can promote the survival or induce the homeostatic proliferation of the latently infected memory T-cells without triggering activation of the virus, thus inadvertently expanding the reservoir of infected cells (Chomont et al., 2009; Marini et al., 2008).
Compounds that stimulate protein kinase C and NF-κB such as the phorbol ester prostratin and 5-hydroxynaphthalene-1,4-dione (5HN) have been shown to reactivate latent infection in vitro (Kulkosky et al., 2001; Yang et al., 2009). Intriguingly, prostratin also has anti-HIV-1 replication effects (Biancotto et al., 2004; Rullas et al., 2004) and a similar dual effect of the compound on FIV replication has been described in vitro (Chan et al., 2013). Histone deacetylase inhibitors such as valproic acid and suberoylanilide hydroxamic acid (SAHA or Vorinostat) have been shown to reverse HIV latency by remodelling the HIV-repressive nucleosome nuc-1 (Archin et al., 2009; Contreras et al., 2009; Van Lint et al., 1996; Ylisastigui et al., 2004). SAHA is a selective class I and II histone deacetylase inhibitor and is approved as a clinical treatment for cutaneous T-cell lymphoma. It has been used in a number of ex vivo and clinical studies of the latent reservoir (Archin et al., 2012; Shan et al., 2012). However, further research is required to investigate fully the long-term side effects of SAHA in terms of its potential as a mutagen and its ability to reactivate other latent viruses (Archin et al., 2012; Kerr et al., 2010; Wightman et al., 2012). Using a siRNA screen, a novel HIV replication-inhibiting host factor has been identified recently (Zhu et al., 2012). This factor, named bromodomain containing 4, can be inhibited by a small molecule known as JQ1 (Filippakopoulos et al., 2010) and JQ1 has been shown to have HIV-1 latency reversing activity by several laboratories (Banerjee et al., 2012; Li et al., 2013; Zhu et al., 2012). Other compounds and small molecules that have been shown recently to reactivate latent HIV-1 infection include a bacterial protein named HIV-1-reactivating factor (Wolschendorf et al., 2010), the aldehyde dehydrogenase inhibitor disulfiram (Xing et al., 2011) and a number of quinolin-8-ol derivatives (Xing et al., 2012). Curiously, a recent clinical trial has demonstrated that intensification of HAART with the CCR5 antagonist Maraviroc (MVC) caused a reduction in the size of the latently infected T-cell reservoir (Gutiérrez et al., 2011). The mechanism behind this effect of MVC is unknown, but the binding of MVC to CCR5 may lead to the stimulation of cell signalling analogous to the stimulation of the other major HIV-1 co-receptor, CXCR4, by the binding of HIV-1 Env (Wu & Yoder, 2009; Yoder et al., 2008). If the findings of this study are confirmed, a new way of reactivating latently infected cells may have been identified. Moreover, it has been shown recently that fever enhances the activity of Tat, a phenomenon mediated by the heat-shock protein Hsp-90 (Roesch et al., 2012). Using a J-Lat model, this study demonstrated that although hyperthermia by itself cannot reactivate latency, it can enhance the reactivation effect of other treatments such as the co-cultivation of the J-Lat cells with IL-2 supplemented PBMCs (Roesch et al., 2012). This suggests that the artificial induction of fever may be used to boost the effectiveness of any future latency reversing therapies.
Borrowing from the concept of HAART, a combination of different latency reversing agents may be used synergistically to enhance the effect of reactivation therapies (Burnett et al., 2010; Deeks et al., 2012; Reuse et al., 2009). The number of compounds identified is likely to increase thanks to on-going and future high-throughput screens that look for molecules which can stimulate latent HIV-1 to reactivate. One novel screening method described recently can measure the expression of cell-associated viral RNA among latently infected T-cells as soon as the cells are extracted from the patient without the need for further co-culturing (Archin et al., 2012). Using this assay, an increase in viral RNA can be demonstrated amongst the resting T-cells from patients after being treated with a single dose of SAHA. However, an important caveat to this type of experiment is that stimulation of viral transcription, viral protein synthesis or even virion production may not necessarily lead to the destruction of the latently infected cell (see below).
Stimulation of latent virus replication may not lead to the depletion of the viral reservoir
It has been assumed that once the latent provirus is reactivated inside a resting T-cell, the cell would die by HIV-induced cytopathic effects or be killed by the host immune response (Richman et al., 2009). However, this view has recently been challenged (Shan et al., 2012). Stimulation of resting CD4+ T-cells from HAART-treated patients with the SAHA did not reduce the size of the latent reservoir (Shan et al., 2012). Furthermore, latently infected resting T-cells reactivated by SAHA were killed neither by viral cytopathic effects, nor by autologous CD8+ T-cells isolated from the same patients. Only after antigen-specific stimulation of the autologous CD8+ T-cells was efficient killing of the SAHA-reactivated, infected resting CD4+ T-cells restored. The transduction of the survival gene Bcl-2 into the resting T-cells during the establishment of the latent infection assay, as well as the use of modified reporter viruses may have increased the survival rate during the study. However, reactivation by SAHA of the latent wild-type virus within unmodified resting T-cells isolated from patients also did not lead to a contraction of the latent virus reservoir. These findings showed that any future therapeutic regime to eliminate the latent reservoir would require the boosting of anti-HIV cytotoxic T-lymphocyte (CTL) responses, which would likely be in a state of exhaustion after years of chronic activation (Trautmann et al., 2006). In addition to stimulating the CD8+ T-cells with viral antigens and cytokines, inhibiting the function of immunoregulatory molecule such as PD-1 may be another option for the restoration of full CTL function in the patient against HIV-1 (Eichbaum, 2011). Also, it is known that resting T-cells are less vulnerable to cell death compared with their activated counterparts (van Leeuwen et al., 2009). Would the use of drugs that stimulate cellular activation, such as prostratin, lead to the death of the reactivated infected T-cells? Alternatively, is it possible to use novel technologies such as nanoparticles (Peer et al., 2007), intrabodies (Pérez-Martínez et al., 2010) or RNA aptamers (Burnett & Rossi, 2012) to target the reactivated infected T-cells for destruction (Fig. 2)?
Fig. 2.
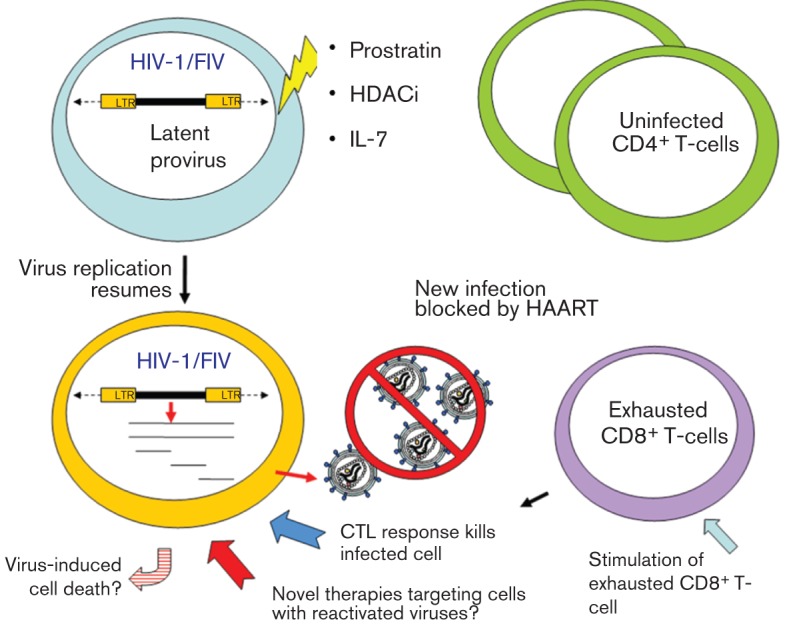
A theoretical scheme to eliminate the latent HIV-1 reservoir within the resting T-cell population. Virus replication in latently infected cells could be reactivated by, for example, treatments with prostratin, histone deacetylase inhibitors (HDACi) or IL-7. Meanwhile the CTL responses of the patient could be restored by stimulation with viral antigens and cytokines. Although the reactivation of the latent virus may not lead to the apoptosis of the infected cell as previously assumed, the restored CTL response or the use of novel drug-delivery technologies may allow the specific targeting of the infected cells for destruction.
Another observation that may be a cause for concern among the ever growing literature on latency-reversing compounds is that even the most promising molecules such as prostratin, SAHA and JQ1 cannot reliably stimulate productive infection from all HAART-treated patients’ samples, despite being very successful in reactivating latent viruses from in vitro models (Contreras et al., 2009; Kulkosky et al., 2001; Zhu et al., 2012). The variable performances of these compounds may be due to sampling errors as a result of the fact that there are so few latently infected cells within the patient (hence the need for in vitro model systems), or the underlying activation status of the cells, as in the case for prostratin (Chan et al., 2012; Kulkosky et al., 2001). Alternatively, this may indicate that the current in vitro models does not represent all the subset of CD4+ T-cells that are latently infected. Also can we assume that our current methods of handling CD4+ T-cells accurately reproduce in vivo conditions? Nevertheless, the potential for false negatives and false positives in the current assays demands further research into the basic molecular biology of HIV-1, T-cell biology and improvements to existing HIV-latency models.
Conclusion
After more than two decades of research we are only beginning to appreciate the full complexity of the problem of HIV-1 persistence and latency. Recent research suggests that there are multiple reservoirs of replication-competent virus which contribute to viral persistence. To achieve a sterilizing cure of HIV-1 requires significant disruption or even elimination of all these reservoirs. In addition, there are still many unanswered questions regarding HIV-1 latency remaining. For example, what is the source of the persistent low-level viraemia? What is the contribution of direct infection of resting T-cells to the overall size of the viral reservoir? Can gene therapy lead to a functional cure of HIV-1? How do we eliminate the infected T-cells once they are reactivated? Research into novel small animal models of HIV-1 latency such as the Rag2−/−γC−/− mouse or FIV-infection of cats may speed up the drug development process but their relevance to the clinic needs to be established. Further research into these issues is needed urgently in order to stop the global HIV/AIDS epidemic, which continues to be a serious global threat to public health almost 30 years after its discovery.
Acknowledgements
The authors would like to thank the School of Veterinary Medicine, University of Glasgow and the Wellcome Trust for funding.
References
- Ackley C. D., Yamamoto J. K., Levy N., Pedersen N. C., Cooper M. D. (1990). Immunologic abnormalities in pathogen-free cats experimentally infected with feline immunodeficiency virus. J Virol 64, 5652–5655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexaki A., Wigdahl B. (2008). HIV-1 infection of bone marrow hematopoietic progenitor cells and their role in trafficking and viral dissemination. PLoS Pathog 4, e1000215 10.1371/journal.ppat.1000215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allers K., Hütter G., Hofmann J., Loddenkemper C., Rieger K., Thiel E., Schneider T. (2011). Evidence for the cure of HIV infection by CCR5Δ32/Δ32 stem cell transplantation. Blood 117, 2791–2799 10.1182/blood-2010-09-309591 [DOI] [PubMed] [Google Scholar]
- Anderson J., Li M.-J., Palmer B., Remling L., Li S., Yam P., Yee J.-K., Rossi J., Zaia J., Akkina R. (2007). Safety and efficacy of a lentiviral vector containing three anti-HIV genes–CCR5 ribozyme, tat-rev siRNA, and TAR decoy–in SCID-hu mouse-derived T cells. Mol Ther 15, 1182–1188 [DOI] [PubMed] [Google Scholar]
- Archin N. M., Espeseth A., Parker D., Cheema M., Hazuda D., Margolis D. M. (2009). Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retroviruses 25, 207–212 10.1089/aid.2008.0191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archin N. M., Liberty A. L., Kashuba A. D., Choudhary S. K., Kuruc J. D., Crooks A. M., Parker D. C., Anderson E. M., Kearney M. F. & other authors (2012). Administration of Vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487, 482–485 10.1038/nature11286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assogba B. D., Leavell S., Porter K., Burkhard M. J. (2007). Mucosal administration of low-dose cell-associated feline immunodeficiency virus promotes viral latency. J Infect Dis 195, 1184–1188 10.1086/512861 [DOI] [PubMed] [Google Scholar]
- Bai J., Gorantla S., Banda N., Cagnon L., Rossi J., Akkina R. (2000). Characterization of anti-CCR5 ribozyme-transduced CD34+ hematopoietic progenitor cells in vitro and in a SCID-hu mouse model in vivo. Mol Ther 1, 244–254 10.1006/mthe.2000.0038 [DOI] [PubMed] [Google Scholar]
- Bailey J. R., Sedaghat A. R., Kieffer T., Brennan T., Lee P. K., Wind-Rotolo M., Haggerty C. M., Kamireddi A. R., Liu Y. & other authors (2006). Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J Virol 80, 6441–6457 10.1128/JVI.00591-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldauf H. M., Pan X., Erikson E., Schmidt S., Daddacha W., Burggraf M., Schenkova K., Ambiel I., Wabnitz G. & other authors (2012). SAMHD1 restricts HIV-1 infection in resting CD4+ T cells. Nat Med 18, 1682–1689 10.1038/nm.2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee C., Archin N., Michaels D., Belkina A. C., Denis G. V., Bradner J., Sebastiani P., Margolis D. M., Montano M. (2012). BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukoc Biol 92, 1147–1154 10.1189/jlb.0312165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlough J. E., Ackley C. D., George J. W., Levy N., Acevedo R., Moore P. F., Rideout B. A., Cooper M. D., Pedersen N. C. (1991). Acquired immune dysfunction in cats with experimentally induced feline immunodeficiency virus infection: comparison of short-term and long-term infections. J Acquir Immune Defic Syndr 4, 219–227 [PubMed] [Google Scholar]
- Beatty J. A., Willett B. J., Gault E. A., Jarrett O. (1996). A longitudinal study of feline immunodeficiency virus-specific cytotoxic T lymphocytes in experimentally infected cats, using antigen-specific induction. J Virol 70, 6199–6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebenek K., Abbotts J., Wilson S. H., Kunkel T. A. (1993). Error-prone polymerization by HIV-1 reverse transcriptase. Contribution of template-primer misalignment, miscoding, and termination probability to mutational hot spots. J Biol Chem 268, 10324–10334 [PubMed] [Google Scholar]
- Benkirane M., Chun R. F., Xiao H., Ogryzko V. V., Howard B. H., Nakatani Y., Jeang K. T. (1998). Activation of integrated provirus requires histone acetyltransferase. p300 and P/CAF are coactivators for HIV-1 Tat. J Biol Chem 273, 24898–24905 10.1074/jbc.273.38.24898 [DOI] [PubMed] [Google Scholar]
- Berard M., Tough D. F. (2002). Qualitative differences between naïve and memory T cells. Immunology 106, 127–138 10.1046/j.1365-2567.2002.01447.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best B. M., Letendre S. L., Koopmans P., Rossi S. S., Clifford D. B., Collier A. C., Gelman B. B., Marra C. M., McArthur J. C. & other authors (2012). Low cerebrospinal fluid concentrations of the nucleotide HIV reverse transcriptase inhibitor, tenofovir. J Acquir Immune Defic Syndr 59, 376–381 10.1097/QAI.0b013e318247ec54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancotto A., Grivel J.-C., Gondois-Rey F., Bettendroffer L., Vigne R., Brown S., Margolis L. B., Hirsch I. (2004). Dual role of prostratin in inhibition of infection and reactivation of human immunodeficiency virus from latency in primary blood lymphocytes and lymphoid tissue. J Virol 78, 10507–10515 10.1128/JVI.78.19.10507-10515.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazkova J., Trejbalova K., Gondois-Rey F., Halfon P., Philibert P., Guiguen A., Verdin E., Olive D., Van Lint C. & other authors (2009). CpG methylation controls reactivation of HIV from latency. PLoS Pathog 5, e1000554 10.1371/journal.ppat.1000554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleul C. C., Wu L. J., Hoxie J. A., Springer T. A., Mackay C. R. (1997). The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci U S A 94, 1925–1930 10.1073/pnas.94.5.1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boberg A., Bråve A., Johansson S., Wahren B., Hinkula J., Rollman E. (2008). Murine models for HIV vaccination and challenge. Expert Rev Vaccines 7, 117–130 10.1586/14760584.7.1.117 [DOI] [PubMed] [Google Scholar]
- Bosque A., Planelles V. (2009). Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 113, 58–65 10.1182/blood-2008-07-168393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosque A., Famiglietti M., Weyrich A. S., Goulston C., Planelles V. (2011). Homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PLoS Pathog 7, e1002288 10.1371/journal.ppat.1002288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley J. M., Paiardini M. (2011). Immunodeficiency lentiviral infections in natural and non-natural hosts. Blood 118, 847–854 10.1182/blood-2010-12-325936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley J. M., Schacker T. W., Ruff L. E., Price D. A., Taylor J. H., Beilman G. J., Nguyen P. L., Khoruts A., Larson M. & other authors (2004a). CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med 200, 749–759 10.1084/jem.20040874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley J. M., Hill B. J., Ambrozak D. R., Price D. A., Guenaga F. J., Casazza J. P., Kuruppu J., Yazdani J., Migueles S. A. & other authors (2004b). T-cell subsets that harbor human immunodeficiency virus (HIV) in vivo: implications for HIV pathogenesis. J Virol 78, 1160–1168 10.1128/JVI.78.3.1160-1168.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan T. P., Woods J. O., Sedaghat A. R., Siliciano J. D., Siliciano R. F., Wilke C. O. (2009). Analysis of human immunodeficiency virus type 1 viremia and provirus in resting CD4+ T cells reveals a novel source of residual viremia in patients on antiretroviral therapy. J Virol 83, 8470–8481 10.1128/JVI.02568-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks D. G., Kitchen S. G., Kitchen C. M. R., Scripture-Adams D. D., Zack J. A. (2001). Generation of HIV latency during thymopoiesis. Nat Med 7, 459–464 10.1038/86531 [DOI] [PubMed] [Google Scholar]
- Bukrinsky M. I., Stanwick T. L., Dempsey M. P., Stevenson M. (1991). Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science 254, 423–427 10.1126/science.1925601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke B., Brown H. J., Marsden M. D., Bristol G., Vatakis D. N., Zack J. A. (2007). Primary cell model for activation-inducible human immunodeficiency virus. J Virol 81, 7424–7434 10.1128/JVI.02838-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett J. C., Rossi J. J. (2012). RNA-based therapeutics: current progress and future prospects. Chem Biol 19, 60–71 10.1016/j.chembiol.2011.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett J. C., Lim K. I., Calafi A., Rossi J. J., Schaffer D. V., Arkin A. P. (2010). Combinatorial latency reactivation for HIV-1 subtypes and variants. J Virol 84, 5958–5974 10.1128/JVI.00161-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzón M. J., Massanella M., Llibre J. M., Esteve A., Dahl V., Puertas M. C., Gatell J. M., Domingo P., Paredes R. & other authors (2010). HIV-1 replication and immune dynamics are affected by Raltegravir intensification of HAART-suppressed subjects. Nat Med 16, 460–465 10.1038/nm.2111 [DOI] [PubMed] [Google Scholar]
- Cabrita G. J. M., Ferreira B. S., da Silva C. L., Gonçalves R., Almeida-Porada G., Cabral J. M. S. (2003). Hematopoietic stem cells: from the bone to the bioreactor. Trends Biotechnol 21, 233–240 10.1016/S0167-7799(03)00076-3 [DOI] [PubMed] [Google Scholar]
- Canestri A., Lescure F.-X., Jaureguiberry S., Moulignier A., Amiel C., Marcelin A. G., Peytavin G., Tubiana R., Pialoux G., Katlama C. (2010). Discordance between cerebral spinal fluid and plasma HIV replication in patients with neurological symptoms who are receiving suppressive antiretroviral therapy. Clin Infect Dis 50, 773–778 10.1086/650538 [DOI] [PubMed] [Google Scholar]
- Carter C. C., Onafuwa-Nuga A., McNamara L. A., Riddell J., IV, Bixby D., Savona M. R., Collins K. L. (2010). HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med 16, 446–451 10.1038/nm.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter C. C., McNamara L. A., Onafuwa-Nuga A., Shackleton M., Riddell J., IV, Bixby D., Savona M. R., Morrison S. J., Collins K. L. (2011). HIV-1 utilizes the CXCR4 chemokine receptor to infect multipotent hematopoietic stem and progenitor cells. Cell Host Microbe 9, 223–234 10.1016/j.chom.2011.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahroudi A., Bosinger S. E., Vanderford T. H., Paiardini M., Silvestri G. (2012). Natural SIV hosts: showing AIDS the door. Science 335, 1188–1193 10.1126/science.1217550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C. N., McMonagle E. L., Hosie M. J., Willett B. J. (2013). Prostratin exhibits both replication enhancing and inhibiting effects on FIV infection of feline CD4+ T-cells. Virus Res. 171, 121–128 10.1016/j.virusres.2012.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. F., Mu Y., Greene W. C. (2002). Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J 21, 6539–6548 10.1093/emboj/cdf660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang K., Rice A. P. (2012). MicroRNA-mediated restriction of HIV-1 in resting CD4+ T cells and monocytes. Viruses 4, 1390–1409 10.3390/v4091390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe H., Farzan M., Sun Y., Sullivan N., Rollins B., Ponath P. D., Wu L., Mackay C. R., LaRosa G. & other authors (1996). The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85, 1135–1148 10.1016/S0092-8674(00)81313-6 [DOI] [PubMed] [Google Scholar]
- Chomont N., El-Far M., Ancuta P., Trautmann L., Procopio F. A., Yassine-Diab B., Boucher G., Boulassel M.-R., Ghattas G. & other authors (2009). HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15, 893–900 10.1038/nm.1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary S. K., Rezk N. L., Ince W. L., Cheema M., Zhang L., Su L., Swanstrom R., Kashuba A. D., Margolis D. M. (2009). Suppression of human immunodeficiency virus type 1 (HIV-1) viremia with reverse transcriptase and integrase inhibitors, CD4+ T-cell recovery, and viral rebound upon interruption of therapy in a new model for HIV treatment in the humanized Rag2–/–γc–/– mouse. J Virol 83, 8254–8258 10.1128/JVI.00580-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary S. K., Archin N. M., Cheema M., Dahl N. P., Garcia J. V., Margolis D. M. (2012). Latent HIV-1 infection of resting CD4+ T cells in the humanized Rag2−/− γc−/− mouse. J Virol 86, 114–120 10.1128/JVI.05590-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun T. W., Finzi D., Margolick J., Chadwick K., Schwartz D., Siliciano R. F. (1995). In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med 1, 1284–1290 10.1038/nm1295-1284 [DOI] [PubMed] [Google Scholar]
- Chun T. W., Carruth L., Finzi D., Shen X., DiGiuseppe J. A., Taylor H., Hermankova M., Chadwick K., Margolick J. & other authors (1997). Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387, 183–188 10.1038/387183a0 [DOI] [PubMed] [Google Scholar]
- Chun T. W., Engel D., Mizell S. B., Ehler L. A., Fauci A. S. (1998). Induction of HIV-1 replication in latently infected CD4+ T cells using a combination of cytokines. J Exp Med 188, 83–91 10.1084/jem.188.1.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun T. W., Davey R. T., Jr, Engel D., Lane H. C., Fauci A. S. (1999). Re-emergence of HIV after stopping therapy. Nature 401, 874–875 10.1038/44755 [DOI] [PubMed] [Google Scholar]
- Chun T. W., Nickle D. C., Justement J. S., Large D., Semerjian A., Curlin M. E., O’Shea M. A., Hallahan C. W., Daucher M. & other authors (2005). HIV-infected individuals receiving effective antiviral therapy for extended periods of time continually replenish their viral reservoir. J Clin Invest 115, 3250–3255 10.1172/JCI26197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun T.-W., Nickle D. C., Justement J. S., Meyers J. H., Roby G., Hallahan C. W., Kottilil S., Moir S., Mican J. M. & other authors (2008). Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J Infect Dis 197, 714–720 10.1086/527324 [DOI] [PubMed] [Google Scholar]
- Churchill M. J., Gorry P. R., Cowley D., Lal L., Sonza S., Purcell D. F. J., Thompson K. A., Gabuzda D., McArthur J. C. & other authors (2006). Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J Neurovirol 12, 146–152 10.1080/13550280600748946 [DOI] [PubMed] [Google Scholar]
- Cicala C., Arthos J., Censoplano N., Cruz C., Chung E., Martinelli E., Lempicki R. A., Natarajan V., VanRyk D. & other authors (2006). HIV-1 gp120 induces NFAT nuclear translocation in resting CD4+ T-cells. Virology 345, 105–114 10.1016/j.virol.2005.09.052 [DOI] [PubMed] [Google Scholar]
- Cillo A., Krishnan A., Mitsuyasu R., McMahon D., Li S., Rossi J., Zaia J., Mellors J. W. (2012). Plasma viremia and cellular HIV-1 DNA persist despite autologous hematopoietic stem cell transplantation for AIDS-related lymphoma. In CROI. Seattle, USA: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffer P. J., Burgering B. M. (2004). Forkhead-box transcription factors and their role in the immune system. Nat Rev Immunol 4, 889–899 10.1038/nri1488 [DOI] [PubMed] [Google Scholar]
- Coiras M., López-Huertas M. R., Pérez-Olmeda M., Alcamí J. (2009). Understanding HIV-1 latency provides clues for the eradication of long-term reservoirs. Nat Rev Microbiol 7, 798–812 10.1038/nrmicro2223 [DOI] [PubMed] [Google Scholar]
- Coleman C. M., Wu L. (2009). HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology 6, 51. 10.1186/1742-4690-6-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin L., Van Lint C. (2009). Molecular control of HIV-1 postintegration latency: implications for the development of new therapeutic strategies. Retrovirology 6, 111. 10.1186/1742-4690-6-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor R. I., Sheridan K. E., Ceradini D., Choe S., Landau N. R. (1997). Change in coreceptor use correlates with disease progression in HIV-1–infected individuals. J Exp Med 185, 621–628 10.1084/jem.185.4.621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras X., Schweneker M., Chen C.-S., McCune J. M., Deeks S. G., Martin J., Peterlin B. M. (2009). Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J Biol Chem 284, 6782–6789 10.1074/jbc.M807898200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull J. J., Romerio F., Sun J. M., Volker J. L., Galvin K. M., Davie J. R., Shi Y., Hansen U., Margolis D. M. (2000). The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol 74, 6790–6799 10.1128/JVI.74.15.6790-6799.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Arminio A., Sabin C. A., Phillips A. N., Reiss P., Weber R., Kirk O., El-Sadr W., De Wit S., Mateu S. & other authors (2004). Cardio- and cerebrovascular events in HIV-infected persons. AIDS 18, 1811–1817 10.1097/00002030-200409030-00010 [DOI] [PubMed] [Google Scholar]
- Dalgleish A. G., Beverley P. C., Clapham P. R., Crawford D. H., Greaves M. F., Weiss R. A. (1984). The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312, 763–767 10.1038/312763a0 [DOI] [PubMed] [Google Scholar]
- Davey R. T., Jr, Bhat N., Yoder C., Chun T. W., Metcalf J. A., Dewar R., Natarajan V., Lempicki R. A., Adelsberger J. W. & other authors (1999). HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci U S A 96, 15109–15114 10.1073/pnas.96.26.15109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis B. R., Schwartz D. H., Marx J. C., Johnson C. E., Berry J. M., Lyding J., Merigan T. C., Zander A. (1991). Absent or rare human immunodeficiency virus infection of bone marrow stem/progenitor cells in vivo. J Virol 65, 1985–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M., Carrington M., Winkler C., Huttley G. A., Smith M. W., Allikmets R., Goedert J. J., Buchbinder S. P., Vittinghoff E. & other authors (1996). Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 273, 1856–1862 10.1126/science.273.5283.1856 [DOI] [PubMed] [Google Scholar]
- Deeks S. G. (2011). HIV infection, inflammation, immunosenescence, and aging. Annu Rev Med 62, 141–155 . 10.1146/annurev-med-042909-093756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks S. G., Autran B., Berkhout B., Benkirane M., Cairns S., Chomont N., Chun T. W., Churchill M., Di Mascio M. & other authors (2012). Towards an HIV cure: a global scientific strategy. Nat Rev Immunol 12, 607–614 10.1038/nri3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H., Liu R., Ellmeier W., Choe S., Unutmaz D., Burkhart M., Di Marzio P., Marmon S., Sutton R. E. & other authors (1996). Identification of a major co-receptor for primary isolates of HIV-1. Nature 381, 661–666 10.1038/381661a0 [DOI] [PubMed] [Google Scholar]
- Descours B., Cribier A., Chable-Bessia C., Ayinde D., Rice G., Crow Y., Yatim A., Schwartz O., Laguette N., Benkirane M. (2012). SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4+ T-cells. Retrovirology 9, 87 10.1186/1742-4690-9-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Mascio M., Srinivasula S., Bhattacharjee A., Cheng L., Martiniova L., Herscovitch P., Lertora J., Kiesewetter D. (2009). Antiretroviral tissue kinetics: in vivo imaging using positron emission tomography. Antimicrob Agents Chemother 53, 4086–4095 10.1128/AAC.00419-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinoso J. B., Rabi S. A., Blankson J. N., Gama L., Mankowski J. L., Siliciano R. F., Zink M. C., Clements J. E. (2009a). A simian immunodeficiency virus-infected macaque model to study viral reservoirs that persist during highly active antiretroviral therapy. J Virol 83, 9247–9257 10.1128/JVI.00840-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinoso J. B., Kim S. Y., Wiegand A. M., Palmer S. E., Gange S. J., Cranmer L., O’Shea A., Callender M., Spivak A. & other authors (2009b). Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc Natl Acad Sci U S A 106, 9403–9408 10.1073/pnas.0903107106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douek D. C., Roederer M., Koup R. A. (2009). Emerging concepts in the immunopathogenesis of AIDS. Annu Rev Med 60, 471–484 10.1146/annurev.med.60.041807.123549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand C. M., Ghiaur G., Siliciano J. D., Rabi S. A., Eisele E. E., Salgado M., Shan L., Lai J. F., Zhang H. & other authors (2012). HIV-1 DNA is detected in bone marrow populations containing CD4+ T cells but is not found in purified CD34+ hematopoietic progenitor cells in most patients on antiretroviral therapy. J Infect Dis 205, 1014–1018 10.1093/infdis/jir884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egberink H. F., Keldermans C. E., Koolen M. J., Horzinek M. C. (1992). Humoral immune response to feline immunodeficiency virus in cats with experimentally induced and naturally acquired infections. Am J Vet Res 53, 1133–1138 [PubMed] [Google Scholar]
- Eichbaum Q. (2011). PD-1 signaling in HIV and chronic viral infection–potential for therapeutic intervention? Curr Med Chem 18, 3971–3980 10.2174/092986711796957239 [DOI] [PubMed] [Google Scholar]
- Eisele E., Siliciano R. F. (2012). Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 37, 377–388 10.1016/j.immuni.2012.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Else L. J., Taylor S., Back D. J., Khoo S. H. (2011). Pharmacokinetics of antiretroviral drugs in anatomical sanctuary sites: the male and female genital tract. Antivir Ther 16, 1149–1167 10.3851/IMP1919 [DOI] [PubMed] [Google Scholar]
- Feng Y., Broder C. C., Kennedy P. E., Berger E. A. (1996). HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272, 872–877 10.1126/science.272.5263.872 [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W. B., Fedorov O., Morse E. M., Keates T., Hickman T. T. & other authors (2010). Selective inhibition of BET bromodomains. Nature 468, 1067–1073 10.1038/nature09504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi D., Hermankova M., Pierson T., Carruth L. M., Buck C., Chaisson R. E., Quinn T. C., Chadwick K., Margolick J. & other authors (1997). Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300 10.1126/science.278.5341.1295 [DOI] [PubMed] [Google Scholar]
- Flynn J. N., Dunham S., Mueller A., Cannon C., Jarrett O. (2002). Involvement of cytolytic and non-cytolytic T cells in the control of feline immunodeficiency virus infection. Vet Immunol Immunopathol 85, 159–170 10.1016/S0165-2427(01)00425-1 [DOI] [PubMed] [Google Scholar]
- Folks T. M., Justement J., Kinter A., Dinarello C. A., Fauci A. S. (1987). Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 238, 800–802 10.1126/science.3313729 [DOI] [PubMed] [Google Scholar]
- Folks T. M., Kessler S. W., Orenstein J. M., Justement J. S., Jaffe E. S., Fauci A. S. (1988). Infection and replication of HIV-1 in purified progenitor cells of normal human bone marrow. Science 242, 919–922 10.1126/science.2460922 [DOI] [PubMed] [Google Scholar]
- Folks T. M., Clouse K. A., Justement J., Rabson A., Duh E., Kehrl J. H., Fauci A. S. (1989). Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T-cell clone. Proc Natl Acad Sci U S A 86, 2365–2368 10.1073/pnas.86.7.2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel R., Lombardo A., Arens A., Miller J. C., Genovese P., Kaeppel C., Nowrouzi A., Bartholomae C. C., Wang J. & other authors (2011). An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 29, 816–823 10.1038/nbt.1948 [DOI] [PubMed] [Google Scholar]
- Gandhi R. T., Zheng L., Bosch R. J., Chan E. S., Margolis D. M., Read S., Kallungal B., Palmer S., Medvik K. & other authors (2010). The effect of Raltegravir intensification on low-level residual viremia in HIV-infected patients on antiretroviral therapy: a randomized controlled trial. PLoS Med 7, e1000321 10.1371/journal.pmed.1000321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner S., Markovits P., Markovitz D. M., Kaplan M. H., Gallo R. C., Popovic M. (1986). The role of mononuclear phagocytes in HTLV-III/LAV infection. Science 233, 215–219 10.1126/science.3014648 [DOI] [PubMed] [Google Scholar]
- Ghose R., Liou L. Y., Herrmann C. H., Rice A. P. (2001). Induction of TAK (cyclin T1/P-TEFb) in purified resting CD4+ T lymphocytes by combination of cytokines. J Virol 75, 11336–11343 10.1128/JVI.75.23.11336-11343.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman J. P., Blundell M. P., Lopes L., Kinnon C., Di Santo J. P., Thrasher A. J. (1998). Enhanced human cell engraftment in mice deficient in RAG2 and the common cytokine receptor gamma chain. Br J Haematol 103, 335–342 10.1046/j.1365-2141.1998.00980.x [DOI] [PubMed] [Google Scholar]
- González-Scarano F., Martín-García J. (2005). The neuropathogenesis of AIDS. Nat Rev Immunol 5, 69–81 10.1038/nri1527 [DOI] [PubMed] [Google Scholar]
- Greger I. H., Demarchi F., Giacca M., Proudfoot N. J. (1998). Transcriptional interference perturbs the binding of Sp1 to the HIV-1 promoter. Nucleic Acids Res 26, 1294–1301 10.1093/nar/26.5.1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez C., Díaz L., Vallejo A., Hernández-Novoa B., Abad M., Madrid N., Dahl V., Rubio R., Moreno A. M. & other authors (2011). Intensification of antiretroviral therapy with a CCR5 antagonist in patients with chronic HIV-1 infection: effect on T cells latently infected. PLoS ONE 6, e27864 10.1371/journal.pone.0027864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaland R. E., Yu W., Rice A. P. (2005). Identification of LKLF-regulated genes in quiescent CD4+ T lymphocytes. Mol Immunol 42, 627–641 10.1016/j.molimm.2004.09.012 [DOI] [PubMed] [Google Scholar]
- Halfon P., Giorgetti C., Khiri H., Pénaranda G., Terriou P., Porcu-Buisson G., Chabert-Orsini V. (2010). Semen may harbor HIV despite effective HAART: another piece in the puzzle. PLoS ONE 5, e10569 10.1371/journal.pone.0010569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Lassen K., Monie D., Sedaghat A. R., Shimoji S., Liu X., Pierson T. C., Margolick J. B., Siliciano R. F., Siliciano J. D. (2004). Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. J Virol 78, 6122–6133 10.1128/JVI.78.12.6122-6133.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Wind-Rotolo M., Yang H. C., Siliciano J. D., Siliciano R. F. (2007). Experimental approaches to the study of HIV-1 latency. Nat Rev Microbiol 5, 95–106 10.1038/nrmicro1580 [DOI] [PubMed] [Google Scholar]
- Han Y., Lin Y. B., An W., Xu J., Yang H. C., O’Connell K., Dordai D., Boeke J. D., Siliciano J. D., Siliciano R. F. (2008). Orientation-dependent regulation of integrated HIV-1 expression by host gene transcriptional readthrough. Cell Host Microbe 4, 134–146 10.1016/j.chom.2008.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano H., Hayes T. L., Dahl V., Sinclair E., Lee T. H., Hoh R., Lampiris H., Hunt P. W., Palmer S. & other authors (2011). A randomized, controlled trial of Raltegravir intensification in antiretroviral-treated, HIV-infected patients with a suboptimal CD4+ T cell response. J Infect Dis 203, 960–968 10.1093/infdis/jiq138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht R., Stover J., Bollinger L., Muhib F., Case K., de Ferranti D. (2010). Financing of HIV/AIDS programme scale-up in low-income and middle-income countries, 2009-31. Lancet 376, 1254–1260 10.1016/S0140-6736(10)61255-X [DOI] [PubMed] [Google Scholar]
- Hermankova M., Siliciano J. D., Zhou Y., Monie D., Chadwick K., Margolick J. B., Quinn T. C., Siliciano R. F. (2003). Analysis of human immunodeficiency virus type 1 gene expression in latently infected resting CD4+ T lymphocytes in vivo. J Virol 77, 7383–7392 10.1128/JVI.77.13.7383-7392.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselton R. M., Greiner D. L., Mordes J. P., Rajan T. V., Sullivan J. L., Shultz L. D. (1995). High levels of human peripheral blood mononuclear cell engraftment and enhanced susceptibility to human immunodeficiency virus type 1 infection in NOD/LtSz-scid/scid mice. J Infect Dis 172, 974–982 10.1093/infdis/172.4.974 [DOI] [PubMed] [Google Scholar]
- Hirsch V. M., Fuerst T. R., Sutter G., Carroll M. W., Yang L. C., Goldstein S., Piatak M., Jr, Elkins W. R., Alvord W. G. & other authors (1996). Patterns of viral replication correlate with outcome in simian immunodeficiency virus (SIV)-infected macaques: effect of prior immunization with a trivalent SIV vaccine in modified vaccinia virus Ankara. J Virol 70, 3741–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch V. M., Lifson J. D., Kuah-Teh J. (2000). Simian immunodeficiency virus infection of monkeys as a model system for the study of AIDS pathogenesis, treatment, and prevention. In Advances in Pharmacology, vol. 49, pp. 437–477 Academic Press; 10.1016/S1054-3589(00)49034-4 [DOI] [PubMed] [Google Scholar]
- Ho D. D., Rota T. R., Hirsch M. S. (1986). Infection of monocyte/macrophages by human T lymphotropic virus type III. J Clin Invest 77, 1712–1715 10.1172/JCI112491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho D. D., Neumann A. U., Perelson A. S., Chen W., Leonard J. M., Markowitz M. (1995). Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373, 123–126 10.1038/373123a0 [DOI] [PubMed] [Google Scholar]
- Holt N., Wang J., Kim K., Friedman G., Wang X., Taupin V., Crooks G. M., Kohn D. B., Gregory P. D. & other authors (2010). Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol 28, 839–847 10.1038/nbt.1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosie M. J., Pajek D., Samman A., Willett B. J. (2011). Feline immunodeficiency virus (FIV) neutralization: a review. Viruses 3, 1870–1890 10.3390/v3101870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard K. E., Reckling S. K., Egan E. A., Dean G. A. (2010). Acute mucosal pathogenesis of feline immunodeficiency virus is independent of viral dose in vaginally infected cats. Retrovirology 7, 2. 10.1186/1742-4690-7-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hütter G., Nowak D., Mossner M., Ganepola S., Müssig A., Allers K., Schneider T., Hofmann J., Kücherer C. & other authors (2009). Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med 360, 692–698 10.1056/NEJMoa0802905 [DOI] [PubMed] [Google Scholar]
- Joos B., Fischer M., Kuster H., Pillai S. K., Wong J. K., Böni J., Hirschel B., Weber R., Trkola A., Günthard H. F., Swiss HIV Cohort Study (2008). HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc Natl Acad Sci U S A 105, 16725–16730 10.1073/pnas.0804192105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A., Bisgrove D., Verdin E. (2003). HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 22, 1868–1877 10.1093/emboj/cdg188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefsson L., Eriksson S., Sinclair E., Ho T., Killian M., Epling L., Shao W., Lewis B., Bacchetti P. & other authors (2012). Hematopoietic precursor cells isolated from patients on long-term suppressive HIV therapy did not contain HIV-1 DNA. J Infect Dis 206, 28–34 10.1093/infdis/jis301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A., Vahlenkamp T. W., Garg H., Tompkins W. A. F., Tompkins M. B. (2004). Preferential replication of FIV in activated CD4+CD25+T cells independent of cellular proliferation. Virology 321, 307–322 10.1016/j.virol.2004.01.014 [DOI] [PubMed] [Google Scholar]
- Joshi A., Garg H., Tompkins M. B., Tompkins W. A. (2005). Different thresholds of T cell activation regulate FIV infection of CD4+CD25+ and CD4+CD25- cells. Virology 335, 212–221 10.1016/j.virol.2005.02.016 [DOI] [PubMed] [Google Scholar]
- June C., Pablo T., Stein D., Mitsuyasu R., Lalezari J., Wang S., Lee G., Levine B., Tang W. & other authors (2012). Induction of acquired CCR5 deficiency with zinc finger nuclease-modified autologous CD4 T cells (SB-728-T) correlates with increases in CD4 count and effects on viral load in HIV-infected subjects. In 19th Conference on Retroviruses and Opportunistic Infections (CROI) [Google Scholar]
- Kauder S. E., Bosque A., Lindqvist A., Planelles V., Verdin E. (2009). Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog 5, e1000495 10.1371/journal.ppat.1000495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr J. S., Galloway S., Lagrutta A., Armstrong M., Miller T., Richon V. M., Andrews P. A. (2010). Nonclinical safety assessment of the histone deacetylase inhibitor Vorinostat. Int J Toxicol 29, 3–19 10.1177/1091581809352111 [DOI] [PubMed] [Google Scholar]
- Kornfeld H., Cruikshank W. W., Pyle S. W., Berman J. S., Center D. M. (1988). Lymphocyte activation by HIV-1 envelope glycoprotein. Nature 335, 445–448 10.1038/335445a0 [DOI] [PubMed] [Google Scholar]
- Kraase M., Sloan R., Klein D., Logan N., McMonagle L., Biek R., Willett B. J., Hosie M. J. (2010). Feline immunodeficiency virus env gene evolution in experimentally infected cats. Vet Immunol Immunopathol 134, 96–106 10.1016/j.vetimm.2009.10.015 [DOI] [PubMed] [Google Scholar]
- Kulkosky J., Culnan D. M., Roman J., Dornadula G., Schnell M., Boyd M. R., Pomerantz R. J. (2001). Prostratin: activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood 98, 3006–3015 10.1182/blood.V98.10.3006 [DOI] [PubMed] [Google Scholar]
- Kumar P., Ban H.-S., Kim S.-S., Wu H., Pearson T., Greiner D. L., Laouar A., Yao J., Haridas V. & other authors (2008). T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell 134, 577–586 10.1016/j.cell.2008.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwara A., Delong A., Rezk N., Hogan J., Burtwell H., Chapman S., Moreira C. C., Kurpewski J., Ingersoll J. & other authors (2008). Antiretroviral drug concentrations and HIV RNA in the genital tract of HIV-infected women receiving long-term highly active antiretroviral therapy. Clin Infect Dis 46, 719–725 10.1086/527387 [DOI] [PubMed] [Google Scholar]
- Kwon H. S., Brent M. M., Getachew R., Jayakumar P., Chen L. F., Schnolzer M., McBurney M. W., Marmorstein R., Greene W. C., Ott M. (2008). Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe 3, 158–167 10.1016/j.chom.2008.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen K. G., Bailey J. R., Siliciano R. F. (2004). Analysis of human immunodeficiency virus type 1 transcriptional elongation in resting CD4+ T cells in vivo. J Virol 78, 9105–9114 10.1128/JVI.78.17.9105-9114.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen K. G., Ramyar K. X., Bailey J. R., Zhou Y., Siliciano R. F. (2006). Nuclear retention of multiply spliced HIV-1 RNA in resting CD4+ T cells. PLoS Pathog 2, e68 10.1371/journal.ppat.0020068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launay O., Tod M., Tschöpe I., Si-Mohamed A., Bélarbi L., Charpentier C., Goujard C., Taburet A. M., Lortholary O. & other authors (2011). Residual HIV-1 RNA and HIV-1 DNA production in the genital tract reservoir of women treated with HAART: the prospective ANRS EP24 GYNODYN study. Antivir Ther 16, 843–852 10.3851/IMP1856 [DOI] [PubMed] [Google Scholar]
- Le Douce V., Herbein G., Rohr O., Schwartz C. (2010). Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology 7, 32. 10.1186/1742-4690-7-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy Y., Lacabaratz C., Weiss L., Viard J. P., Goujard C., Lelièvre J. D., Boué F., Molina J. M., Rouzioux C. & other authors (2009). Enhanced T cell recovery in HIV-1-infected adults through IL-7 treatment. J Clin Invest 119, 997–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy Y., Sereti I., Tambussi G., Routy J. P., Lelievre J. D., Delfraissy J. F., Molina J. M., Fischl M., Goujard C. & other authors (2012). Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clin Infect Dis 55, 291–300 10.1093/cid/cis383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin S. R., Evans V. A., Elliott J. H., Spire B., Chomont N. (2011). Finding a cure for HIV: will it ever be achievable? J Int AIDS Soc 14, 4. 10.1186/1758-2652-14-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Guo J., Wu Y., Zhou Q. (2013). The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res 41, 277–287 10.1093/nar/gks976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusic M., Marcello A., Cereseto A., Giacca M. (2003). Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J 22, 6550–6561 10.1093/emboj/cdg631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansky L. M., Temin H. M. (1995). Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol 69, 5087–5094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcello A. (2006). Latency: the hidden HIV-1 challenge. Retrovirology 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis L., Shattock R. (2006). Selective transmission of CCR5-utilizing HIV-1: the ‘gatekeeper’ problem resolved? Nat Rev Microbiol 4, 312–317 [DOI] [PubMed] [Google Scholar]
- Marini A., Harper J. M., Romerio F. (2008). An in vitro system to model the establishment and reactivation of HIV-1 latency. J Immunol 181, 7713–7720 [DOI] [PubMed] [Google Scholar]
- Marsden M. D., Zack J. A. (2009). Eradication of HIV: current challenges and new directions. J Antimicrob Chemother 63, 7–10 10.1093/jac/dkn455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbonye U., Karn J. (2011). Control of HIV latency by epigenetic and non-epigenetic mechanisms. Curr HIV Res 9, 554–567 10.2174/157016211798998736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnel S. J., Sparger E. E., Luciw P. A., Murphy B. G. (2012). Pharmacologic reactivation of latent feline immunodeficiency virus ex vivo in peripheral CD4+ T-lymphocytes. Virus Res 170, 174–179 10.1016/j.virusres.2012.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon D., Jones J., Wiegand A., Gange S. J., Kearney M., Palmer S., McNulty S., Metcalf J. A., Acosta E. & other authors (2010). Short-course raltegravir intensification does not reduce persistent low-level viremia in patients with HIV-1 suppression during receipt of combination antiretroviral therapy. Clin Infect Dis 50, 912–919 10.1086/650749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara L. A., Collins K. L. (2011). Hematopoietic stem/precursor cells as HIV reservoirs. Curr Opin HIV AIDS 6, 43–48 10.1097/COH.0b013e32834086b3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton T., Lim H. B., Montgomery D., Rockway T., Tang H., Cheng X. H., Lu L. J., Mo H. M., Kohlbrenner W. E., Molla A. (2004). Inhibition of human immunodeficiency virus type I integrase by naphthamidines and 2-aminobenzimidazoles. Antiviral Res 64, 35–45 10.1016/j.antiviral.2004.04.007 [DOI] [PubMed] [Google Scholar]
- Mowat A. M., Viney J. L. (1997). The anatomical basis of intestinal immunity. Immunol Rev 156, 145–166 10.1111/j.1600-065X.1997.tb00966.x [DOI] [PubMed] [Google Scholar]
- Murphy B., Vapniarsky N., Hillman C., Castillo D., McDonnel S., Moore P., Luciw P. A., Sparger E. E. (2012). FIV establishes a latent infection in feline peripheral blood CD4+ T lymphocytes in vivo during the asymptomatic phase of infection. Retrovirology 9, 12 10.1186/1742-4690-9-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal T. F., Holland H. K., Baum C. M., Villinger F., Ansari A. A., Saral R., Wingard J. R., Fleming W. H. (1995). CD34+ progenitor cells from asymptomatic patients are not a major reservoir for human immunodeficiency virus-1. Blood 86, 1749–1756 [PubMed] [Google Scholar]
- Nicholson J. K. A., Cross G. D., Callaway C. S., McDougal J. S. (1986). In vitro infection of human monocytes with human T lymphotropic virus type III/lymphadenopathy-associated virus (HTLV-III/LAV). J Immunol 137, 323–329 [PubMed] [Google Scholar]
- North T. W., Higgins J., Deere J. D., Hayes T. L., Villalobos A., Adamson L., Shacklett B. L., Schinazi R. F., Luciw P. A. (2010). Viral sanctuaries during highly active antiretroviral therapy in a nonhuman primate model for AIDS. J Virol 84, 2913–2922 10.1128/JVI.02356-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotney C., English R. V., Housman J., Davidson M. G., Nasisse M. P., Jeng C. R., Davis W. C., Tompkins M. B. (1990). Lymphocyte population changes in cats naturally infected with feline immunodeficiency virus. AIDS 4, 1213–1218 10.1097/00002030-199012000-00005 [DOI] [PubMed] [Google Scholar]
- O’Brien S. J., Moore J. P. (2000). The effect of genetic variation in chemokines and their receptors on HIV transmission and progression to AIDS. Immunol Rev 177, 99–111 10.1034/j.1600-065X.2000.17710.x [DOI] [PubMed] [Google Scholar]
- O’Doherty U., Swiggard W. J., Malim M. H. (2000). Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol 74, 10074–10080 10.1128/JVI.74.21.10074-10080.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski M. A., Chun T. W., Justement S. J., Motola I., Spinelli M. A., Adelsberger J., Ehler L. A., Mizell S. B., Hallahan C. W., Fauci A. S. (1999). Both memory and CD45RA+/CD62L+ naive CD4+ T cells are infected in human immunodeficiency virus type 1-infected individuals. J Virol 73, 6430–6435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald-Richter K., Grill S. M., Leelawong M., Unutmaz D. (2004). HIV infection of primary human T cells is determined by tunable thresholds of T cell activation. Eur J Immunol 34, 1705–1714 10.1002/eji.200424892 [DOI] [PubMed] [Google Scholar]
- Pace M. J., Agosto L., Graf E. H., O’Doherty U. (2011). HIV reservoirs and latency models. Virology 411, 344–354 10.1016/j.virol.2010.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S., Maldarelli F., Wiegand A., Bernstein B., Hanna G. J., Brun S. C., Kempf D. J., Mellors J. W., Coffin J. M., King M. S. (2008). Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A 105, 3879–3884 10.1073/pnas.0800050105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S., Josefsson L., Coffin J. M. (2011). HIV reservoirs and the possibility of a cure for HIV infection. J Intern Med 270, 550–560 10.1111/j.1365-2796.2011.02457.x [DOI] [PubMed] [Google Scholar]
- Parada C. A., Roeder R. G. (1996). Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature 384, 375–378 10.1038/384375a0 [DOI] [PubMed] [Google Scholar]
- Pattanayak V., Ramirez C. L., Joung J. K., Liu D. R. (2011). Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods 8, 765–770 10.1038/nmeth.1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen N. C., Ho E. W., Brown M. L., Yamamoto J. K. (1987). Isolation of a T-lymphotropic virus from domestic cats with an immunodeficiency-like syndrome. Science 235, 790–793 10.1126/science.3643650 [DOI] [PubMed] [Google Scholar]
- Peer D., Karp J. M., Hong S., Farokhzad O. C., Margalit R., Langer R. (2007). Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol 2, 751–760 10.1038/nnano.2007.387 [DOI] [PubMed] [Google Scholar]
- Perez E. E., Wang J., Miller J. C., Jouvenot Y., Kim K. A., Liu O., Wang N., Lee G., Bartsevich V. V. & other authors (2008). Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26, 808–816 10.1038/nbt1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Martínez D., Tanaka T., Rabbitts T. H. (2010). Intracellular antibodies and cancer: new technologies offer therapeutic opportunities. Bioessays 32, 589–598 10.1002/bies.201000009 [DOI] [PubMed] [Google Scholar]
- Pierson T., Hoffman T. L., Blankson J., Finzi D., Chadwick K., Margolick J. B., Buck C., Siliciano J. D., Doms R. W., Siliciano R. F. (2000). Characterization of chemokine receptor utilization of viruses in the latent reservoir for human immunodeficiency virus type 1. J Virol 74, 7824–7833 10.1128/JVI.74.17.7824-7833.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson T. C., Zhou Y., Kieffer T. L., Ruff C. T., Buck C., Siliciano R. F. (2002). Molecular characterization of preintegration latency in human immunodeficiency virus type 1 infection. J Virol 76, 8518–8531 10.1128/JVI.76.17.8518-8513.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins J. M., Jurriaans S., van Praag R. M., Blaak H., van Rij R., Schellekens P. T., ten Berge I. J., Yong S. L., Fox C. H. & other authors (1999). Immuno-activation with anti-CD3 and recombinant human IL-2 in HIV-1-infected patients on potent antiretroviral therapy. AIDS 13, 2405–2410 10.1097/00002030-199912030-00012 [DOI] [PubMed] [Google Scholar]
- Ramratnam B., Mittler J. E., Zhang L., Boden D., Hurley A., Fang F., Macken C. A., Perelson A. S., Markowitz M., Ho D. D. (2000). The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat Med 6, 82–85 10.1038/71577 [DOI] [PubMed] [Google Scholar]
- Reuse S., Calao M., Kabeya K., Guiguen A., Gatot J. S., Quivy V., Vanhulle C., Lamine A., Vaira D. & other authors (2009). Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: implications for treatment of latent infection. PLoS ONE 4, e6093 10.1371/journal.pone.0006093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman D. D., Margolis D. M., Delaney M., Greene W. C., Hazuda D., Pomerantz R. J. (2009). The challenge of finding a cure for HIV infection. Science 323, 1304–1307 10.1126/science.1165706 [DOI] [PubMed] [Google Scholar]
- Roesch F., Meziane O., Kula A., Nisole S., Porrot F., Anderson I., Mammano F., Fassati A., Marcello A. & other authors (2012). Hyperthermia stimulates HIV-1 replication. PLoS Pathog 8, e1002792 10.1371/journal.ppat.1002792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M. I., Medina K. L., Garrett K., Kolar G., Comp P. C., Shultz L. D., Capra J. D., Wilson P., Schipul A., Kincade P. W. (2001). Relatively normal human lymphopoiesis but rapid turnover of newly formed B cells in transplanted nonobese diabetic/SCID mice. J Immunol 167, 3033–3042 [DOI] [PubMed] [Google Scholar]
- Ruff C. T., Ray S. C., Kwon P., Zinn R., Pendleton A., Hutton N., Ashworth R., Gange S., Quinn T. C. & other authors (2002). Persistence of wild-type virus and lack of temporal structure in the latent reservoir for human immunodeficiency virus type 1 in pediatric patients with extensive antiretroviral exposure. J Virol 76, 9481–9492 10.1128/JVI.76.18.9481-9492.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rullas J., Bermejo M., García-Pérez J., Beltán M., González N., Hezareh M., Brown S. J., Alcamí J. (2004). Prostratin induces HIV activation and downregulates HIV receptors in peripheral blood lymphocytes. Antivir Ther 9, 545–554 [PubMed] [Google Scholar]
- Sahu G. K., Lee K., Ji J. X., Braciale V., Baron S., Cloyd M. W. (2006). A novel in vitro system to generate and study latently HIV-infected long-lived normal CD4+ T-lymphocytes. Virology 355, 127–137 10.1016/j.virol.2006.07.020 [DOI] [PubMed] [Google Scholar]
- Sahu G. K., Paar D., Frost S. D. W., Smith M. M., Weaver S., Cloyd M. W. (2009). Low-level plasma HIVs in patients on prolonged suppressive highly active antiretroviral therapy are produced mostly by cells other than CD4 T-cells. J Med Virol 81, 9–15 10.1002/jmv.21366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh S., Solomon A., Wightman F., Xhilaga M., Cameron P. U., Lewin S. R. (2007). CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: a novel model of HIV-1 latency. Blood 110, 4161–4164 10.1182/blood-2007-06-097907 [DOI] [PubMed] [Google Scholar]
- Saleh S., Wightman F., Ramanayake S., Alexander M., Kumar N., Khoury G., Pereira C., Purcell D., Cameron P. U., Lewin S. R. (2011). Expression and reactivation of HIV in a chemokine induced model of HIV latency in primary resting CD4+ T cells. Retrovirology 8, 80 10.1186/1742-4690-8-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnittman S. M., Psallidopoulos M. C., Lane H. C., Thompson L., Baseler M., Massari F., Fox C. H., Salzman N. P., Fauci A. S. (1989). The reservoir for HIV-1 in human peripheral blood is a T cell that maintains expression of CD4. Science 245, 305–308 10.1126/science.2665081 [DOI] [PubMed] [Google Scholar]
- Schröder A. R., Shinn P., Chen H., Berry C., Ecker J. R., Bushman F. (2002). HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110, 521–529 10.1016/S0092-8674(02)00864-4 [DOI] [PubMed] [Google Scholar]
- Scripture-Adams D. D., Brooks D. G., Korin Y. D., Zack J. A. (2002). Interleukin-7 induces expression of latent human immunodeficiency virus type 1 with minimal effects on T-cell phenotype. J Virol 76, 13077–13082 10.1128/JVI.76.24.13077-13082.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan L., Deng K., Shroff N. S., Durand C. M., Rabi S. A., Yang H. C., Zhang H., Margolick J. B., Blankson J. N., Siliciano R. F. (2012). Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 36, 491–501 10.1016/j.immuni.2012.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen A. D., Zink M. C., Mankowski J. L., Chadwick K., Margolick J. B., Carruth L. M., Li M., Clements J. E., Siliciano R. F. (2003). Resting CD4+ T lymphocytes but not thymocytes provide a latent viral reservoir in a simian immunodeficiency virus-Macaca nemestrina model of human immunodeficiency virus type 1-infected patients on highly active antiretroviral therapy. J Virol 77, 4938–4949 10.1128/JVI.77.8.4938-4949.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojima M., Miyazawa T., Ikeda Y., McMonagle E. L., Haining H., Akashi H., Takeuchi Y., Hosie M. J., Willett B. J. (2004). Use of CD134 as a primary receptor by the feline immunodeficiency virus. Science 303, 1192–1195 10.1126/science.1092124 [DOI] [PubMed] [Google Scholar]
- Siliciano, R. F. & Greene, W. C. (2011). HIV Latency. Cold Spring Harbor Perspectives in Medicine 1. [DOI] [PMC free article] [PubMed]
- Siliciano J. D., Siliciano R. F. (2010). Biomarkers of HIV replication. Curr Opin HIV AIDS 5, 491–497 10.1097/COH.0b013e32833f206f [DOI] [PubMed] [Google Scholar]
- Siliciano J. D., Kajdas J., Finzi D., Quinn T. C., Chadwick K., Margolick J. B., Kovacs C., Gange S. J., Siliciano R. F. (2003). Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med 9, 727–728 10.1038/nm880 [DOI] [PubMed] [Google Scholar]
- Sloan R. D., Wainberg M. A. (2011). The role of unintegrated DNA in HIV infection. Retrovirology 8, 52 10.1186/1742-4690-8-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S. K., Kessler S. W., Justement J. S., Schnittman S. M., Greenhouse J. J., Brown C. C., Musongela L., Musey K., Kapita B., Fauci A. S. (1992). CD34+ bone marrow cells are infected with HIV in a subset of seropositive individuals. J Immunol 149, 689–697 [PubMed] [Google Scholar]
- Stevenson M., Stanwick T. L., Dempsey M. P., Lamonica C. A. (1990). HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J 9, 1551–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svarovskaia E. S., Barr R., Zhang X. C., Pais G. C. G., Marchand C., Pommier Y., Burke T. R., Jr, Pathak V. K. (2004). Azido-containing diketo acid derivatives inhibit human immunodeficiency virus type 1 integrase in vivo and influence the frequency of deletions at two-long-terminal-repeat-circle junctions. J Virol 78, 3210–3222 10.1128/JVI.78.7.3210-3222.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan C. H., Bühler B., Tschan M. P., Barbas C. F., Torbett B. E. (2006). T-cell protection and enrichment through lentiviral CCR5 intrabody gene delivery. Gene Ther 13, 1480–1492 10.1038/sj.gt.3302801 [DOI] [PubMed] [Google Scholar]
- Tobin N. H., Learn G. H., Holte S. E., Wang Y., Melvin A. J., McKernan J. L., Pawluk D. M., Mohan K. M., Lewis P. F. & other authors (2005). Evidence that low-level viremias during effective highly active antiretroviral therapy result from two processes: expression of archival virus and replication of virus. J Virol 79, 9625–9634 10.1128/JVI.79.15.9625-9634.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompkins M. B., Tompkins W. A. (2008). Lentivirus-induced immune dysregulation. Vet Immunol Immunopathol 123, 45–55 10.1016/j.vetimm.2008.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traggiai E., Chicha L., Mazzucchelli L., Bronz L., Piffaretti J. C., Lanzavecchia A., Manz M. G. (2004). Development of a human adaptive immune system in cord blood cell-transplanted mice. Science 304, 104–107 10.1126/science.1093933 [DOI] [PubMed] [Google Scholar]
- Trautmann L., Janbazian L., Chomont N., Said E. A., Gimmig S., Bessette B., Boulassel M.-R., Delwart E., Sepulveda H. & other authors (2006). Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med 12, 1198–1202 10.1038/nm1482 [DOI] [PubMed] [Google Scholar]
- Tyagi M., Pearson R. J., Karn J. (2010). Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol 84, 6425–6437 10.1128/JVI.01519-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzachanis D., Lafuente E. M., Li L., Boussiotis V. A. (2004). Intrinsic and extrinsic regulation of T lymphocyte quiescence. Leuk Lymphoma 45, 1959–1967 10.1080/1042819042000219494 [DOI] [PubMed] [Google Scholar]
- Urnov F. D., Miller J. C., Lee Y.-L., Beausejour C. M., Rock J. M., Augustus S., Jamieson A. C., Porteus M. H., Gregory P. D., Holmes M. C. (2005). Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435, 646–651 10.1038/nature03556 [DOI] [PubMed] [Google Scholar]
- Van Duyne R., Pedati C., Guendel I., Carpio L., Kehn-Hall K., Saifuddin M., Kashanchi F. (2009). The utilization of humanized mouse models for the study of human retroviral infections. Retrovirology 6, 76. 10.1186/1742-4690-6-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Grevenynghe J., Procopio F. A., He Z., Chomont N., Riou C., Zhang Y., Gimmig S., Boucher G., Wilkinson P. & other authors (2008). Transcription factor FOXO3a controls the persistence of memory CD4+ T cells during HIV infection. Nat Med 14, 266–274 10.1038/nm1728 [DOI] [PubMed] [Google Scholar]
- van Leeuwen E. M. M., Sprent J., Surh C. D. (2009). Generation and maintenance of memory CD4+ T cells. Curr Opin Immunol 21, 167–172 10.1016/j.coi.2009.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C., Emiliani S., Ott M., Verdin E. (1996). Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J 15, 1112–1120 [PMC free article] [PubMed] [Google Scholar]
- Vatakis D. N., Bristol G., Wilkinson T. A., Chow S. A., Zack J. A. (2007). Immediate activation fails to rescue efficient human immunodeficiency virus replication in quiescent CD4+ T cells. J Virol 81, 3574–3582 10.1128/JVI.02569-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatakis D. N., Kim S., Kim N., Chow S. A., Zack J. A. (2009). Human immunodeficiency virus integration efficiency and site selection in quiescent CD4+ T cells. J Virol 83, 6222–6233 10.1128/JVI.00356-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatakis D. N., Nixon C. C., Zack J. A. (2010). Quiescent T cells and HIV: an unresolved relationship. Immunol Res 48, 110–121 10.1007/s12026-010-8171-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E., Paras P., Jr, Van Lint C. (1993). Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J 12, 3249–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volberding P. A., Deeks S. G. (2010). Antiretroviral therapy and management of HIV infection. Lancet 376, 49–62 10.1016/S0140-6736(10)60676-9 [DOI] [PubMed] [Google Scholar]
- Wang F. X., Xu Y., Sullivan J., Souder E., Argyris E. G., Acheampong E. A., Fisher J., Sierra M., Thomson M. M. & other authors (2005). IL-7 is a potent and proviral strain-specific inducer of latent HIV-1 cellular reservoirs of infected individuals on virally suppressive HAART. J Clin Invest 115, 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman D., Rabin R. L., Arthos J., Rubbert A., Dybul M., Swofford R., Venkatesan S., Farber J. M., Fauci A. S. (1997). Macrophage-tropic HIV and SIV envelope proteins induce a signal through the CCR5 chemokine receptor. Nature 389, 981–985 10.1038/40173 [DOI] [PubMed] [Google Scholar]
- Wightman F., Solomon A., Khoury G., Green J. A., Gray L., Gorry P. R., Ho Y. S., Saksena N. K., Hoy J. & other authors (2010). Both CD31+ and CD31⁻ naive CD4+ T cells are persistent HIV type 1-infected reservoirs in individuals receiving antiretroviral therapy. J Infect Dis 202, 1738–1748 10.1086/656721 [DOI] [PubMed] [Google Scholar]
- Wightman F., Ellenberg P., Churchill M., Lewin S. R. (2012). HDAC inhibitors in HIV. Immunol Cell Biol 90, 47–54 10.1038/icb.2011.95 [DOI] [PubMed] [Google Scholar]
- Wilen C. B., Wang J., Tilton J. C., Miller J. C., Kim K. A., Rebar E. J., Sherrill-Mix S. A., Patro S. C., Secreto A. J. & other authors (2011). Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog 7, e1002020 10.1371/journal.ppat.1002020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett B. J., Picard L., Hosie M. J., Turner J. D., Adema K., Clapham P. R. (1997). Shared usage of the chemokine receptor CXCR4 by the feline and human immunodeficiency viruses. J Virol 71, 6407–6415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S. A., Greene W. C. (2007). Regulation of HIV-1 latency by T-cell activation. Cytokine 39, 63–74 10.1016/j.cyto.2007.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S. A., Kwon H., Chen L. F., Greene W. C. (2007). Sustained induction of NF-κB is required for efficient expression of latent human immunodeficiency virus type 1. J Virol 81, 6043–6056 10.1128/JVI.02074-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolschendorf F., Duverger A., Jones J., Wagner F. H., Huff J., Benjamin W. H., Saag M. S., Niederweis M., Kutsch O. (2010). Hit-and-run stimulation: a novel concept to reactivate latent HIV-1 infection without cytokine gene induction. J Virol 84, 8712–8720 10.1128/JVI.00523-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. T., Yoder A. (2009). Chemokine coreceptor signaling in HIV-1 infection and pathogenesis. PLoS Pathog 5, e1000520 10.1371/journal.ppat.1000520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L. J., Paxton W. A., Kassam N., Ruffing N., Rottman J. B., Sullivan N., Choe H., Sodroski J., Newman W. & other authors (1997). CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J Exp Med 185, 1681–1692 10.1084/jem.185.9.1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing S., Bullen C. K., Shroff N. S., Shan L., Yang H. C., Manucci J. L., Bhat S., Zhang H., Margolick J. B. & other authors (2011). Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J Virol 85, 6060–6064 10.1128/JVI.02033-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing S. F., Bhat S., Shroff N. S., Zhang H., Lopez J. A., Margolick J. B., Liu J. O., Siliciano R. F. (2012). Novel structurally related compounds reactivate latent HIV-1 in a bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J Antimicrob Chemother 67, 398–403 10.1093/jac/dkr496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto J. K., Sparger E., Ho E. W., Andersen P. R., O’Connor T. P., Mandell C. P., Lowenstine L., Munn R., Pedersen N. C. (1988). Pathogenesis of experimentally induced feline immunodeficiency virus infection in cats. Am J Vet Res 49, 1246–1258 [PubMed] [Google Scholar]
- Yamamoto J. K., Hansen H., Ho E. W., Morishita T. Y., Okuda T., Sawa T. R., Nakamura R. M., Pedersen N. C. (1989). Epidemiologic and clinical aspects of feline immunodeficiency virus infection in cats from the continental United States and Canada and possible mode of transmission. J Am Vet Med Assoc 194, 213–220 [PubMed] [Google Scholar]
- Yang H. C. (2011). Primary cell models of HIV latency. Curr Opin HIV AIDS 6, 62–67 10.1097/COH.0b013e3283412568 [DOI] [PubMed] [Google Scholar]
- Yang H. C., Xing S. F., Shan L., O’Connell K., Dinoso J., Shen A. D., Zhou Y., Shrum C. K., Han Y. F. & other authors (2009). Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J Clin Invest 119, 3473–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung F., Hoberg J. E., Ramsey C. S., Keller M. D., Jones D. R., Frye R. A., Mayo M. W. (2004). Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23, 2369–2380 10.1038/sj.emboj.7600244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylisastigui L., Archin N. M., Lehrman G., Bosch R. J., Margolis D. M. (2004). Coaxing HIV-1 from resting CD4 T cells: histone deacetylase inhibition allows latent viral expression. AIDS 18, 1101–1108 10.1097/00002030-200405210-00003 [DOI] [PubMed] [Google Scholar]
- Yoder A., Yu D. Y., Dong L., Iyer S. R., Xu X. H., Kelly J., Liu J., Wang W. F., Vorster P. J. & other authors (2008). HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell 134, 782–792 10.1016/j.cell.2008.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J., Wang J., Crain K., Fearns C., Kim K. A., Hua K. L., Gregory P. D., Holmes M. C., Torbett B. E. (2012). Zinc-finger nuclease editing of human cxcr4 promotes HIV-1 CD4+ T cell resistance and enrichment. Mol Ther 20, 849–859 10.1038/mt.2011.310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukl S. A., Shergill A. K., McQuaid K., Gianella S., Lampiris H., Hare C. B., Pandori M., Sinclair E., Günthard H. F. & other authors (2010). Effect of Raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS 24, 2451–2460 10.1097/QAD.0b013e32833ef7bb [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf I., Fruman D. A. (2003). Regulation of quiescence in lymphocytes. Trends Immunol 24, 380–386 10.1016/S1471-4906(03)00141-8 [DOI] [PubMed] [Google Scholar]
- Zagury D., Bernard J., Leonard R., Cheynier R., Feldman M., Sarin P. S., Gallo R. C. (1986). Long-term cultures of HTLV-III–infected T cells: a model of cytopathology of T-cell depletion in AIDS. Science 231, 850–853 10.1126/science.2418502 [DOI] [PubMed] [Google Scholar]
- Zhou Y., Zhang H., Siliciano J. D., Siliciano R. F. (2005). Kinetics of human immunodeficiency virus type 1 decay following entry into resting CD4+ T cells. J Virol 79, 2199–2210 10.1128/JVI.79.4.2199-2210.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T., Mo H., Wang N., Nam D. S., Cao Y., Koup R. A., Ho D. D. (1993). Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 261, 1179–1181 10.1126/science.8356453 [DOI] [PubMed] [Google Scholar]
- Zhu J., Gaiha G. D., John S. P., Pertel T., Chin C. R., Gao G., Qu H., Walker B. D., Elledge S. J., Brass A. L. (2012). Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep 2, 807–816 10.1016/j.celrep.2012.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]