

Abstract
Ionizing radiation (IR) causes not only acute tissue damage but also residual bone marrow (BM) suppression. The induction of residual BM injury is primarily attributable to the induction of reactive oxygen species (ROS) pressure in hematopoietic cells. In this study, we examined if SB431542, a transforming growth factor β1 (TGFβ1) inhibitor, can mitigate IR-induced BM suppression in vitro. Our results showed that treatment with SB431542 protected mice bone marrow mononuclear cells (BMMNCs), hematopoietic progenitor cells (HPCs) and hematopoietic stem cells (HSCs) from IR-induced suppression using cell viability assays, clonogenic assays and competitive repopulation assays. Moreover, expression of gene-related ROS production in hematopoietic cells was analyzed. The expression of NOX1, NOX2 and NOX4 was increased in irradiated BMMNCs, and that of NOX2 and NOX4 was reduced by SB431542 treatment. Therefore, the results from this study suggest that SB431542, a TGFβ1 inhibitor, alleviates IR-induced BM suppression at least in part via inhibiting IR-induced NOX2 and NOX4 expression.
Keywords: TGFβ1, ionizing radiation, bone marrow suppression, NADPH oxidase
INTRODUCTION
Bone marrow (BM) suppression is the most common dose-limiting side-effect of conventional cancer therapy using ionizing radiation (IR) [1, 2] and is the primary cause of death after accidental exposure to a high dose of IR. The myelosuppression not only worsens the outcome of cancer treatment but also adversely affects the quality of life of cancer patients. However, the mechanisms through which ionizing radiation (IR) induces BM injury are not well understood, nor has an effective treatment been developed to mitigate the injury.
Transforming growth factor β1 (TGFβ1) is a pleiotropic cytokine involved in many hematopoietic cells activities, including growth, differentiation, quiescence, apoptosis and mobilization [3, 4]. TGFβ1 is secreted as a latent precursor molecule (LTGFβ) bound to LTGFβ-binding protein, LTBP [5]. LTGFβ can be activated by IR in the extracellular space of irradiated tissue as well as proteolytic processes [6, 7]. Activated TGF-β1 can activate TGF-β type I and type II receptors, which are responsible for activating intracellular mediators such as Smad proteins and p38 mitogen-activated protein kinase (MAPK) [8, 9].
Previous work has shown that IR-induced oxidative stress in hematopoietic cells via the NADPH oxidase (NOX) pathway at least partly contributes to IR-induced BM failure [10]. Mitochondria have been widely accepted to be the main source of cellular-derived reactive oxygen species (ROS). However, recent studies demonstrate that cells can also produce ROS deliberately through a family of tightly regulated NOXs that are homologues of the phagocyte oxidase [11, 12]. In lung fibroblasts, TGFβ-induced NADH oxidase production of hydrogen peroxide begins 8 h after TGFβ stimulation with peak activity at 16 h [13]. In fetal rat hepatocytes, it is demonstrated that TGFβ-induced ROS occurred 4 h after TGFβ treatment [14]. In addition, it was proved TGFβ also plays core roles in up-regulation of NOXs in hepatocytes, kidney myofibroblasts, aortic smooth muscle cells, etc. at least in vitro [15–18].
The aim of the present work was to analyze whether IR-induced BM suppression could be ameliorated by inhibiting TGFβ1, and to study the role of NOXs in this.
MATERIALS AND METHODS
Reagents
Anti-Mouse-CD45.1-percp-cy5.5 (clone A20, Ly5.1), Anti-Mouse-CD45.2-PE (clone 104, Ly5.2), biotin-conjugated anti-Mouse-CD4 (clone GK1.5), anti-Mouse-CD45R/B220 (clone RA3-6B2), anti-Mouse-Ly6G/Gr-1 (clone RB6-8C5), anti-Mouse-CD11b (clone M1/70) and APC-Cy7-conjugated streptavidin were obtained from eBioscience (San Diego, CA, USA). SB431542 was obtained from Sigma (St. Louis, MO, USA).
Mice
Male C57BL/6 mice were purchased from the Institute of Laboratory Animal Sciences (PUMC, Beijing, China) and housed five to a cage at an Animal Care-certified animal facility in the Institute of Radiation Medicine, PUMC. They received food and water ad libitum. All of the mice were used at approximately 8–10 weeks of age. The Institutional Animal Care and Use Committee of PUMC approved all experimental procedures used in this study.
Treatment of BMMNCs with IR and SB431542
The femora and tibiae were harvested from the mice immediately after they were euthanized with CO2. BMMNCs, isolated as described previously [1, 2] and incubated (1 × 106/ml in complete medium) with SB431542 (1 × 10−3 M–1 × 10−9 M) or with 0.2% dimethylsulfoxide (DMSO) (vehicle control) at 37°C for 60 min. Then, cells were exposed to 0, 1, 2 or 4 Gy irradiation generated in an Exposure Instrument Cammacell-40 137Cesium-irradiator (Atomic Energy, Lin, CA, USA) at a rate of 0.76 Gy/min. Cells were incubated at 37°C, with 5% CO2 and 100% humidity for various times as indicated in individual experiments.
Cell viability assays
The cells (1 × 105 cells/well in 100 µl medium) were plated in a 96-well plate and were cultured for 18 h. Cell viability was monitored using the luminescent-based CellTiter Glo TM (Promega, Madison, WI, USA) according to the manufacturer's recommended protocols [19]. Luminescence of each well was read using the Infinite M200 multimode microplate reader (TECAN, Switzerland). Cell viability was normalized as a percentage of the untreated cells. Each experiment was done in triplicate and the results were averaged.
Clonogenic assays
CFU-GM was analyzed using MethoCult M3534 medium (Stem Cell Technologies, Vancouver, BC, Canada). BM-MNCs incubated with SB431542 and irradiated as described above were suspended in Methocult M3534 medium at 2 × 104 or 1 × 105 viable cells/ml and seeded in wells of 24-well plates. The plates were incubated for 7 days. Colonies of ≥50 cells were scored under an inverted microscope on Day 7, and results were expressed as the number of CFU-GM per 105 cells.
Competitive repopulation assay (CRA)
Competitive repopulation assays were performed using the Ly5 congenic mouse system as described previously [10, 20]. After incubation with SB431542 (1 μM) or exposure to irradiation (2 Gy) as described above, donor cells (C57BL/6-Ly-5.1 mice, 1 × 105 BMMNCs) were mixed with 1 × 105 competitive BM-MNCs pooled from three C57BL/6-Ly5.1/Ly5.2 hybrid mice. Cells were transplanted into lethally irradiated (9.0 Gy IR) C57BL/6-Ly-5.2 mice (seven recipients/group) by lateral canthus-vein injection. For analysis of engraftment, peripheral blood was obtained from the medial canthus using heparin-coated micropipettes (Drummond Scientific, Broomall, PA, USA) at 2 months after transplantation from all the recipients. After red blood cells had been lysed with 0.15 M NH4Cl solution, the blood samples were stained with FITC-conjugated anti-CD45.1, PE-conjugated anti-CD45.2, PerCP-conjugated anti-B220, APC-conjugated anti-CD3, PE/Cy7-conjugated Anti-Gr-1 and CD11b, and were analyzed with a LSR II flow cytometer (BD Bioscience, San Jose, CA, USA).
Quantification Real-time PCR assays
BM-MNCs were incubated with SB431542 or exposed to irradiation (2 Gy) as described above and cells were incubated for 4 or 24 h. Total RNA was extracted from the BMMNCs using TRIzol reagent (Applied Biosystems, Grand Island, NY, USA) following the manufacturer's protocol. First-strand cDNA was synthesized from total RNA using RNA PCR Kit (AWV) Ver. 3.0 (Takara Bio Inc., Shiga, Japan) according to the manufacturer's protocol. PCR primers for the TβRII, NOX1, NOX2, NOX4 and the housekeeping gene GAPDH were obtained from Sangon Biotech (Shanghai, China). The sequences of primers used in this study were: TβRII, 5'-TAC TCT GGA GAC GGT TTG-3' (forward) and 5'-TGC TGG TGG TGT ATT CTT-3' (reverse); NOX1, 5'-TCG ACA CAC AGG AAT CAG GA-3' (forward) and 5'-TTA CAC GAG AGA AAT TCT TGG G-3' (reverse); NOX2, 5'-TGC AGT GCT ATC ATC CAA GC-3' (forward) and 5'-CTT TCT CAG GGG TTC CAG TG-3' (reverse); NOX4, 5'-GAT TTC TGG ACC TTT GTG CCT TT-3' (forward) and 5'-TGA TGG TGA CAG GTT TGT TGC T-3' (reverse); GAPDH, 5'-TGA AGG TCG GTG TGA ACG GAT TTG GC-3' (forward) and 5'-CAT GTA GGC CAT GAG GTC CAC CAC-3' (reverse). cDNA samples were mixed with primers and SYBR Master Mix (ABI Bioscience Inc.) in a total volume of 25 μl. All samples were analyzed in triplicate using an ABI Prism 7500 Sequence Detection System (Applied Biosystems-Life Technologies). Thermal cycling conditions were 2 min at 50°C and 10 min at 95°C followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. The threshold cycle (CT) values for each reaction were determined and averaged using TaqMan SDS analysis software (Applied Biosystems-Life Technologies). The changes in target genes expression were calculated using the comparative CT method (fold changes =2[-ΔΔCT]) as described previously [21].
Western blot assays
BM-MNCs were incubated with SB431542 or exposed to irradiation (2 Gy) as described above and cells were incubated for 24 h. Harvested cells were lysed in M-PER mammalian protein extraction reagent (Thermo Scientific, Rockford, IL, USA). The protein concentration was estimated using the bicinchoninic acid protein assay kit (Beyotime Institute of Biotechnology, Jiangsu, China). Cell lysates (50 μg) were loaded onto 5–10% gradient polyacrylamide gels. Proteins were electroblotted onto polyvinylidene difluoride membranes (Millipore, MA, USA) and immunolabeled using 1:400 dilutions of antibodies against TβRII, NOX2, NOX4 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and 1:2000 dilutions of antibodies against β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The enhanced chemiluminescence plus reagent (Boster Biotechnology Co., Wuhan, China) was used for chemiluminescent signal detection. Quantitative analysis was carried out using Quantity One version 4.6.2 (http://www.bio-rad.com).
Statistical analysis
The data were analyzed by analysis of variance (ANOVA). In the event that ANOVA justified post hoc comparisons between group means, these were conducted using the Student–Newman–Keuls test for multiple comparisons. For experiments in which only single experimental and control groups were used, group differences were examined using the unpaired Student's t test. Differences were considered significant at P < 0.05. All of these analyses were done using SPSS 16.0 software (SPSS Inc.).
RESULTS
SB431542 protected BMMNCs from irradiation injury in vitro
Luminescence assays were performed to evaluated cell viability which manifested drugs' radiation protective effect on BMMNCs. Fig. 1A depicts the SB431542 toxicity, in which SB431542 at concentrations higher than 10 μM inhibited the bone marrow cell viability. Therefore, SB431542 at concentrations lower than 10 μM was chosen for the subsequent experiments. As shown in Fig. 1B and C, compared with the sham irradiation group, BMMNC's viabilities decreased significantly after irradiation exposure (1 Gy for 30.28%, P < 0.01; 4 Gy for 51.94%, P < 0.01). Treatment with SB431542 (1 µM) increased cell viabilities of irradiated BMMNCs (1 Gy for 13.25%, P < 0.01; 4 Gy for 20.81%, P < 0.01). SB431542 treatment displays a protective effect on irradiated BMMNCs at 4 Gy better than at 1 Gy.
Fig. 1.

SB431542 reduced IR-induced suppression of BMMNC viability. The cells were sham-irradiated as a control or sublethally irradiated with 1–4 Gy IR after receiving vehicle or SB431542 treatment and were cultured for 18 h. Cell viability was monitored as described in the text. (A) Cell toxicity assays; (B) cells treated with 1 Gy irradiation; (C) cells treated with 4 Gy irradiation. Date were expressed as relative viability as mean ± SE (n = 6).
SB431542 raised the ability of forming colonies of CFU-GM
Clonogenic assays were performed to evaluate viability of HPCs affected by IR and SB431542. As shown in Fig. 2, SB431542 was capable of increasing CFU-GM in sham-irradiated cells, whereas the cells treated with different dose of irradiation (1–4 Gy) exhibited a diminished ability to form CFU-GM by 42.1-89.5% (P < 0.01) and treatment with SB431542 (10 µM–0.1 µM) enhanced the ability to form CFU-GM by 60.0–292.4% (P < 0.01) significantly. These results suggest SB431542 could ameliorate IR-induced mice HPC injury.
Fig. 2.

SB431542 reduces IR-induced suppression of HPC clonogenic function. Mice BMMNCs were sham-irradiated as a control or sublethally irradiated with 1–4 Gy IR after receiving vehicle or SB431542 treatment. Clonogenic function of HPCs in BM-MNCs was analyzed by CFC assay. Colonies of ≥50 cells were scored under an inverted microscope on Day 7, and results were expressed as the number of CFU-GM per 105 cells. Data are presented as mean ± SE. *P < 0.01 vs. control, n = 6.
SB431542 enhances long-term and multi-lineage engraftment of irradiated HSCs
Because long-term and multi-lineage engraftment is a gold standard to measure HSC function, we performed a competitive repopulation assay to validate whether IR-induced HSC function decline could be ameliorated by SB431542 treatment. As shown in Fig. 3, mice receiving donor cells exposed to irradiation with vehicle treatment showed a substantial decrease in donor cell engraftment in all the lineages 2 months after transplantation. Treated with SB431542 (1 μM), the donor cell engraftment increased 23.1% at 2 months, 16.7% of B cells, 18.8% of T cells and 6.7% of myeloid cells derived from donor cells. These findings indicate that SB431542 treatment indeed preserved the function of HSCs after irradiation exposure, resulting in enhanced long-term and multi-lineage engraftment after BM transplantation.
Fig. 3.

SB431542 reduces IR-induced suppression of HSC long-term engraftment after transplantation. Donor BMMNCs, treated with IR (2 Gy) after receiving vehicle or SB431542 (1 µM), were mixed with competitive cells. Cells were transplanted into receptor mice as described in the text, and donor cell engraftment was analyzed 2 months after transplantation. The data are expressed as means ± SE of percentage of donor-derived cells as: (A) leukocytes (CD45.1 + cells), (B) B cells (CD45.1 + B220 + cells), (C) T cells (CD45.1 + CD3 + cells) and (D) myeloid cells (CD45.1 + CD11b + and/or Gr-1 + granulocyte–monocyte–macrophage) in the peripheral blood (n = 7 recipient mice/group).
Irradiation increased TβRII expression in BMMNCs
TGFβ1 can be activated by ionizing irradiation in the extracellular space of irradiated tissue as well as proteolytic processes [6, 7]. To confirm irradiation exposure could up-regulate TGFβ1 activity in bone marrow hematopoietic cells, TβRII expression in BMMNCs exposed to irradiation was analyzed. As shown in Fig. 4, after irradiation exposure, TβRII mRNA and protein expression were up-regulated. These results suggest irradiation might activate the TGFβ pathway partly by over-expression of TβRII.
Fig. 4.
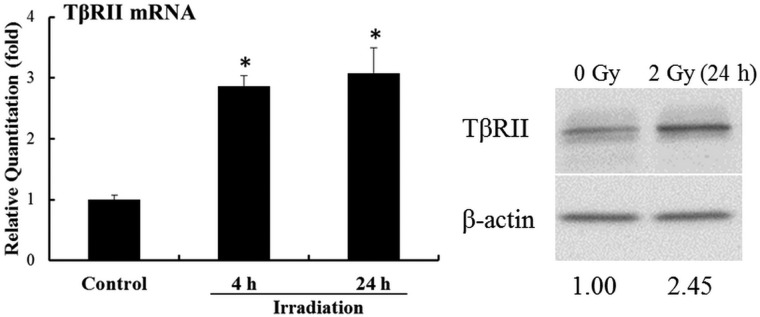
IR induces expression of TβRII in BMMNCs. The cells were sham-irradiated as a control or irradiated with 2.0 Gy IR. They were incubated for 4–24 h and analyzed for the mRNA and protein expression of TβRII by qRT-PCR and western blot assays. The levels of TβRII mRNA expression are expressed as means ± SE of fold changes compared with the control (n = 3). *P < 0.05 vs. control.
SB431542 induced down-regulation of NOX expression
Previous work has shown that IR-induced ROS stress in hematopoietic cells via up-regulating NOX4 at least partly contributes to IR-induced BM failure [10]. In this study, NOX1, NOX2 and NOX4 expression in BMMNCs treated with irradiation and SB431542 was investigated. As shown in Fig. 5, irradiation exposure up-regulated NOX1 mRNA expression significantly by 4.07-fold (P < 0.01) at 4 h, and by 1.61-fold (P < 0.05) at 24 h. After irradiation, NOX2 mRNA expression was up-regulated by irradiation (1.56-fold, 4 h, P < 0.01; 1.26-fold, 24 h, P < 0.05). Compared with sham-irradiated cells, a consistently higher level (about 2-fold) of NOX4 mRNA expression of irradiated cells was observed from 4–24 h. Treated with SB431542 (1 μM), NOX2 and NOX4 mRNA expression was down-regulated, but not NOX1. These data, verified by western blots assays as shown in Fig. 5, suggest radiation exposure up-regulates NOX expression in BMMNCs, which at least partly contributes to oxidative stress in the BM hematopoietic system. And SB431542, a TGFβ1 inhibitor, could reduce IR-induced up-regulation of NOX2 and NOX4 expression.
Fig. 5.

SB431542 inhibits IR-induced expression of NOX2 and NOX4 in BMMNCs. The cells were sham-irradiated as a control or irradiated with 2.0 Gy IR after receiving vehicle or SB431542 (1 µM) treatment. They were incubated for 4–24 h and analyzed for the mRNA and protein expression of NOX1, NOX2 and NOX4 using qRT-PCR and western blot assays. (A) The levels of NOX1 mRNA and protein expression; (B) the levels of NOX2 mRNA and protein expression; (C) the levels of NOX4 mRNA and protein expression. Results of mRNA expression are expressed as means ± SE of fold changes compared with those of the control (n = 3). *P < 0.05 vs. control, #P < 0.05 vs. vehicle.
DISCUSSION
In spite of the extensive study of TGFβ1 inhibitor to prevent fibroblast activity [22, 23], the pharmacological effect of SB431542 on IR-induced BM suppression has not been well investigated. In this study, we examined whether SB431542 can mitigate IR-induced BM injury in vitro. Our results showed that IR increased TβRII mRNA expression in BMMNCs, which provides additional evidence that IR could up-regulate TGFβ1 activity. Then, it was found that treatment with SB431542 significantly alleviated depressed clonogenic function of HPCs and HSCs induced by IR. It was also observed that expression of NOX1, NOX2 and NOX4 in BMMNCs was up-regulated by IR and that of NOX2 and NOX4 was ameliorated by treatment with SB431542. Therefore, results from this study demonstrate that SB431542, a wildly used TGFβ1 inhibitor, has the potential to be used as a therapeutic agent to mitigate IR-induced BM suppression in part via inhibition of NOX expression.
Increasing evidence suggests that IR-induced injury is caused, at least in part, by induction of oxidative stress. This suggestion is in agreement with our previous findings [1, 10], which demonstrate IR-induced up-regulation of NOXs in hematopoietic cells at least partly contribute to IR-induced BM failure [10], and using antioxidants such as N-acetylcysteine (NAC), Mn(III) meso-tetrakis-(N-ethylpyridinium-2-yl) porphyrin (MnTE) and resveratrol effectively ameliorates IR-induced BM failure in mice [10, 20, 24]. Elegant work by Bondi et al. [17] showed that TGFβ1 increases both the activity of NADPH oxidase and expression of NOX2 and NOX4 in kidney, indicating that this growth factor induces production of ROS, which could be effectively decreased by inhibiting TβRI. It has also been reported that treatment with TGFβ1 increases NOX4 expression and activity in human aortic smooth muscle cells [13], pulmonary fibroblasts [25], airway smooth muscle [26] and in hepatocytes [27]. Our work showed that IR increased NOX1, NOX2 and NOX4 expression in mice BMMNCs, and treatment with SB431542 decreased NOX2 and NOX4 expression after IR. These data suggest inhibiting TGFβ1 could attenuate IR-induced ROS stress via the NOX pathway, which at least partly protects mouse hematopoietic function from IR damage. However, SB431542 functions only as a radiation protectant to reduce IR-induced BM failure, because in our preliminary cell viability assays, we found that post-IR treatment with SB431542 had no significant effect on the activity of the BMMNCs exposed to IR.
Radiation-induced fibrosis, an important side-effect in the treatment of cancer, plays a core role in injury of normal tissues or organs, which not only adversely affects the quality of life of cancer patients but also worsens the outcome of cancer treatment [23]. Increasing evidence shows that TGFβ1 is the major cytokine responsible for the regulation of fibroblast proliferation and differentiation [28, 29]. IR not only induces long-term TGFβ1 overexpression owing in part to oxidative stress and an inflammatory response, but also actives LTGFβ in the extracellular space of irradiated tissue as well as proteolytic processes [6, 7]. Many studies suggest inhibiting TGFβ1 could protect lungs, kidneys, blood vessels, liver, skin, etc. from radiotherapy-induced injury [30–33]. In addition, TGF-β1 inhibitor can attenuate tumor cell migration [34] and enhance sensitivity to radiation therapy in pancreatic cancer, glioblastoma and breast cancer [35–37]. TGF-β1 antibody was tested in a phase III clinical trial for breast cancers, prostate cancers, colon cancers and renal carcinoma [33]. Therefore, SB431542 has the potential to increase the therapeutic efficacy of radiotherapy not only by reducing normal tissue injury, but also by inhibiting tumor growth.
In summary, our study shows that SB431542 ameliorates IR-induced BM suppression. These findings provide a better understanding that TGF-β1 inhibitor may have the potential to be used as a therapy for other oxidative stress-related organ injury.
FUNDING
This study was supported by Wu Jieping Medical Foundation (No 320.6750.12342), National Natural Science Foundation of China (No 81129020 and 81072237) & Tianjin Natural Science Foundation (11JCZDJC19100).
REFERENCES
- 1.Wang Y, Schulte BA, LaRue AC, et al. Total body irradiation selectively induces murine hematopoietic stem cell senescence. Blood. 2006;107:358–66. doi: 10.1182/blood-2005-04-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meng A, Wang Y, Van Zant G, et al. Ionizing radiation and busulfan induce premature senescence in murine bone marrow hematopoietic cells. Cancer Res. 2003;63:5414–19. [PubMed] [Google Scholar]
- 3.Ruscetti FW, Akel S, Bartelmez SH. Autocrine transforming growth factor-beta regulation of hematopoiesis: many outcomes that depend on the context. Oncogene. 2005;24:5751–63. doi: 10.1038/sj.onc.1208921. [DOI] [PubMed] [Google Scholar]
- 4.Soderberg SS, Karlsson G, Karlsson S. Complex and context dependent regulation of hematopoiesis by TGF-beta superfamily signaling. Ann New York Acad Sci. 2009;1176:55–69. doi: 10.1111/j.1749-6632.2009.04569.x. [DOI] [PubMed] [Google Scholar]
- 5.Munger JS, Harpel JG, Gleizes PE, et al. Latent transforming growth factor-beta: structural features and mechanisms of activation. Kidney International. 1997;51:1376–82. doi: 10.1038/ki.1997.188. [DOI] [PubMed] [Google Scholar]
- 6.Jobling MF, Mott JD, Finnegan MT, et al. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiation Res. 2006;166:839–48. doi: 10.1667/RR0695.1. [DOI] [PubMed] [Google Scholar]
- 7.Ehrhart EJ, Segarini P, Tsang ML, et al. Latent transforming growth factor beta1 activation in situ: quantitative and functional evidence after low-dose gamma-irradiation. FASEB J. 1997;11:991–1002. doi: 10.1096/fasebj.11.12.9337152. [DOI] [PubMed] [Google Scholar]
- 8.Watanabe H, de Caestecker MP, Yamada Y. Transcriptional cross-talk between Smad, ERK1/2, and p38 mitogen-activated protein kinase pathways regulates transforming growth factor-beta-induced aggrecan gene expression in chondrogenic ATDC5 cells. J Biol Chem. 2001;276:14466–73. doi: 10.1074/jbc.M005724200. [DOI] [PubMed] [Google Scholar]
- 9.Yano H, Hamanaka R, Nakamura M, et al. Smad, but not MAPK, pathway mediates the expression of type I collagen in radiation induced fibrosis. Biochemical Biophys Res Comm. 2012;418:457–63. doi: 10.1016/j.bbrc.2012.01.039. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Liu L, Pazhanisamy SK, et al. Total body irradiation causes residual bone marrow injury by induction of persistent oxidative stress in murine hematopoietic stem cells. Free Radical Biol Med. 2010;48:348–56. doi: 10.1016/j.freeradbiomed.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 12.Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radical Biol Med. 2007;43:332–47. doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thannickal VJ, Fanburg BL. Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor beta 1. J Biol Chem. 1995;270:30334–8. doi: 10.1074/jbc.270.51.30334. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez A, Alvarez AM, Benito M, et al. Cycloheximide prevents apoptosis, reactive oxygen species production, and glutathione depletion induced by transforming growth factor beta in fetal rat hepatocytes in primary culture. Hepatology. 1997;26:935–43. doi: 10.1002/hep.510260420. [DOI] [PubMed] [Google Scholar]
- 15.Martin-Garrido A, Brown DI, Lyle AN, et al. NADPH oxidase 4 mediates TGF-beta-induced smooth muscle alpha-actin via p38MAPK and serum response factor. Free Radical Biol Med. 2011;50:354–62. doi: 10.1016/j.freeradbiomed.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caja L, Sancho P, Bertran E, et al. Overactivation of the MEK/ERK pathway in liver tumor cells confers resistance to TGF-{beta}-induced cell death through impairing up-regulation of the NADPH oxidase NOX4. Cancer Res. 2009;69:7595–602. doi: 10.1158/0008-5472.CAN-09-1482. [DOI] [PubMed] [Google Scholar]
- 17.Bondi CD, Manickam N, Lee DY, et al. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J Am Soc Nephrol. 2010;21:93–102. doi: 10.1681/ASN.2009020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Black D, Lyman S, Qian T, et al. Transforming growth factor beta mediates hepatocyte apoptosis through Smad3 generation of reactive oxygen species. Biochimie. 2007;89:1464–73. doi: 10.1016/j.biochi.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noah JW, Severson W, Noah DL, et al. A cell-based luminescence assay is effective for high-throughput screening of potential influenza antivirals. Antiviral Res. 2007;73:50–9. doi: 10.1016/j.antiviral.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Li H, Wang Y, Pazhanisamy SK, et al. Mn(III) meso-tetrakis-(N-ethylpyridinium-2-yl) porphyrin mitigates total body irradiation-induced long-term bone marrow suppression. Free Radical Biol Med. 2011;51:30–7. doi: 10.1016/j.freeradbiomed.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang H, Li J, Wang YY, et al. Retinoblastoma 94 enhances radiation treatment of esophageal squamous cell carcinoma in vitro and in vivo. J Radiat Res. 2012;53:117–24. doi: 10.1269/jrr.11051. [DOI] [PubMed] [Google Scholar]
- 22.Andarawewa KL, Paupert J, Pal A, et al. New rationales for using TGFbeta inhibitors in radiotherapy. Int J Radiat Biol. 2007;83:803–11. doi: 10.1080/09553000701711063. [DOI] [PubMed] [Google Scholar]
- 23.Pohlers D, Brenmoehl J, Loffler I, et al. TGF-beta and fibrosis in different organs–molecular pathway imprints. Biochim Biophys Acta. 2009;1792:746–56. doi: 10.1016/j.bbadis.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Zhai Z, Wang Y, et al. Resveratrol ameliorates ionizing irradiation-induced long-term hematopoietic stem cell injury in mice. Free Radical Biol Med. 2013;54:40–50. doi: 10.1016/j.freeradbiomed.2012.10.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amara N, Goven D, Prost F, et al. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax. 2010;65:733–8. doi: 10.1136/thx.2009.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michaeloudes C, Sukkar MB, Khorasani NM, et al. TGF-beta regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells. Am J Physiol Lung Cellular Molecular Physiol. 2011;300:L295–304. doi: 10.1152/ajplung.00134.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carmona-Cuenca I, Roncero C, Sancho P, et al. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J Hepatol. 2008;49:965–76. doi: 10.1016/j.jhep.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 28.Yano H, Hamanaka R, Nakamura M, et al. Smad, but not MAPK, pathway mediates the expression of type I collagen in radiation induced fibrosis. Biochem Biophys Res Comm. 2012;418:457–63. doi: 10.1016/j.bbrc.2012.01.039. [DOI] [PubMed] [Google Scholar]
- 29.O'Sullivan B, Levin W. Late radiation-related fibrosis: pathogenesis, manifestations, and current management. Sem Radiat Oncol. 2003;13:274–89. doi: 10.1016/S1053-4296(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 30.Reeves A, Zagurovskaya M, Gupta S, et al. Inhibition of transforming growth factor-beta signaling in normal lung epithelial cells confers resistance to ionizing radiation. Int J Radiat Oncol Biology Phys. 2007;68:187–95. doi: 10.1016/j.ijrobp.2006.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scharpfenecker M, Floot B, Russell NS, et al. Endoglin haploinsufficiency reduces radiation-induced fibrosis and telangiectasia formation in mouse kidneys. Radiotherapy Oncol. 2009;92:484–91. doi: 10.1016/j.radonc.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Kudo K, Abe Y, et al. Inhibition of transforming growth factor-beta, hypoxia-inducible factor-1alpha and vascular endothelial growth factor reduced late rectal injury induced by irradiation. J Radiat Res. 2009;50:233–9. doi: 10.1269/jrr.08112. [DOI] [PubMed] [Google Scholar]
- 33.Anscher MS. Targeting the TGF-beta1 pathway to prevent normal tissue injury after cancer therapy. Oncologist. 2010;15:350–9. doi: 10.1634/theoncologist.2009-S101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jakowlew SB. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006;25:435–57. doi: 10.1007/s10555-006-9006-2. [DOI] [PubMed] [Google Scholar]
- 35.Ahmed MM, Alcock RA, Chendil D, et al. Restoration of transforming growth factor-beta signaling enhances radiosensitivity by altering the Bcl-2/Bax ratio in the p53 mutant pancreatic cancer cell line MIA PaCa-2. J Biol Chem. 2002;277:2234–46. doi: 10.1074/jbc.M110168200. [DOI] [PubMed] [Google Scholar]
- 36.Zhang M, Kleber S, Rohrich M, et al. Blockade of TGF-beta signaling by the TGFbetaR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011;71:7155–67. doi: 10.1158/0008-5472.CAN-11-1212. [DOI] [PubMed] [Google Scholar]
- 37.Bouquet F, Pal A, Pilones KA, et al. TGFbeta1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin Cancer Res. 2011;17:6754–65. doi: 10.1158/1078-0432.CCR-11-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]