

Abstract
miR-34a is transcriptionally induced by the tumor suppressor gene p53, which is often downregulated in non-small cell lung cancer (NSCLC). To address whether the downstream signal of miR-34a is sufficient to induce apoptosis and to alter cellular radiosensitivity, a chemical synthetic miR-34a mimic was delivered into A549 and H1299 cells, with or without co-treatment of γ-irradiation. Results showed that ectopic expression of miR-34a induced dose-dependent cell growth inhibition and apoptosis in a p53-independent manner in both NSCLC cell lines. Interestingly, LyGDI was discovered as a new target gene of miR-34a, and downregulation of LyGDI promoted Rac1 activation and membrane translocation, resulting in cell apoptosis. Furthermore, restoration of miR-34a indirectly reduced cyclooxygenase-2 (COX-2) expression. Taken together, these results demonstrate that restoration of miR-34a expression enhances radiation-induced apoptosis, partly by suppressing the LyGDI signaling pathway, and miR-34a could possibly be used as a radiosensitizer for non-small cell lung cancer therapy.
Keywords: miR-34a, non-small cell lung cancer, radiosensitivity, LyGDI, Rac1, COX-2
INTRODUCTION
miRNAs represent an abundant class of 21-nucleotide-long, non-coding RNAs involved in post-transcriptional control of gene expression. In association with the Dicer/Argonaute complex, miRNAs bind to complementary sites in the 3′-untranslated region of target mRNAs and cause translational downregulation and/or degradation of target mRNAs, thereby inhibiting gene expression [1, 2]. The influence of miRNAs on gene expression is predicted to be widespread, with more than 60% of human protein coding genes being subjected to regulation by miRNAs [3]. Among the p53-regulated miRNAs, miR-34a seems to display the most pronounced induction by p53 [4–8]. Ectopic expression of miR-34a induces apoptosis, senescence, cell cycle arrest and inhibition of cellular migration and invasion. Therefore, miR-34a may be an important mediator of the tumor suppressive activities of p53 [9–11].
miR-34a is a member of the miR-34 family, which is composed of miR-34a, miR-34b and miR-34c. The miR-34a gene is located on chromosome 1p36.22 in a region that has previously been associated with various cancers [12]. More recently, the epigenetic inactivation of miR-34a has been identified in many common tumor types (lung, breast, colon, kidney, bladder, pancreatic cancer and melanoma) and also in cell lines derived from those tumors. Besides p53 mutation or functional inhibition of the expression of miR-34a, there is evidence that aberrant CpG methylation of the miR-34a promoter can result in concomitant loss of miR-34a expression [13]. The importance of miR-34a in cancer is now firmly established and restoration of functional miR-34 can inhibit various cancer cell growth and induce apoptosis. For example, ectopic expression of miR-34a inhibits p53-deficient gastric cancer cell growth and induces chemosensitization and apoptosis [14]. Furthermore, chemically synthesized miR-34a was shown to block tumor growth in non-small cell lung cancer (NSCLC) in vivo [15], and over-expression of miR-34a in bulk or purified CD44(+) prostate cancer cells inhibited clonogenic expansion, tumor regeneration and metastasis [16]. Likewise, transient expression of miR-34a in glioma strongly inhibited in vivo glioma xenograft growth [17] and targeted expression of miR-34a sensitized medulloblastoma cells to various classes of chemotherapeutic agents, including mitomycin C and cisplatin [18]. Finally, over-expression of miR-34a conferred resistance in docetaxel-sensitive MCF-7 cells [19]. These results suggest that re-introduction of miR-34a not only inhibits cell growth but also enhances the drug sensitivity of tumor cells under both in vitro and in vivo conditions. While miR-34a may be used as an adjuvant in cancer therapy, the mechanism of restoration of miR-34a expression on radiation sensitivity is not clear.
It is well accepted that wild-type p53 is one of the key factors affecting the radio-sensitivity of cancer cells [20], and the p53-mutated or p53-deficient cancer cells respond poorly to radiation [21–22]. miR-34a is a target of p53, and the repressed regulation of SIRT1 by miR-34a is part of a positive feedback loop to p53. As a result, p53 deacetylation by SIRT1 is decreased and leads to increased transcription of p53 targets, such as PUMA. Together with the downregulation of Bcl-2 and other antiapoptotic proteins [5], miR-34 activation promotes apoptosis. In most cancer cells, the interaction between p53 and miR-34a is disrupted. As a result, the induction of apoptosis is diminished after the DNA damages induced by chemotherapy or radiation [11]. There is evidence that apoptosis induced by the re-introduction of miR-34a is dependent on p53 to some extent [5, 23]. Since SIRT1 is an NAD-dependent deacetylase, which has been shown to inhibit several pro-apoptotic proteins [24], we propose that restoration of miR-34a will promote p53-mediated apoptosis.
A number of mRNAs have been proved to be direct miR-34a targets, which encode factors required for G1/S transition (c-MYC, E2F, CDK4, CDK6), anti-apoptotic proteins (Bcl2, SIRT1), and proteins involved in tumor invasion (c-MET) [5]. However, it is likely that miR-34a may regulate additional yet unconfirmed targets, because networks analyses by bioinformatics suggest that several hundred mRNAs match the miR-34a seed sequence, such as the mRNA of LyGDI. Thus, identifying new targets of miR-34a is a hot topic, which is crucial for elucidating the role and mechanism of miR-34a in cancer biology [25].
So far, there is little understanding of how cellular miR-34a expression affects the response of NSCLC cells to radiation, and ultimately clinical outcome. Since miR-34a targets bcl-2 as well as hundreds of additional genes, it is essential to identify the downstream targets in NSCLC. In this study, we hypothesized that restoration of miR-34a expression in NSCLC cells would enhance their radiosensitivity and improve the clinical outcome of this often-fatal cancer.
MATERIALS AND METHODS
Cell lines, antibodies, miRNA and irradiation
A549 and H1299 lung cancer cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin (100 units/ml), and streptomycin (100 μg/ml). Antibodies specific to human Ly-GDI (c-20), COX-2 (M-19), Rac1 (c-14), Caspase-3 (S17), and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Hsa-miR-34a (miR-34a) mimics and negative control miRNA were obtained from Shanghai Gene Pharma Co. Ltd (China, Shanghai). The sequence for miR-34a mimics is 5′-uggcagugucuuagcugguugu-3′, and the negative control miRNA(NC) sequence is 5′-uucuccgaacgugucacgutt-3′. Cells were treated with 60Co γ-ray irradiation (IR) (Radiation Center of Soochow University, Suzhou, China) at a dose rate of 1 Gy/min.
miR-34a mimic and negative control miRNA(NC) transfection
Both A549 and H1299 cell lines were seeded at 3000/well into 12-well plates and incubated 24 h before transfection. miR-34a mimics and negative control miRNA(NC) in 200 μl of serum-free, antibiotic-free medium were mixed with 5 μl of Lipofectamine 2000 transfection reagent (Invitrogen, China), and was then dissolved in 200 μl of the same medium and allowed to stand at room temperature for 20 min. The resulting 400 μl of transfection solutions were added to each well containing 0.6 ml of medium. After 4 h incubation, the cultures were replaced with 1 ml fresh medium supplemented with 10% FBS and antibiotics. For western blot, cells were seeded at 1 × 105 in 6-well plates or 3 × 105 in 6-cm dishes and collected after an additional 48 h incubation [14].
Cell proliferation
The proliferation of non-small cell lung cancer A549 and H1299 cells were examined by MTT assay. Cells were seeded at 3000/well into 96-well plates and incubated overnight, then transfected with miR-34a mimics (5–30 nM) and negative control miRNA(30 nM), respectively. After 4 h incubation, the cultures were replaced with 0.5 ml fresh medium supplemented with 10% FBS and antibiotics. After cells were incubated for another 48 h, 20 μl of MTT(3-(4,5-dimethylthiazole)-2, 5-diphenyltetrazoliumbromide) labeling reagent was added into each well prior to incubating at 37°C for 4 h. Reaction was stopped by adding 100 μl solubilization solution and incubating the plates at 37°C overnight. The plates were read on a microplate reader (Model 680, Bio-Rad) at a wavelength of 560 nm with a reference wavelength of 655 nm. The percentage cell proliferation for each group was calculated by adjusting the negative control group to 100% [26]. Cell proliferation was calculated using the following equation:
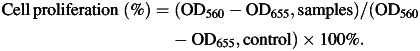 |
Western blot analysis
Cells were harvested and washed twice with cold PBS at 4°C, then were lysed with Laemmli buffer, resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and transferred to Millipore membranes (Millipore, Billerica, MA, USA). After blocking, the membranes were then probed with a primary antibody, followed by a peroxidase-conjugated secondary antibody. Immunoreactive proteins were visualized using ECL Plus reagents (Amersham Biosciences, Little Chalfont, UK) [27].
Ionizing radiation and colony-forming assay
A549 and H1299 cells were seeded at 5000/well into 6-well and transfected with miR-34a mimics (30 nM) or negative control miRNA (30 nM) respectively. After 4 h incubation, cells were irradiated at 0, 2, 4, or 6 Gy with 60Co γ-ray, respectively. Two hours after IR, cells were trypsinized, counted, and seeded in 6-cm dishes with 5 ml medium at appropriate cell numbers (100–500/dish). After a further 12-day incubation, the cell colonies were washed with cold phosphate saline buffer, fixed with 75% methanol, and stained with 0.5% crystal violet. Colonies containing more than 50 cells were counted using a dissecting microscope. The surviving fraction was then calculated as follows: mean number of colonies/(number of cells inoculated × plating efficiency), where plating efficiency is defined as the mean number of colonies/number of cells inoculated for untreated controls. And cell survival curves were generated using GraphPad Prism 5.0 (GraphPad Software Inc., La Jolla, CA) based on the multitarget/single-hit model S = 1 − (1 − e−D/D0)N)(L–Q), and then D0 was calculated [28].
Total RNA extraction and quantitative real-time RT-PCR
Total RNA was extracted from A549 and H1299 cells using the Trizol (Invitrogen) agent. SuperScript One-Step reverse transcription-PCR (RT-PCR) kit (Invitrogen) was used to quantify mRNA levels as per the instructions of the manufacturer. Primers used for human LyGDI were as follows: forward: 5′-ATGGATCCATGACTGAAAAAGCCCCAGA-3′ and reverse : 5′-ATGAATTCTCATTCTGTCCACTCCTTCT-3′. GAPDH was used as an internal control, forward: 5′-TCCCTGAGCTGAACGGGAAG-3′ reverse : 5′-GGAGGAGTGGGTGTCGCTGT-3′. Samples were analyzed with the ABI PRISM 7000 sequence detection system (Applied BioSystems). All PCRs were performed in triplicate. The relative quantitative method was used for the quantitative analysis. The calibrator was the averaged ΔCt from the untreated cells and GAPDH internal control [29].
Immunofluorescence observation and miR-34a mimics transfection
Stably expressed GFP fused LyGDI A549 cells were maintained in G418 culture medium. Cells were transfected with 30 nM miR-34a mimics and 30 nM negative control miRNA. GFP-fused LyGDI cells were observed 48 h later by fluorescence inverted microscope.
Apoptosis analysis
Annexin V staining was performed as the apoptosis assay. A549 cells were seeded in 60-mm culture dishes at 3 × 105 cells per dish, and treated with PBS, 2 Gy 60Co γ-ray IR alone, 30 nM miR-34a transfection and 30 nM miR-34a transfection plus 2 Gy IR. Cells were then harvested by trypsinization 48 h after treatment, and were washed with cold PBS three times, fixed with 75% methanol on ice, stained with annexin V (BioVision, Branch of Shang Hai, China) according to the manufacturer's instructions, and kept in a dark place 1 h before being analyzed by flow cytometry. The percentages of annexin V-positive cells were calculated as the cell apoptotic rate [30].
Membrane and cytosol fractions protein extraction
A549 cells were transfected with 30 nM miR-34a or treated with 5 µM aspirin, γ-ray ionizing radiation and co-treatment, respectively, then the cells were collected after 48 h culture and treated with lysis buffer (20 mM Tris-HCl, pH7.5, 100 mM NaCl, 5 mM EDTA, 2 mM PMSF, 1 × protease inhibitor) at 4°C for 30 min. The samples were centrifuged at 500 × g at 4°C for 10 min, and the pellets were dissolved in lysis buffer plus 0.1% (w/v) Triton X-100 for the membrane fractions. The supernatants were re-centrifuged at 15 000 rpm at 4°C for 20 min, and the supernatants were saved as cytosolic fractions [31].
Drug treatment
A549 cells were seeded at 3 × 105 in 60mm dishes 24 h before drug treatment. The aspirin (ASA) was purchased from Sigma Company (St Louis, MO). Stock solutions of mM ASA were dissolved in DMSO and diluted in culture medium to 5 μM for cell treatment. Cell apoptosis assay was used to determine the single or the combined effect of miR-34a transfection, aspirin treatment and IR. Each treatment had triplicates. The data represented at least three independent experiments.
Statistical analysis
Data are presented as means with 95% confidence intervals of at least three independent experiments, and data are presented as mean ± standard deviation (SD). The Student's t-test was used to compare the means of the different groups. A P-value of <0.05 was considered statistically significant.
RESULTS
Restoration of miR-34a expression inhibited cell growth and enhanced the apoptotic sensitivity in non-small lung cancer cells
Several reports have shown that expression of miR-34a was low in A549 and H1299 NSCLC cells [32], which was also confirmed by us (data not shown). miR-34a mimic and negative control miRNA(NC) were transfected into A549 (p53 + /+) and H1299 (p53–/–) cells to observe the cell proliferation and apoptosis induced by miR-34a over-expression. As expected, both cell lines showed dose-dependent cell growth inhibition after 48 h transfection with 10–30 nM miR-34a (Fig. 1A and B). It is interesting that the ectopic expression of miR-34a-induced cell growth inhibition is irrelevant with p53 status. Since bcl-2 is one of the target genes of miR-34a, we checked the expression level of Bcl-2 and Puma using Western blotting in both cell lines. Our results demonstrated that the expression of antiapoptotic protein Bcl-2 was decreased while the expression of apoptotic Puma was upregulated in both miR-34a mimic transfection cells (Fig. 1B and C). These results suggest that forced expression of miR-34a inhibits the cell proliferation through inducing apoptosis in a p53-independent manner in NSCLC. To test whether miR-34a mimic transfection could sensitize A549 and H1299 cells to IR or not, we transfected 30 nM miR-34a mimic and 30 nM NC into both cell lines which were then treated with γ-IR at 0, 2, 4 and 6 Gy, respectively. Cell survival curves were measured using a colony-forming assay. As shown in Fig. 1E and F, the D0 of A549 cells was 1.502, 1.495 and 1.215 Gy, respectively for radiation alone, NC transfection plus radiation, and miR-34a transfection plus radiation. And the D0 of H1299 cells was 1.609, 1.595 and 1.265 Gy, respectively. This indicates that restoration of miR-34a expression enhances the radiosensitivity of NSCLC cells.
Fig. 1.

Restoration of miR-34a expression inhibited cell growth and enhanced irradiation sensitivity in NSCLC cells. (A) (B) Restoration of miR-34a inhibited the growth of A549 (left) and H1299 (right) cells. (MTT assay). (C) (D) Western blot to detect the restoration of miR-34a induced apoptosis in A549 (left) and H1299 (right) cells. Actin was used as the loading control. (D) (E) Restoration of miR-34a enhanced the irradiation-induced apoptotic sensitivity. The clonogenic forming of A549 (left) and H1299 (right) cells was depicted using cell survival curves.
Restoration of miR-34a expression downregulated LyGDI gene expression
Computational miRNA target prediction suggested that LyGDI was one of the target genes of miR-34a (http://cbio.mskcc.org/cgi-bin/mirnaviewer/mirnaviewer.pl). There is one defined miR-34a target site at 83 to the start of the LyGDI 3' UTR, as shown in Fig. 2A. To test whether miR-34a could suppress the LyGDI expression, we introduced negative control miRNA(NC) and miR-34a mimics into both A549 and H1299 cells and then measured the LyGDI expression. Significant inhibition of LyGDI expression was found in both cell lines by real-time RT-PCR (Fig. 2B and C) and by western blot (Fig. 2D and E). Previously we transfected GFP-fused LyGDI using EGFP plasmids into A549 cells and got a stable cell line. Once the miR-34a mimic was introduced into those cells, the green GFP fluorescence gradually disappeared (Fig. 2F), indicating that LyGDI is a target gene of miR-34a.
Fig. 2.

Restoration of miR-34a expression downregulated LyGDI gene expression. (A) Target sequence of miR-34a in LyGDI 3′UTR was predicted by TargetScan. (B) (C) The expression of LyGDI mRNA in miR-34a mimics (30 nM) and NC (negative control miRNA) transfected A549 cells and H1299 cells was measured by real-time RT-PCR. The relative LyGDI expression levels were normalized against GAPDH and presented as mean ± SD from triplicate experiments. (D) (E) The protein levels of LyGDI were also examined at 48 h by Western blot in 30 nM of NC or miR-34a mimics transfected A549 (left) and H1299 (right) cells, respectively. (F) GFP-fused LyGDI A549 cells were observed after transfection with 30 nM of NC or miR-34a mimics 48 h later by fluorescence inverted microscope.
Downregulation of LyGDI expression by miR-34a promoted Rac1 activation and membrane distribution
LyGDI (also called RhoGDI2,D4-GDI, RhoGDIβ) is one of the key regulators of RhoGTPases. It mainly binds with Rac1, Rac3 and CDC42. Normally, LyGDI maintains Rho family GTPases in an inactive state in the cytosol and shuttles them between cytosol and membrane [27]. Zhang et al. reported that silencing of LyGDI by siRNA interference abrogated tumor growth and lung metastasis of otherwise highly invasive MDA-MB-231 breast cancer cells. LyGDI-depleted cells underwent rapid apoptosis (anoikis), which was known to hinder metastasis [33].
Next we sought to investigate whether or not downregulation of LyGDI by miR-34a would affect cell apoptosis and its interaction with Rac1. We introduced the miR-34a mimic into A549 cells and/or treated with IR. After 48 h proteins form, and membrane and cytosolic parts were collected. No Rac1 expression change was found in cytosolic parts after miR-34a mimic transfection and/or 2 Gy IR treatment. However, active Rac1 was found in the membrane fraction in miR-34a transfection alone, and miR-34a plus IR samples with downregulated LyGDI and activated Caspase-3 (Fig. 3A). Apoptosis also increased in those two samples (Fig. 3B). These data suggests that the apoptosis induced by miR-34a- mediated down-regulation of LyGDI may be related to Rac1 membrane distribution and activation.
Fig. 3.

Downregulation of LyGDI expression by miR-34a-promoted Rac1 activation and membrane distribution. (A) The protein expression of LyGDI, Rac1, active Rac1 and Caspase-3 in A549 cells was resolved with western blot after treatment with PBS, 2 Gy IR alone, 30 nM miR-34a transfection and 30 nM miR-34a transfection plus 2 Gy IR after 48 h. β-actin was used as the loading control. (B) Apoptosis as a percentage of A549 cells was measured using annexin V staining analyzed by flow cytometry.
Downregulation of LyGDI expression by miR-34a suppressed COX-2 expression and enhanced IR-induced apoptosis
The role of COX-2 (Cyclooxygenase-2) in carcinogenesis has been recently described [34]. It could regulate Ras, Akt as well as EGFR signal pathways, and affect cells growth, cell adhesion, migration, cell invasion, cell radiation sensitivity and so on. In our unpublished data, upregulated LyGDI promoted NSCLC cell migration through regulating COX-2 expression. Others also reported that LyGDI cooperated with Vav-1 to induce nuclear translocation of specific NFAT-1 forms, so that LyGDI upregulation activates COX-2 gene transcription in breast MDA-MB-231 cancer cells. COX-2 is thought to be one of the targeting genes of LyGDI [35]. To address whether ectopic expression of miR-34a would indirectly affect COX-2 expression and change the radiation sensitivity, A549 cells were transfected with miR-34a mimics (30 nM) alone, 2 Gy IR, 5 µM aspirin (ASA, a COX-2 inhibitor) treatment, and co-treatment with miR-34a transfection, 2 Gy IR as well as ASA, respectively. As shown in Fig. 4A, corresponding to LyGDI downregulated expression, COX-2 expression was reduced in miR-34a mimics transfection alone, ASA treatment as well as co-treatment, respectively. Moreover, the apoptosis assay demonstrated that cell death increased dramatically in those treatments (Fig. 4B), indicating that downregulated LyGDI expression by miR-34a could suppress COX-2 expression and enhance IR-induced apoptosis in NSCLC cells.
Fig. 4.

Downregulation of LyGDI expression by miR-34a-suppressed COX-2 expression and enhanced radiation-induced apoptosis. (A) The protein expression of LyGDI and COX-2 were resolved by western blot after treatment with PBS, 2 Gy IR alone, 30 nM miR-34a transfection, 5 μM aspirin (ASA) treatment and co-treatment as indicated after 48 h. β-actin was the loading control. (B) Apoptosis as a percentage of A549 cells was measured using annexin V staining analyzed by flow cytometry.
DISCUSSION
miR-34a is transcriptionally induced by the tumor suppressor gene p53, which is often downregulated in various human cancer types, including lung cancer. Also, low miR-34a expression level correlates with a high probability of relapse in NSCLC patients [36]. Overexpression of miR-34a induces apoptosis, senescence and cell cycle arrest, and inhibits migration and invasion. Thus, miR-34a displays an antiproliferative phenotype in numerous cancer cell types. We found that ectopic expression of miR-34a inhibited cell growth and induced apoptosis in NSCLC, which was independent of p53 status, suggesting that downstream of the miR-34a pathway signals were sufficient to induce apoptosis and block NSCLC cell growth. Moreover, miR-34a enhances radiation sensitivity in NSCLC cells. Ours results indicate that miR-34a might be used as a sensitizer for NSCLC radiotherapy.
Till now, many miR-34a target genes have been identified [25], including those involved in G1 arrest (E2F3, cyclinE2, CDK4, CDK6, C-Myc, N-Myc), apoptosis (bcl-2, Survivin), p53 activity (Sirt1, MTA2, YY1), metastasis (AXL, c-Met), WNT signaling (Wnt1, LEF1) and glycolysis (LDHA). These data suggest that miR-34a might be involved in many cell physiological functions. We predicated and further confirmed that LyGDI is a new miR-34a target gene. There were multiple lines of evidence showing that LyGDI expression was related to tumor invasion and anti-radiation. LyGDI was found upregulated in highly invasive breast cancer cells and gastric tumors. Our previous data also showed that LyGDI was upregulated in clinical samples of NSCLC patients, and its expression was related to metastasis and anti-radiation-induced apoptosis [29, 37–39]. In addition, the nuclear translocation of truncated LyGDI, or downregulation of LyGDI, enhanced the apoptotic process [40, 41]. Disruption of LyGDI using small interfering RNA (siRNA) effectively blocked the motility and invasive of potential tumor cells in vitro and in vivo. Furthermore, silencing of LyGDI resulted in constitutive Rac1 activation and translocation from cytosol to cellular membrane compartments and in sustained activation of p38 and JNK kinases [33]. Consistent with those investigators, we found that ectopic expression of miR-34a downregulated LyGDI expression, and thus promoted Rac1 activation and its membrane translocation. This is a new signal pathway regulated by miR-34a.
Moreover, COX-2, a major regulator gene of invasion and metastasis in breast cancer cells, had also been identified as a target gene of LyGDI [35]. Our previous data confirmed that LyGDI increased the expression level of COX-2 in A549 lung cancer cells. In this study, we did not find any evidence that miR-34a could directly regulate the expression of COX-2 (data not shown), but restoration of miR-34a downregulated both LyGDI and COX-2 protein expression. Downregulation of COX-2 expression by Cyclooxygenase inhibitors showed a radiation-enhancing effect on COX-2-positive human cancer cells, indicating that COX-2 is a radiosensitive-related gene [42], but the mechanism was unclear. The role of Cox-2 in cell signal pathways such as the cell cycle checkpoint pathway, may need to be investigated.
mi-34a is a DNA damage-responsive gene, its expression is also affected by IR. Kato M et al. show that Caenorhabditis elegans with loss-of-function mutations in the mir-34 gene has an abnormal cellular survival response to radiation, those animals are highly radiosensitive in the soma and radioresistant in the germline. Their findings show a role for miR-34a in both apoptotic and non-apoptotic cell death in vivo. Further it has been confirmed in vitro in breast cancer cells. They imply that anti-miR-34 is a radiosensitizing agent in p53-mutant breast cancer, although the mechanism is not clear as yet. The author stated that the ‘impact of miR-34 on cell survival largely depends on the cell's innate mode of cell death post-irradiation’ [43]. On the contrary, Ji et al. found that miR-34 restoration significantly inhibited pancreatic cancer cell (MiaPaCa2 and BxPC3) clonogenic cell growth and invasion, induced apoptosis and G1 and G2/M arrest in cell cycle, and sensitized the cells to chemotherapy and radiation. It may hold significant promise as a novel molecular therapy for human pancreatic cancer with loss of p53-miR34 [44]. Others reported that low expression of miR-34a in chronic lymphocytic leukemia (CLL) was associated with p53 inactivation but also chemotherapy-refractory disease, impaired DNA damage response, and apoptosis-resistance irrespective of 17p deletion/TP53 mutation [45]. Those results suggest the role of miR-34a is still controversial. We have shown that the response of cells to IR depends on the IR dose and miR-34a expression level. Like P53, low expression goes on transcriptional function to repair DNA damage after post-irradiation, however, high expression will induce apoptosis directly. Elucidation of the mechanisms underlying miR-34a regulation and its role in radiosensitivity need further study.
CONCLUSION
In conclusion, our study showed that restoration of miR-34a expression inhibited cell growth and induced apoptosis in NSCLC cells, which was independent of p53 status. Moreover, LyGDI was discovered as a new target gene of miR-34a. Downregulation of LyGDI expression promoted Rac1 activation and membrane translocation leading to cell apoptosis. Furthermore, restoration of miR-34a indirectly reduced COX-2 expression. Altogether, restoring expression of miR-34a enhanced IR-induced apoptosis, indicating that miR-34a might be used as a sensitizer for NSCLC radiotherapy.
FUNDING
This research work was supported by the Nature Science Foundation of China (Project No. 81071878), a Key International Cooperation Project (No. 81020108028), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
REFERENCES
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 3.Friedman RC, Farh KK, Burge CB, et al. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bommer GT, Gerin I, Feng Y, K , et al. p53–mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 5.Chang TC, Wentzel EA, Kent OA, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–52. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He L, He X, Lim LP, et al. A microRNA component of the 53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raver-Shapira N, Marciano E, Meiri E, et al. Transcriptional activation of miR-34a contributes to pp53–mediated apoptosis. Mol Cell. 2007;26:731–43. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 8.Tarasov V, Jung P, Verdoodt B, et al. Differential regulation of microRNAs by pp53 revealed by massively parallel sequencing: miR-34a is a pp53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–93. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- 9.Hermeking H. Cancer Cell. 2007;12:414–8. doi: 10.1016/j.ccr.2007.10.028. pp53 enters the microRNA world. [DOI] [PubMed] [Google Scholar]
- 10.He X, He L, Hannon GJ. The guardian's little helper: microRNAs in the pp53 tumor suppressor network. Cancer Res. 2007;67:11099–101. doi: 10.1158/0008-5472.CAN-07-2672. [DOI] [PubMed] [Google Scholar]
- 11.Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193–9. doi: 10.1038/cdd.2009.56. [DOI] [PubMed] [Google Scholar]
- 12.Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lodygin D, Tarasov V, Epanchintsev A, et al. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7:2591–600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- 14.Ji Q, Hao X, Meng Y, et al. Restoration of tumor suppressor miR-34 inhibits human pp53–mutant gastric cancer tumorspheres. BMC Cancer. 2008;8:266. doi: 10.1186/1471-2407-8-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiggins JF, Ruffino L, Kelnar K, et al. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010;70:5923–30. doi: 10.1158/0008-5472.CAN-10-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu C, Kelnar K, Liu B, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211–5. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Guessous F, Zhang Y, et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009;69:7569–76. doi: 10.1158/0008-5472.CAN-09-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weeraratne SD, Amani V, Neiss A, et al. miR-34a confers chemosensitivity through modulation of MAGE-A and pp53 in medulloblastoma. Neuro Oncol. 2011;13:165–75. doi: 10.1093/neuonc/noq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kastl L, Brown I, Schofield AC. miRNA-34a is associated with docetaxel resistance in human breast cancer cells. Breast Cancer Res Treat. 2012;131:445–54. doi: 10.1007/s10549-011-1424-3. [DOI] [PubMed] [Google Scholar]
- 20.El-Deiry WS. The role of p53 in chemosensitivity and radiosensitivity. Oncogene. 2003;22:7486–95. doi: 10.1038/sj.onc.1206949. [DOI] [PubMed] [Google Scholar]
- 21.Maebayashi K, Mitsuhashi N, Takahashi T, et al. p53 mutation decreased radiosensitivity in rat yolk sac tumor cell lines. Int J Radiat Oncol Biol Phys. 1999;44:677–82. doi: 10.1016/s0360-3016(99)00025-5. [DOI] [PubMed] [Google Scholar]
- 22.Shin HJ, Kim JY, Hampson L, et al. Human papillomavirus 16 E6 increases the radiosensitivity of 53–mutated cervical cancer cells, associated with up-regulation of aurora A. Int J Radiat Biol. 2010;86:769–79. doi: 10.3109/09553002.2010.484477. [DOI] [PubMed] [Google Scholar]
- 23.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. 2008;105:13421–6. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujita Y, Kojima K, Hamada N, et al. Effects of miR-34a on cell growth and chemoresistance in prostate cancer PC3 cells. Biochem Biophys Res Commun. 2008;377:114–9. doi: 10.1016/j.bbrc.2008.09.086. [DOI] [PubMed] [Google Scholar]
- 25.Kaller M, Liffers ST, Oeljeklaus S, et al. Genome-wide characterization of miR-34a induced changes in protein and mRNA expression by a combined pulsed SILAC and microarray analysis. Mol Cell Proteomics. 2011;10:1–16. doi: 10.1074/mcp.M111.010462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arora H, Qureshi R, Jin S, et al. miR-9 and let-7g enhance the sensitivity to ionizing radiation by suppression of NFkB1. Exp Mol Med. 2011;43:298–304. doi: 10.3858/emm.2011.43.5.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou X, Suto S, Ota T, et al. Nuclear translocation of cleaved LyGDI dissociated from rho and rac during p53–dependent ionizing radiation-induced thymic apoptosis in vitro. Radiat Res. 2004;162:287–95. doi: 10.1667/rr3220. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Yang H, Gu K, et al. In vitro and in vivo study of a nanoliposomal cisplatin as a radiosensitizer. Int J Nanomedicine. 2011;6:437–44. doi: 10.2147/IJN.S15997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Zhang B. D4-GDI, a Rho GTPase regulator, promotes breast cancer cell invasiveness. Cancer Res. 2006;66:5592–8. doi: 10.1158/0008-5472.CAN-05-4004. [DOI] [PubMed] [Google Scholar]
- 30.He C, Xiong J, Xu X, et al. Functional elucidation of MiR-34 in osteosarcoma cells and primary tumor samples. Biochem Biophys Res Commun. 2009;388:35–40. doi: 10.1016/j.bbrc.2009.07.101. [DOI] [PubMed] [Google Scholar]
- 31.Huang CY, Yang LC, Liu KY, et al. ZAK negatively regulates RhoGDIbeta-induced Rac1-mediated hypertrophic growth and cell migration. J Biomed Sci. 2009;16:56. doi: 10.1186/1423-0127-16-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bandi N, Vassella E. miR-34a, miR-15a/16 are co-regulated in non-small cell lung cancer and control cell cycle progression in a synergistic and Rb-dependent manner. Mol Cancer. 2011;10:55. doi: 10.1186/1476-4598-10-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Rivera Rosado LA, Moon SY, et al. Silencing of D4-GDI inhibits growth and invasive behavior in MDA-MB-231 cells by activation of Rac-dependent p38 and JNK signaling. J Biol Chem. 2009;284:12956–65. doi: 10.1074/jbc.M807845200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghosh N, Chaki R, Mandal V, et al. COX-2 as a target for cancer chemotherapy. Pharmacol Rep. 2010;62:233–44. doi: 10.1016/s1734-1140(10)70262-0. [DOI] [PubMed] [Google Scholar]
- 35.Schunke D, Span P, Ronneburg H, et al. Cyclooxygenase-2 is a target gene of rho GDP dissociation inhibitor beta in breast cancer cells. Cancer Res. 2007;67:10694–702. doi: 10.1158/0008-5472.CAN-07-1621. [DOI] [PubMed] [Google Scholar]
- 36.Gallardo E, Navarro A, Viñolas N, et al. miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis. 2009;30:1903–9. doi: 10.1093/carcin/bgp219. [DOI] [PubMed] [Google Scholar]
- 37.Cho HJ, Baek KE, Park SM, et al. RhoGDI2 expression is associated with tumor growth and malignant progression of gastric cancer. Clin Cancer Res. 2009;15:2612–9. doi: 10.1158/1078-0432.CCR-08-2192. [DOI] [PubMed] [Google Scholar]
- 38.Moon HG, Jeong SH, Ju YT, et al. Up-regulation of RhoGDI2 in human breast cancer and its prognostic implications. Cancer Res Treat. 2010;42:151–6. doi: 10.4143/crt.2010.42.3.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao Li-li, Guang-xin duan, Dong Yu-jin, et al. Gene transfection of LyGDI into A549 cell line and its effect on cell growth and sensitivity to chemotherapy and radiotherapy. Journal of Soochow University (Medical Science Edition, Chinese) 2011;31:870–3. [Google Scholar]
- 40.MR Choi, M Groot, HC Drexler. Functional implications of caspase-mediated RhoGDI2 processing during apoptosis of HL60 and K562 leukemia cells. Apoptosis. 2007;12:2025–35. doi: 10.1007/s10495-007-0121-5. [DOI] [PubMed] [Google Scholar]
- 41.Zhou X, Xu YX. LyGDI expression in HeLa cell increased its radio-sensitivity during irradiation induced apoptosis. China J Radio Med Prot. 2006;26(462–4) (in Chinese) [Google Scholar]
- 42.You Keun Shin, Ji Sun Park, Hyun Seok Kim, et al. Radiosensitivity enhancement by celecoxib, a cyclooxygenase (COX)-2 selective inhibitor, via COX-2-dependent cell cycle regulation on human cancer cells expressing differential COX-2 levels. Cancer Res. 2005;65:9501–9. doi: 10.1158/0008-5472.CAN-05-0220. [DOI] [PubMed] [Google Scholar]
- 43.Kato M, Paranjape T, Muller RU, et al. The mir-34 microRNA is required for the DNA damage response in vivo in C.elegans and in vitro in human breast cancer cells. Oncogene. 2009;28:2419–24. doi: 10.1038/onc.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ji Q, Hao X, Zhang M, et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One. 2009;4:e6816. doi: 10.1371/journal.pone.0006816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zenz T, Mohr J, Eldering E, et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood. 2009;113:3801–8. doi: 10.1182/blood-2008-08-172254. [DOI] [PubMed] [Google Scholar]