

Abstract
Photochemical activation of a deoxyribozyme with peroxidase activity was achieved by the synthesis and incorporation of a caged deoxyguanosine.
The oxidation of organic and inorganic substrates by peroxides in a cellular context is catalyzed by peroxidases.1 Well-studied members of this class of proteins are horseradish peroxidase, cytochrome c peroxidase, and glutathione peroxidase.1 Moreover, antibodies with peroxidase activity have been developed.2 All these catalysts are composed of a polypeptide and a metalloporphyrin cofactor, hemin and derivatives of hemin. The Sen group evolved a DNA aptamer which binds to hemin with a high affinity (0.9 μM binding constant) and, most importantly, exhibits a significant peroxidase activity.3-5 We and others have previously reported the light triggering of a variety of DNA functions through the installation of light-removable caging groups, including RNA cleavage, DNA transcription, amplification and polymerization, and DNA binding.6,7 Thus we became interested in the photochemical regulation of peroxidase activity through the installation of light-removable protecting groups (caging groups) on the deoxyoligonucleotide.
The peroxidase deoxyribozyme PS2.M evolved by Sen et al. is an 18-mer oligonucleotide of the sequence 5′-GTGGGTAGGGCGGGTTGG-3′ which is assumed to fold into the G-quadruplex structure D1 (Fig. 1) (a complementary structure with flipped strands in the front has been postulated by Sen et al. as well; not shown).4 Due to the prevalence of deoxyguanosines in D1, we investigated the ability to apply our NPOM caging strategy6,8,9 to the purine base of dG thus disrupting an essential hydrogen bond. The synthesis of the caged deoxyguanosine phosphoramidite 6 (Scheme 1) commences with the synthesis of the acetyl and dimethylformamidine protected deoxyguanosine 2 from deoxyguanosine (1) in 61% yield. Caging of 2 with 6-nitropiperonyloxymethylene chloride (NPOM-Cl, synthesized in 4 steps from commercially available material9) was achieved in 79% (DBU, DMF, rt) furnishing 3. The diol 4 was obtained in 92% yield through removal of the acetate groups with K2CO3 in MeOH. Selective tritylation of the primary hydroxy group with DMT-Cl in pyridine delivered 5 in 77% yield. The synthesis of 6 was completed through activation as the phosphoramidite under classical conditions (2-cyanoethyl-diisopropyl-chloro phosphoramidite, DCM, DIPEA, 82% yield).
Fig. 1.
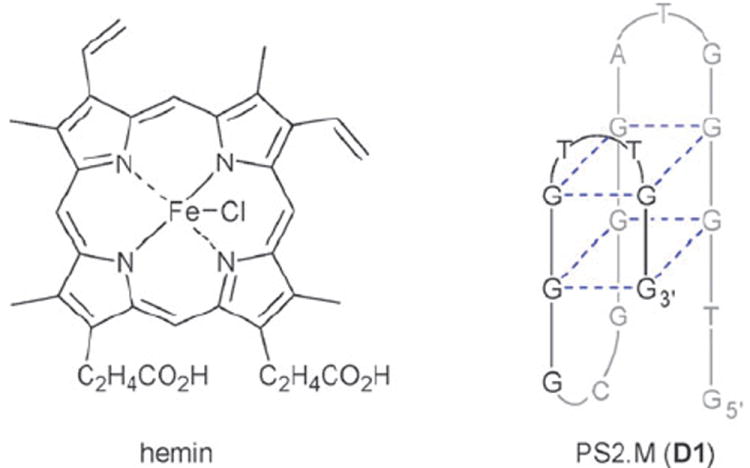
Structure of hemin and schematic of one of the G-quadruplex structures proposed for PS2.M (D1).
Scheme 1.
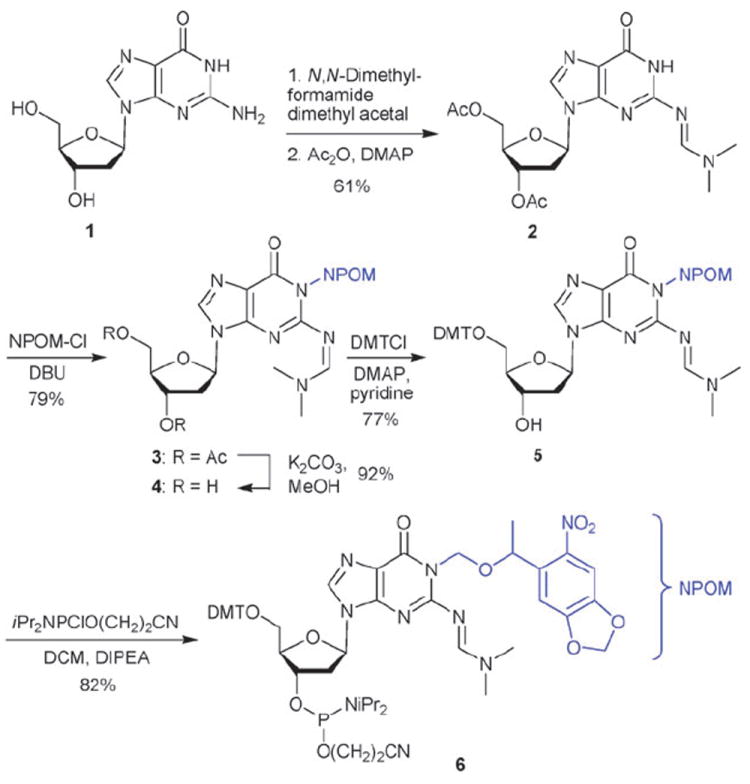
Synthesis of the NPOM-caged deoxyguanosine phosphoramidite 6.
The stability of 6 to DNA synthesis conditions and its rapid decaging through irradiation with UV light of 365 nm (ε365 = 4102 cm−1 M−1) was first verified (data not shown). Using standard DNA synthesis conditions6 6 has been incorporated at several guanosine positions of the peroxidase deoxyribozyme PS2.M (D1) providing the caged mutants D2–D7 (Table 1).
Table 1.
Synthesized peroxidase deoxyribozymes D1–D7 and their catalytic activity before and after UV irradiation.
| |||
|---|---|---|---|
| Sequence | ν(−UV) | ν(+UV) | |
|
| |||
| D1 | 5’-GTGGGTAGGGCGGGTTGG-3’ | 1.04 ± 0.02 | 1.16 ± 0.06 |
| D2 | 5’-GTGGG*TAGGGCGGGTTGG-3’ | ND | 1.00 ± 0.04 |
| D3 | 5’-GTGGGTAGG*GCGGGTTGG-3’ | ND | 1.18 ± 0.13 |
| D4 | 5’-GTGGGTAGGG*CGGGTTGG-3’ | ND | 0.98 ± 0.12 |
| D5 | 5’-GTGGGTAGGGCG*GGTTGG-3’ | ND | 1.15 ± 0.04 |
| D6 | 5’-GTGGGTAGGG*CG*GGTTGG-3’ | ND | 0.84 ± 0.05 |
| D7 | 5’-GTGGGTAGGGCGGGTTG*G-3’ | ND | 1.11 ± 0.09 |
G* indicates a caged deoxyguanosine.
ν = initial velocity in Abs min−1.
ND = not detectable.
In order to conduct the peroxidase-catalyzed reactions, the oligonucleotide was briefly heated to 90 °C followed by a slow cooling period to induce proper folding.3 Complexation of the hemin cofactor (1 equiv.) was achieved through incubation for 40 min at room temperature. The oxidation reactions were then initiated through the addition of an excess of 2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) followed by H2O2. The reaction progress was monitored by measuring the absorbance of the ABTS radical cation at 414 nm (Fig. 2).3
Fig. 2.
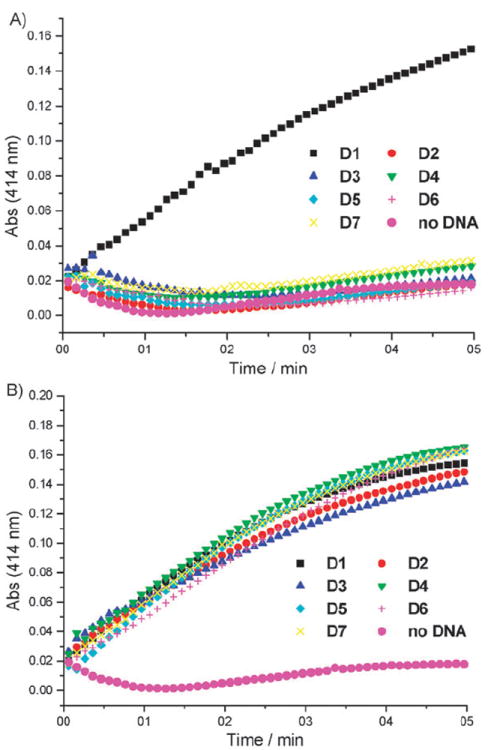
Oxidation of ABTS to the corresponding radical cation by the wild type DNAzyme D1, the caged DNAzymes D2–D7, and just hemin in the absence of any DNA (A) before and (B) after UV irradiation. Average of three experiments.
As expected, the wild type DNAzyme PS2.M (D1) displayed prominent peroxidase activity regardless of UV irradiation. None of the caged DNAzymes D2–D7 exhibited catalytic activity prior to light irradiation, regardless of the position of the caged nucleotide within the DNAzyme sequence demonstrating the ability to stringently regulate DNA function through the installation of a single NPOM caging group. The decaging was achieved by irradiating the DNAzyme solutions for 5 min (365 nm, handheld UV lamp, 23 W) after the heating–cooling period, but prior to the addition of 20 KH buffer.§ After light activation, the catalytic activity of decaged DNAzymes was almost quantitatively restored as determined by calculating reaction velocities from the initial, linear slope (Table 1).
In order to further elucidate why the caging of any of the six dG positions investigated in peroxidase PS2.M inhibited its catalytic activity, we measured the structural organisation of the non-caged oligonucleotide D1 and the caged oligonucleotides D2–D7 by circular dichroism spectroscopy (CD).10,11 Initial CD measurements were taken in the reaction buffer at room temperature in the absence of hemin and showed that all caged DNAzymes had substantially different secondary structures than the non-caged D1 (Fig. 3).
Fig. 3.
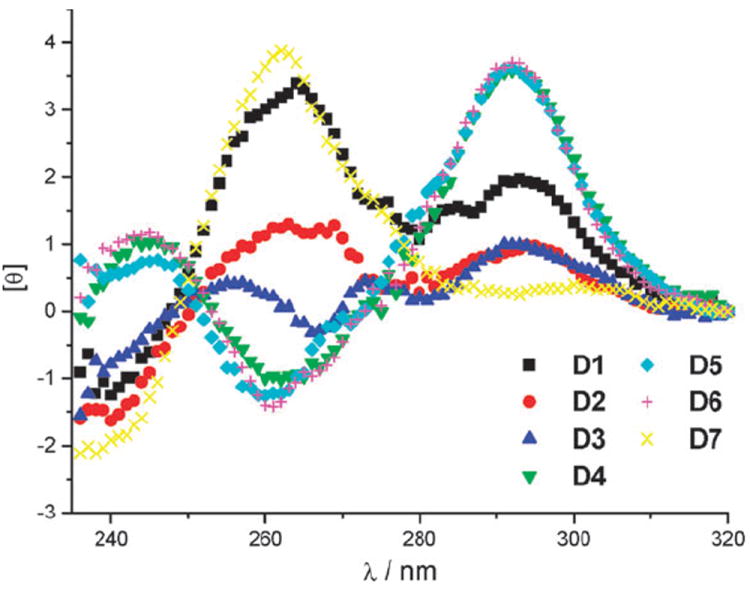
CD spectra of D1 and caged DNAzymes D2–D7 in the absence of hemin.
We were surprised to find that D1 did not exhibit a spectrum consistent with the anti-parallel G-quadruplex structure proposed by Sen et al.4 Based on the CD spectrum shown in Fig. 3, the deoxyoligonucleotide D1 appeared to form a mixture of various secondary structures in solution.10 Thus, we concluded that hemin might play an important role in the formation of a stable quadruplex structure of D1. When CD measurements were taken after DNA incubation with hemin (Fig. 4), the spectrum of D1 showed a significant change, consistent with a parallel rather than an anti-parallel secondary structure, having a large positive peak at 260 nm and a small negative peak at 240 nm.12 On the other hand the caged DNAzymes D2–D7 did not show pronounced changes in their spectra after incubation with hemin, leading to the conclusion that one caging group is sufficient to inhibit G-quadruplex formation and complexation of hemin. However, the CD spectra of caged D4–D6 in the absence (Fig. 3) or presence (Fig. 4) of hemin showed a tendency of the oligonucleotides to form anti-parallel secondary structures, displaying a negative peak at 260 nm and a positive peak at 290 nm. The deoxyoligonucleotide D7 exhibited a (parallel) structure similar to D1, albeit not as pronounced.
Fig. 4.
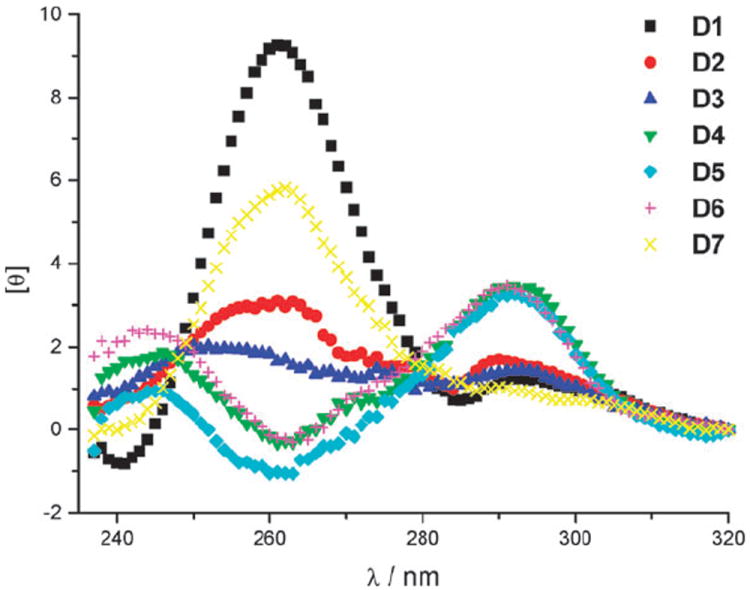
CD spectra of D1 and caged DNAzymes D2–D7 after incubation with hemin.
At first, these observations were in contrast to findings by Heckel et al.,13 who reported that the formation of a three-layer anti-parallel G-quadruplex can only be efficiently inhibited through caging of a deoxyguanosine residue at very select positions which are both located at the core of the sequence and are involved in the formation of the middle layer. In their case, DNA strands caged at the GGG triad closest to the 5′ end, or at an outer layer of the antiparallel G-quadruplex, formed a secondary structure similar to that of the non-caged DNA. However, the findings by Heckel et al. are based on a slightly different oligonucleotide sequence and a different caging strategy, the installation of an ortho-nitrophenylpropyl group on the O-6 position of deoxyguanosine.13
CD spectra of the oligonucleotides D2–D7 after decaging through UV irradiation (5 min, 365 nm, 23 W) followed by incubation with hemin showed that the DNAzymes reform the same secondary structure as observed for D1 (Fig. 5). This is in agreement with the previously observed complete restoration of catalytic activity through irradiation.
Fig. 5.
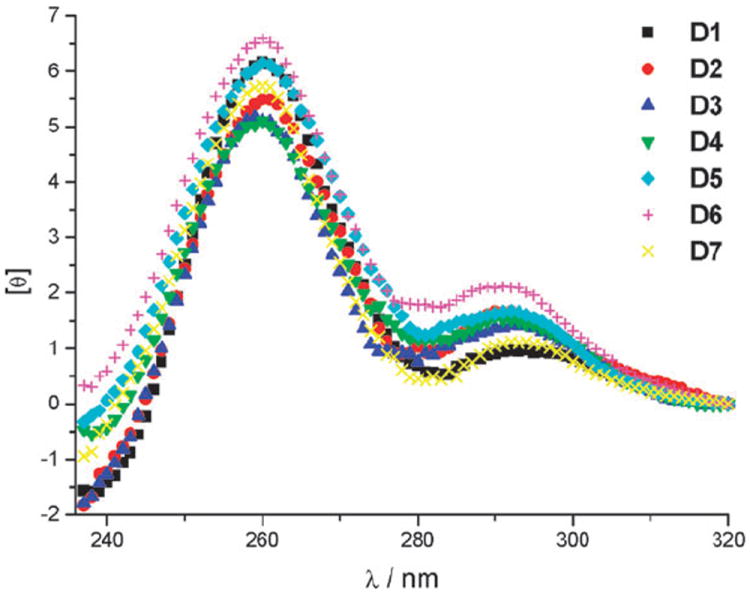
CD spectra of D1 and irradiated (decaged) DNAzymes D2–D7 after incubation with hemin.
The results of the CD measurements led to the conclusion that the initially proposed G-quadruplex structure4,5 was not the predominant D1 secondary structure found under our experimental conditions. Instead of a triple-layer antiparallel G-quadruplex structure, the obtained CD spectra rather are consistent with a parallel G-wire structure proposed by Ohmichi et al. based on CD measurements of a very similar deoxyoligonucleotide sequence (Fig. 6).12 A parallel G-wire structure is also in agreement with our finding that the caged oligonucleotide D2 does not adopt the same secondary structure as D1 even though the caging group is installed on a deoxyguanosine which is not involved in G-quadruplex formation (Fig. 1). Moreover, we conducted dynamic light scattering (DLS) experiments with D1 and D5. These measurements clearly indicate a substantial particle size difference between the two DNA oligomers thus suggesting a highly aggregated species for D1 and a monomeric species for D5 (see Supplementary Information‡). Thus, it can be hypothesized that the active structure of PS2.M (D1) at CD and DLS measurement concentrations (1.0 μM and 5.0 μM, respectively) is a parallel G-wire structure stabilized by hemin intercallation (Fig. 6).
Fig. 6.
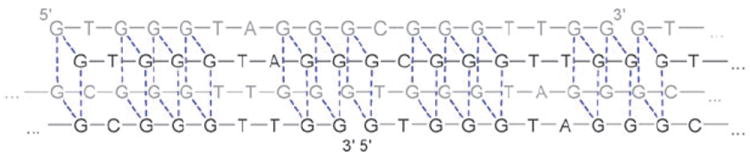
Proposed G-wire structure of the oligonucleotide D1 based on CD and DLS measurements.
Further validation that the caged deoxyribozymes D2–D7 did not bind to hemin came from UV–vis measurements (Fig. 7). Solutions of caged DNAzymes D2, D4 and D7, incubated with hemin, showed no change in the characteristic Soret band (398 nm) as compared to free hemin, while D1 complexed with hemin exhibited a red shift (404 nm) in addition to an increased absorbance.3 Gratifyingly, decaging (5 min, 365 nm, 23 W) enabled hemin intercallation and restoration of the wild-type structure, since the UV–vis spectra displayed the same shift in the Soret band as D1.
Fig. 7.
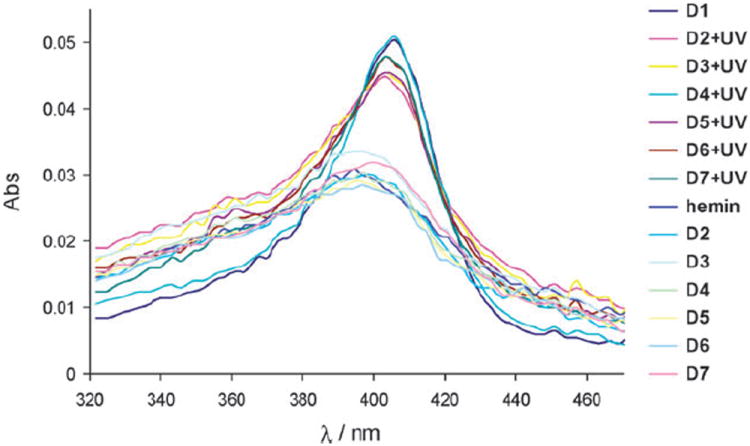
UV–vis spectra of free hemin and hemin incubated with the DNAzymes D1–D7 with and without UV irradiation (5 μM in 20 KH buffer).
In summary, the synthesis of a photocaged deoxyguanosine phosphoramidite using our NPOM caging strategy, and its incorporation into DNA via standard DNA synthesis conditions was accomplished. The developed approach was then applied to the light-triggered activation of a deoxyribozyme with peroxidase activity, and excellent ‘off’ (before irradiation) and ‘on’ (after irradiation) rates were observed. The obtained results demonstrate that in order to disturb the formation of a parallel G-quadruplex (G-wire), the introduction of a single NPOM-caged dG at an arbitrary position within the DNA sequence is sufficient. We believe that the caged guanosine phosphoramidite 6 will find broad application in the photo-chemical regulation of DNA function.
Supplementary Material
Acknowledgments
We thank the Clark lab at NCSU for help with the CD measurements and the Monteiro-Riviere lab for help with the dynamic light scattering experiments. This research was supported in part by research grant no. 5-FY05-1215 from the March of Dimes Birth Defects Foundation. The DNA synthesis was conducted in the Biomolecular Resource Facility of the Comprehensive Cancer Center of Wake Forest University, supported in part by NIH grant P30CA-12197-30. AD is a Beckman Young Investigator and a Cottrell Scholar.
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures and analytical data for the synthesis of the caged deoxyguanosine phosphoramidite; protocols for the measurement of peroxidase activity, CD spectra, UV spectra, and DLS; DNA decaging reactions; copies of 1H NMR spectra for all new compounds.
This article is part of a Molecular BioSystems ‘Emerging Investigators’ issue highlighting the work of outstanding young scientists at the chemical- and systems-biology interfaces.
Irradiation of the DNAzymes (caged or non-caged) in 20 KH (HEPES) buffer led to a significant decrease in catalytic activity. This is probably due to the light sensitivity of HEPES leading to H2O2 formation. The increasead H2O2 concentration thus induces oxidative damage in the DNA and/or oxidation and deactivation of hemin. The same effect was observed when the DNAzymes were diluted in 20 KH buffer that was previously exposed to 365 nm UV light. Consequently, all decagings were conducted in TE buffer.14
References
- 1.(a) Dunford HB. Heme Peroxidases. John Wiley; New York: 1999. [Google Scholar]; (b) Smith AT, Veitch NC. Curr Opin Chem Biol. 1998;2:269–278. doi: 10.1016/s1367-5931(98)80069-0. [DOI] [PubMed] [Google Scholar]
- 2.Yin J, Mills JH, Schultz PG. J Am Chem Soc. 2004;126:3006–3007. doi: 10.1021/ja039198o. [DOI] [PubMed] [Google Scholar]
- 3.Travascio P, Li Y, Sen D. Chem Biol. 1998;5:505–517. doi: 10.1016/s1074-5521(98)90006-0. [DOI] [PubMed] [Google Scholar]
- 4.Lee HW, Chinnapen DJF, Sen D. Pure Appl Chem. 2004;76:1537–1545. [Google Scholar]
- 5.Travascio P, Sen D, Bennet AJ. Can J Chem. 2006;84:613–619. [Google Scholar]
- 6.(a) Young DD, Edwards WF, Lusic H, Lively MO, Deiters A. Chem Commun. 2008:462–464. doi: 10.1039/b715152g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lusic H, Young DD, Lively MO, Deiters A. Org Lett. 2007;9:1903–1906. doi: 10.1021/ol070455u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Heckel A, Buff MC, Raddatz MS, Muller J, Potzsch B, Mayer G. Angew Chem Int Ed. 2006;45:6748–6750. doi: 10.1002/anie.200602346. [DOI] [PubMed] [Google Scholar]; (b) Heckel A, Mayer G. J Am Chem Soc. 2005;127:822–823. doi: 10.1021/ja043285e. [DOI] [PubMed] [Google Scholar]; (c) Krock L, Heckel A. Angew Chem Int Ed. 2005;44:471–473. doi: 10.1002/anie.200461779. [DOI] [PubMed] [Google Scholar]; (d) Tang X, Dmochowski IJ. Org Lett. 2005;7:279–282. doi: 10.1021/ol047729n. [DOI] [PubMed] [Google Scholar]; (e) Tang X, Dmochowski IJ. Angew Chem Int Ed. 2006;45:3523–3526. doi: 10.1002/anie.200600954. [DOI] [PubMed] [Google Scholar]; (f) Tang X, Richards JL, Peritz AE, Dmochowski IJ. Bioorg Med Chem Lett. 2005;15:5303–5306. doi: 10.1016/j.bmcl.2005.08.058. [DOI] [PubMed] [Google Scholar]; (g) Ghosn B, Haselton FR, Gee KR, Monroe WT. Photochem Photobiol. 2005;81:953–959. doi: 10.1562/2004-11-15-RA-373. [DOI] [PubMed] [Google Scholar]; (h) Monroe WT, McQuain MM, Chang MS, Alexander JS, Haselton FR. J Biol Chem. 1999;274:20895–20900. doi: 10.1074/jbc.274.30.20895. [DOI] [PubMed] [Google Scholar]; (i) Ando H, Furuta T, Tsien RY, Okamoto H. Nat Genet. 2001;28:317–325. doi: 10.1038/ng583. [DOI] [PubMed] [Google Scholar]
- 8.Young DD, Deiters A. Bioorg Med Chem Lett. 2006;16:2658–2661. doi: 10.1016/j.bmcl.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 9.Lusic H, Deiters A. Synthesis. 2006:2147–2150. [Google Scholar]
- 10.Balagurumoorthy P, Brahmachari SK, Mohanty D, Bansal M, Sasisekharan V. Nucleic Acids Res. 1992;20:4061–4067. doi: 10.1093/nar/20.15.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakanishi K, Berova N, Woody R. Circular Dichroism: Principles and Applications. 2. Wiley-VCH; New York: 2000. [Google Scholar]
- 12.Sugimoto N, Toda T, Ohmichi T. Chem Commun. 1998:1533–1534. [Google Scholar]
- 13.Mayer G, Krock L, Mikat V, Engeser M, Heckel A. ChemBioChem. 2005;6:1966–1970. doi: 10.1002/cbic.200500198. [DOI] [PubMed] [Google Scholar]
- 14.Lepe-Zuniga JL, Zigler JS, Jr, Gery I. J Immunol Methods. 1987;103:145. doi: 10.1016/0022-1759(87)90253-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.