

Abstract
GB virus type C (GBV-C) is a lymphotropic virus that can cause persistent infection in humans. GBV-C is not associated with any disease, but is associated with reduced mortality in human immunodeficiency virus type 1 (HIV-1)-infected individuals. Related viruses have been isolated from chimpanzees (GBV-Ccpz) and from New World primates (GB virus type A, GBV-A). These viruses are also capable of establishing persistent infection. We determined the nucleotide sequence encoding the envelope glycoprotein (E2) of two GBV-Ccpz isolates obtained from the sera of captive chimpanzees. The deduced GBV-Ccpz E2 protein differed from human GBV-C by 31 % at the amino acid level. Similar to human GBV-C E2, expression of GBV-Ccpz E2 in a tet-off human CD4+ Jurkat T-cell line significantly inhibited the replication of diverse HIV-1 isolates. This anti-HIV-replication effect of GBV-Ccpz E2 protein was reversed by maintaining cells in doxycycline to reduce E2 expression. Previously, we found a 17 aa region within human GBV-C E2 that was sufficient to inhibit HIV-1. Although GBV-Ccpz E2 differed by 3 aa differences in this region, the chimpanzee GBV-C 17mer E2 peptide inhibited HIV-1 replication. Similarly, the GBV-A peptide that aligns with this GBV-C E2 region inhibited HIV-1 replication despite sharing only 5 aa with the human GBV-C E2 sequence. Thus, despite amino acid differences, the peptide region on both the GBV-Ccpz and the GBV-A E2 protein inhibit HIV-1 replication similar to human GBV-C. Consequently, GBV-Ccpz or GBV-A infection of non-human primates may provide an animal model to study GB virus–HIV interactions.
Introduction
GB virus type A (GBV-A) and type C (GBV-C, also called hepatitis G virus) were discovered in 1995, and a closely related chimpanzee variant (GBV-Ccpz) was identified in 1998 (Adams et al., 1998; Birkenmeyer et al., 1998; Simons et al., 1995). GBV-A, GBV-C and a bat GB virus (GBV-D) were recently assigned to a novel genus Pegivirus within the family Flaviviridae by the executive committee of the International Committee on the Taxonomy of Viruses (ICTV) (Stapleton et al., 2011; ICTV, 2012). Although GBV-C was initially thought to cause hepatitis, epidemiological studies failed to identify an association between human GBV-C infection and acute or chronic hepatitis in humans or chimpanzees (Alter, 1997; Polgreen et al., 2003; Stapleton, 2003). GBV-C is lymphotropic, and the virus is found in and produced by T- and B-lymphocytes obtained from infected individuals (Fogeda et al., 1999; George et al., 2003, 2006; Rydze et al., 2012; Xiang et al., 2000). The cellular tropism of GBV-A and GBV-Ccpz has not been elucidated (reviewed by Stapleton et al., 2011). Due to shared modes of transmission, the prevalence of GBV-C in human immunodeficiency virus type 1 (HIV-1)-infected people is high (17–42 %) (Rey et al., 2000; Tillmann et al., 2001; Xiang et al., 2001). Several, though not all studies observed a beneficial association between GBV-C infection and improved survival in HIV-infected individuals (reviewed by Mohr & Stapleton, 2009; Polgreen et al., 2003; Stapleton, 2003). Evidence supporting several potential mechanisms by which GBV-C may influence HIV disease progression have been identified, including direct antiviral effects, altered expression of cytokines, chemokines, HIV entry receptors and modulation of host cell signalling pathways (Bhattarai & Stapleton, 2012a; Bhattarai et al., 2012b; Mohr & Stapleton, 2009).
The GBV-C envelope glycoprotein E2 inhibits HIV-1 replication when it is added to or is expressed within primary or transformed CD4+ T-cells (Jung et al., 2007; Xiang et al., 2012). Similarly, a 17 aa region within the GBV-C E2 protein is sufficient to potently inhibit HIV-1 when delivered to, or expressed within CD4+ T-cells (Xiang et al., 2012). Because this 17mer E2 peptide is part of a larger peptide previously shown to form an amphipathic helix in the presence of lipids and model membranes, it is thought that this region may be involved in GBV-C fusion to host cell membranes (Larios et al., 2005). Another E2 peptide that overlaps with the putative fusion peptide prevents oligomerization of the HIV-1 gp41 membrane proximal ectodomain (MPER) and thus inhibits HIV-1–membrane fusion in an in vitro model (Herrera et al., 2009). These findings raise the possibility of structural or functional mimicry between the putative GBV-C fusion domain and the HIV-1 fusion domain.
Although chimpanzees are permissive for HIV (Fultz et al., 1986) and human GBV-C infection (Bukh et al., 1998), no animal models of GBV-C co-infection with HIV are described. In addition, there are no published reports of in vitro or in vivo interactions between the GBV-Ccpz variant and HIV. Among captive chimpanzees, the prevalence and persistence rates of GBV-Ccpz infection are similar to GBV-C infection in humans, with a low but detectable prevalence of persistent viraemia and a higher prevalence of antibody to GBV-C E2 protein indicating prior infection (Mohr et al., 2011). Only one full-length sequence of GBV-Ccpz is published, and this sequence and the E2 protein coding region share 69 % amino acid identity with human GBV-C isolates (Birkenmeyer et al., 1998; Stapleton et al., 2011). There are no published data that characterize the GBV-Ccpz E2 protein or that examine the potential of GBV-Ccpz or GBV-A E2 protein interactions with HIV. However, if the HIV inhibitory effect of human GBV-C E2 protein is shared with GBV-Ccpz or with the New World primate virus GBV-A, two potential animal models of the interaction between members of the genus Pegivirus and HIV would be possible. Here, we characterize GBV-Ccpz E2 coding sequences from isolates from two infected chimpanzees, and express one of these in a CD4+ T-cell line. HIV-1 replication was inhibited in cells expressing GBV-Ccpz E2 protein, the GBV-Ccpz and the GBV-A 17mer peptides homologous to the human GBV-C 17mer peptide previously shown to inhibit HIV-1 in vitro. The E2 protein and peptide inhibit HIV replication by reducing HIV entry into the cells, and this is independent of HIV receptor expression.
Results
Characterization of the GBV-Ccpz E2 protein coding region
The GBV-Ccpz genome region that aligns to the human predicted GBV-C E2 protein coding region (nt 1167–2318 based on the infectious GBV-C clone, GenBank accession no. AF121950) was amplified from chimpanzee serum using two sets of overlapping primers (see Methods) in nested RT-PCRs as described previously (Xiang et al., 2000). The resultant amplicons were cloned and sequenced as described before (Xiang et al., 2006), and the two newly described GBV-Ccpz E2 nucleotide and deduced amino acid sequences submitted to GenBank (accession nos JX472278 and JX472279 from animals 1855 and Candi, respectively). Phylogenetic analysis of the predicted GBV-Ccpz E2 coding region demonstrated that they were more closely related to the published GBV-Ccpz E2 region than to human GBV-C isolates. Specifically, the E2 coding region of the two isolates differed from each other by only 2 % (nucleotide; Fig. 1a), and 1 % (amino acid; Fig. 1b), or from the published GBV-Ccpz sequence (AF070476) by 10 and 3 %, respectively. In contrast, there was greater divergence between the GBV-Ccpz E2 coding regions and the human GBV-C E2 coding region, with a mean of 37 % nt and 31 % aa difference, respectively. The deduced GBV-Ccpz E2 protein was 375 aa in length and compared with the human GBV-C E2 protein, all three predicted GBV-Ccpz E2 proteins lacked 6 aa at the extreme C terminus. Despite this truncation, the predicted signal peptidase cleavage site was maintained in both the human and the chimpanzee GBV-C E2 proteins (Fig. 1c). All three GBV-Ccpz E2 proteins were predicted to have three glycosylation sites at amino acid positions 37 (N-linked), 105 (N-linked) and 240 (O-linked) (Blom et al., 2004).
Fig. 1.
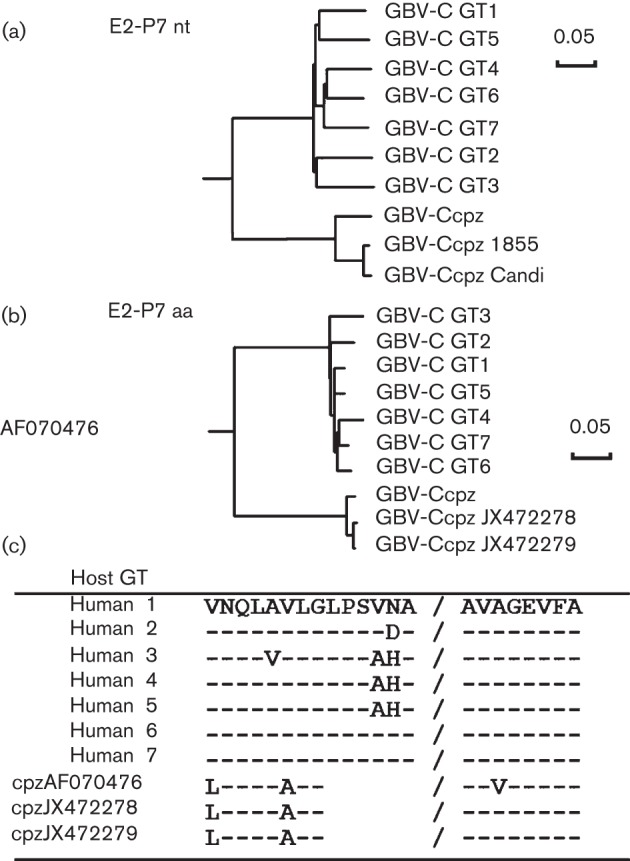
Phylogenetic relationships between human and chimpanzee GBV-C E2 protein coding regions. The nucleotide (nt) (a) and deduced amino acid (aa) (b) sequences of the predicted GBV-C E2 coding region to the predicted signal peptidase cleavage site at the C terminus of E2 and the N terminus of the putative P7 protein (nt 1167–2318 based on GenBank accession no. AF121950) of representative isolates of the seven human genotypes (GT) and the three chimpanzee isolates (cpz; GenBank accession nos JX472278, JX472279 and AF070476 for GBV-Ccpz 1855, GBV-Ccpz Candi and the published complete GBV-Ccpz genome, respectively) are shown. (c) Despite a 6 aa deletion at the C terminus of the predicted GBV-Ccpz E2 protein, the predicted signal peptidase site is retained in the GBV-Ccpz deduced amino acid sequence. Bars, 0.05 nucleotide (a) or amino acid (b) substitutions per site.
GBV-Ccpz E2 protein expression inhibits HIV replication
Expression of the human GBV-C E2 protein in Jurkat cells potently inhibits HIV replication (Xiang et al., 2012). Since there is 31 % difference in amino acid identity between the human and chimpanzee GBV-C virus isolates, it was unclear if the GBV-Ccpz E2 protein would inhibit HIV replication as well. Consequently, the GBV-Ccpz C E2 protein with a C-terminus truncation to remove the transmembrane domain was ligated into a previously described tet-repressible, bicistronic vector (Xiang et al., 2006). The resulting plasmid encoded GBV-Ccpz aa 1–331. The plasmid contains a Kozak sequence directing translation of the GBV-Ccpz E2 reading frame of the JX472278 sequence containing an in-frame polyhistidine tag, followed by stop codons in all three reading frames, and the encephalomyocarditis virus internal ribosomal entry site which directs translation of GFP. Tet-off Jurkat cells were transfected with this plasmid, and a bulk cell line was selected by growing cells in hygromycin and neomycin and sorting for GFP-positive cells using FACS as previously described (Xiang et al., 2006). Previously described Jurkat cell lines that expressed the vector control (with GFP), or the human GBV-C E2 protein coding region in which a frameshift was introduced (FS) to abolish protein expression served as the negative controls, and a Jurkat cell line that expressed E2 protein amplified from a human GBV-C isolate served as the positive control (Xiang et al., 2012).
GBV-Ccpz E2 with C-terminal his-tag expression was demonstrated in GFP-positive cells by immunoblot analysis using mouse polyclonal anti-GBV-C E2 sera, and an anti-His mAb (Fig. 2a). GBV-Ccpz E2 was not detected using the anti-peptide rabbit sera, consistent with the fact that there were three amino acid differences between the chimpanzee GBV-C peptide sequence and the human E2 peptide sequence for rabbit immunization. Both GBV-C E2 and GFP expression were diminished by growing cells in doxycycline (1 µg ml−1) for 5 days, as measured by immunoblot and flow cytometry (Fig. 2).
Fig. 2.
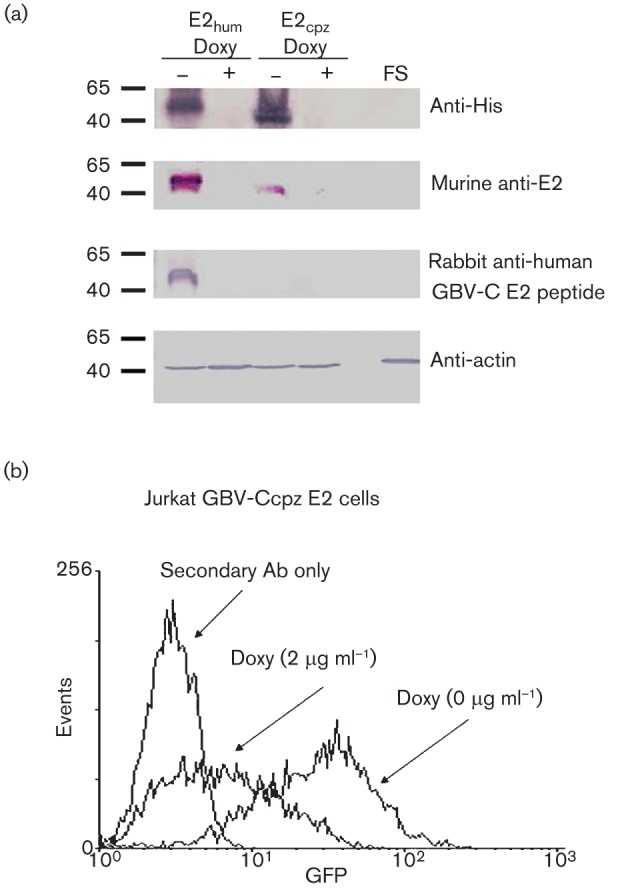
Expression of GBV-Ccpz E2 protein in Jurkat cells. (a) Jurkat cells stably expressing the human and the chimpanzee GBV-C E2 proteins contained immunoreactive proteins detected by anti-penta-His mAb and anti-murine human GBV-C E2 polyclonal antisera. Human GBV-C E2, but not GBV-Ccpz E2, reacted with rabbit anti-E2 peptide (GGAGLTGGFYEPLVRRC) polyclonal antisera. None of the antibodies reacted with cell lysates from the frameshift control Jurkat cell line (FS). Actin was used as a loading control. Following incubation of the Jurkat cells expressing E2 protein in doxycycline (1 µg ml−1) for 5 days, (a) the immunoreactive proteins were not detected, and (b) GFP expression as measured by flow cytometry was reduced.
HIV replication was not detected following infection of Jurkat cells expressing either the chimpanzee GBV-C E2 or the human GBV-C E2 proteins with a clade B, clinical, X4-tropic HIV isolate [catalogue number 1073, NIH AIDS Research and Reference Reagent Program (NARRRP); Fig. 3a]. This anti-HIV replication effect of E2 protein was reversed by maintaining cells in doxycycline to reduce E2 expression (data represent HIV p24 antigen released into culture supernatants from three independent cultures; Fig. 3a). The individual cultures from three independent infections obtained 7 days post-infection (p.i.) were tested, and HIV replication was significantly higher in the FS control cells compared with Jurkat cells expressing either the human or chimpanzee GBV-C E2 proteins, even when maintained in doxycycline (P<0.001; Fig. 3a). Nevertheless, HIV replication was significantly higher in the Jurkat cells expressing GBV-C E2 proteins maintained in doxycycline compared with those without doxycycline (P<0.001), indicating that HIV inhibition correlated with human and chimpanzee GBV-C E2 expression levels. As previously described for the human GBV-C E2 protein-expressing Jurkat cells, there were no differences in cell viability as measured by trypan blue exclusion microscopy (data not shown).
Fig. 3.
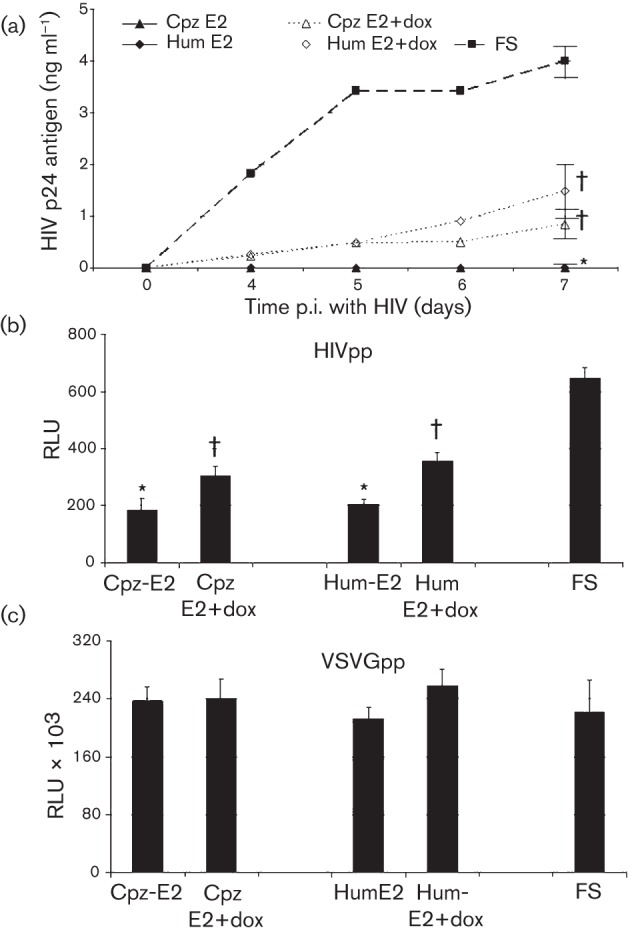
GBV-Ccpz E2 expression inhibits HIV replication. (a) HIV did not replicate in Jurkat cells expressing the human (GBV-C) or chimpanzee (GBV-Ccpz) E2 proteins, but replicated to a significantly greater extent in cells maintained in doxycycline to reduce E2 expression. In contrast, HIV replicated well in Jurkat cells expressing the frameshift (FS) control. (b, c) Similarly, HIV pseudotyped retrovirus particle (HIVpp), but not VSVG pseudotyped particle (VSVGpp) transduction was significantly reduced in Jurkat cells expressing GBV-Ccpz E2 protein. Data are presented as HIV p24 antigen release into culture supernatants (a) or as the relative light units (RLU) (b, c). *, P<0.001 versus FS; †, P<0.01 versus the same cells not maintained in doxycycline. Data shown are means±sd for three independent experiments.
Single cycle replication assays allow assessment of viral envelope specificity, and provide information regarding which step(s) of HIV-1 replication is inhibited. HIV gp160 envelope pseudotyped HIV particles (HIVpp) or vesicular stomatitis virus envelope glycoprotein (VSVG) pseudotyped HIV particles (VSVGpp) containing a luciferase reporter gene were generated in 293T cells as previously described (Mohr et al., 2010; Xiang et al., 2008). The Jurkat cell lines expressing human and chimpanzee GBV-C E2 protein or the FS control were transduced with the HIVpp and VSVGpp, and luciferase activity was measured 96 h later. Consistent with the replication competent studies shown in Fig. 3(a), both human- and chimpanzee-derived GBV-C E2 protein expression inhibited HIVpp transduction in Jurkat cells (P<0.001 compared with the FS control; Fig. 3b). Transduction inhibition was significantly reduced by maintaining the cells in doxycycline. In contrast, there were no significant differences in VSVGpp transduction in the Jurkat cells expressing GBV-C E2 proteins compared to transduction in the FS control Jurkat cells (Fig. 3c). Furthermore, maintaining the cells in doxycycline did not alter VSVGpp transduction. Since HIVpp, but not VSVGpp transduction was inhibited by human and chimpanzee GBV-C E2 protein expression and these particles used the same defective retrovirus backbone, the inhibitory effect is related at least in part to HIVenv-related entry, although post-entry steps might also be inhibited. GBV-C E2 from chimpanzees or humans did not change the surface expression of HIV receptors (CD4, CCR5 and CXCR4) on Jurkat cells by flow cytometry (data not shown), thus HIV entry inhibition was not due to altered receptor expression. The extent of HIV inhibition in the single cycle assay was less than that observed in the infection model (Fig. 3), suggesting that expression of E2 in CD4+ cells effectively prevents cell–cell spread, or that another step besides HIV entry is inhibited by GBV-C E2 expression.
To determine if expression of the chimpanzee GBV-C E2 protein in Jurkat cells inhibits diverse HIV isolates, five additional HIV isolates were studied. Table 1 provides a summary of the characteristics of the viruses studied. These HIV isolates represent both clinical and laboratory derived strains, and include HIV from clades A, B and D. Since GBV-C E2 protein elicits antibodies that cross-react with HIV particles (Mohr et al., 2010), and HIV gp41 T-20 peptide (Fuzeon) react with broadly reactive HIV neutralizing antibodies, it is possible that molecular mimicry between GBV-C and HIV-1 gp41 plays a role in HIV-1 inhibition. To test this hypothesis, two of the HIV isolates studied were resistant to T-20 (Table 1). T-20-resistant viruses were generated by transfecting 293T cells with infectious molecular clones as previously described (Xiang et al., 2012). All HIV isolates examined, including the T-20-resistant HIV strains, were inhibited in Jurkat cells expressing GBV-Ccpz E2 and GBV-Chum E2 as compared with the FS control (Fig. 4; P<0.001 for all).
Table 1. HIV virus strains studied.
| Virus isolate | Derivation | HIV clade | Designation | Catalogue #* |
| AZT Intermediate | Clinical | B | C-B | 1073 |
| HIV 92UG029 | Clinical | A | C-A | 1650 |
| xxHIVLAI | Laboratory | B | L-B | 2969 |
| HIV ELI | Laboratory | D | L-D | 2521 |
| pNL4-3 gp41(36G) V38A, N42D† | Laboratory | B | TB1 | 9488 |
| pNL4-3 gp41(36G) N42S† | Laboratory | B | TB2 | 9495 |
NARRRP catalogue number. All isolates are CXCR4-tropic.
T-20 resistant isolates, amino acid changes noted.
Fig. 4.
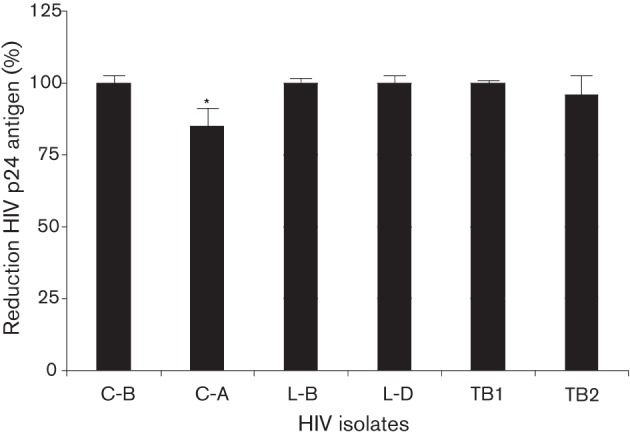
GBV-Ccpz E2 protein expression inhibits diverse HIV isolates. Jurkat cells expressing GBV-Ccpz E2 (1–331 aa) inhibited all six CXCR4-tropic HIV isolates studied. Strains are identified by their clade (B, A or D) or their resistance to T-20 (T) and their derivation as clinical (C) or laboratory (L)-derived strains (see Table 1). HIV inhibition rates are represented as a percentage of the reduction of HIV p24 antigen compared with FS. *, Inhibition was significantly less than other isolates (P<0.001) except isolate B-T2.
GBV-Ccpz and GBV-A E2 peptides are sufficient to inhibit HIV replication
We previously reported that expression of the human GBV-C E2 protein or a peptide motif within E2 (aa 276–292) in CD4+ Jurkat cells significantly inhibited HIV replication in those cells. Furthermore, HIV replication in a CD4+ T-cell line or in primary CD4+ T-cells was inhibited by the addition of a synthetic peptide representing the 276–292 peptide motif (Xiang et al., 2012). Alignment of the three GBV-Ccpz E2 and the GBV-A (GenBank accession no. AF023424) E2 sequences identified homology between the human and chimpanzee GBV-C E2 peptide region, and to a much lesser extent, the GBV-A peptide region. There were two conserved amino acid differences and two additional amino acid polymorphisms in this region in the chimpanzee GBV-C E2 coding region. However, there were ten amino acid differences in the GBV-A coding region (Fig. 5a). Expression of the 17mer GBV-C E2 peptide from both the human and the chimpanzee isolates rendered the Jurkat cells unable to support HIV replication, while Jurkat cells containing the human 17mer amino acids in a scrambled order did not inhibit HIV when compared with HIV replication in the frameshift control cells (Fig. 5b). Expression of the GBV-A 17mer peptide also significantly inhibited HIV replication; however, to a lesser extent than either GBV-C sequence (Fig. 5b). These data suggest that there are critical residues within this peptide motif required for HIV inhibition, and studies to characterize the amino acid required are under way. Neither the GBV-Ccpz or GBV-A 17mer peptides altered cell viability based on trypan blue exclusion microscopy (data not shown). Similar to the near-full-length E2 protein expression, HIVpp were inhibited by the GBV-Ccpz E2 17mer peptide while VSVGpp were not (Fig. 5c, d). Thus, this peptide sequence from human and chimpanzee GBV-C and GBV-A E2 proteins are sufficient to inhibit HIV replication, and the inhibition involves HIV entry into cells. Although GBV-C is associated with reduced surface expression of HIV entry coreceptors CD4, CCR5 and CXCR4 (Maidana Giret et al., 2009; Nattermann et al., 2003; Schwarze-Zander et al., 2010; Xiang et al., 2004, 2006, 2009), none of these receptors were reduced on the surface of Jurkat cells expressing GBV-C E2 proteins or the GBV-A peptide compared to the control cell lines (data not shown). Thus, the HIV entry inhibition is not due to altered HIV entry receptor expression, and studies to further characterize the mechanism of interference are under way.
Fig. 5.

GBV-Ccpz and GBV-A E2 peptide expression is sufficient to inhibit HIV replication. (a) The GBV-C peptide (Hum-E2 17mer) from human isolates previously shown to inhibit HIV is conserved in all seven human GBV-C genotypes (GT); however, there are two to three amino acid differences between human and chimpanzee GBV-C (Cpz-E2 17mer) and ten amino acid differences in the GBV-A sequence (GenBank accession no. AF023424) in this region. NWP, New World primate. (b) The human and chimpanzee GBV-C and the GBV-A peptides inhibited HIV replication in Jurkat cells 7 days p.i. when compared with Jurkat cells expressing the frameshift E2 coding region (FS), while the human peptide expressed in a scrambled order (17mer-SCR) did not. Data represent the mean of three independent infections and the experiment was repeated with similar results. (c, d) Similarly, the Cpz-E2 17mer and the Hum-E2 17mer inhibited HIVpp (c), but not VSVGpp (d) transduction. Data are presented as HIV p24 antigen release into culture supernatants (b) or as the relative light units (RLU) (c, d). *, P<0.001 versus FS.
GBV-Ccpz E2-expressing cells inhibit HIV-1 replication in bystander cells
Since GBV-C is estimated to infect a small number of CD4+ T-cells in infected humans (<5 %) (Xiang et al., 2000; Rydze et al., 2012; Bhattarai & Stapleton, 2012a), expression of E2 protein in cells may not lead to an effect in vivo. To determine if GBV-Ccpz E2 inhibits HIV-1 replication in bystander cells, MT-2 cells were infected with or without co-cultivation (1 : 1 ratio) with GBV-Ccpz E2-expressing Jurkat cells or control cells including parental Jurkat, FS and GBV-Ccpz E2 maintained in doxycycline. GBV-Ccpz E2 significantly reduced HIV replication in MT-2 cells, and this was significantly reduced in GBV-Ccpz E2-expressing cells maintained in doxycycline to reduce E2 expression (P<0.001 for all comparisons; Fig. 6). In contrast, incubation of parental Jurkat cell controls or the GBV-C E2 frameshift (FS) control did not inhibit HIV replication in MT-2 cells. Inhibition was not observed when the GBV-Ccpz E2 Jurkat cells were separated from direct contact with the MT-2 cells in transwells, indicating that cell-to-cell communication may be required for E2 to inhibit HIV replication in bystander cells. Studies to characterize this mechanism are under way.
Fig. 6.
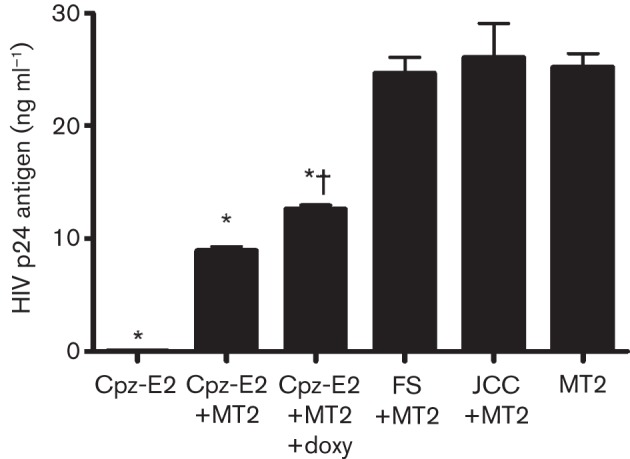
Jurkat cells expressing GBV-Ccpz E2 inhibit HIV replication in bystander cells. HIV replication was significantly less in MT-2 cells incubated with Jurkat cells expressing chimpanzee GBV-C E2 protein (Cpz-E2) compared with MT-2 cells alone, or MT-2 cells incubated with parental Jurkat cell controls (JCC) or Jurkat cells transcribing the frameshift control (FS). *, P<0.001 versus MT-2 cells alone. Maintaining Cpz-E2 in doxycycline (2 µg ml−1) for 5 days prior to co-culture reduced the extent of HIV inhibition. †, P<0.001 compared with Cpz-E2+MT2 cells without doxycycline. Data shown are means±sd for three independent experiments.
Discussion
The natural history and prevalence of chimpanzee GBV-Ccpz in captive chimpanzees is similar to that observed for human GBV-C infection in healthy blood donors (Mohr et al., 2011); however, no experimental data exist characterizing chimpanzee GBV-C or the GBV-Ccpz E2 protein. The two newly reported GBV-Ccpz E2 deduced amino acid sequences differ from the single published GBV-Ccpz E2 by roughly the same amount as do the E2 amino acid sequences of different human GBV-C genotypes (Fig. 1). However, deduced GBV-Ccpz E2 amino acids differ by 31 % from human GBV-C sequences. Despite this sequence heterogeneity, GBV-Ccpz E2 retains the in vitro HIV inhibitory phenotype observed for human GBV-C E2 protein (Xiang et al., 2012). Interestingly, there is a 6 aa deletion at the putative C terminus of the GBV-Ccpz E2, yet the predicted signal peptidase cleavage site was retained.
Like GBV-C E2, GBV-Ccpz E2 inhibited diverse HIV isolates in both replication-competent and single-cycle infections. Expression of the previously mapped GBV-C E2 17mer peptide as well as the homologous peptide regions of GBV-Ccpz and GBV-A E2 were sufficient to inhibit HIV replication, despite considerable amino acid differences in the peptide sequences (Fig. 5a). For the human GBV-C E2 protein, this region was shown to be part of a peptide that forms an amphipathic helix in the presence of lipids, suggesting a role in GBV-C cell fusion (Larios et al., 2005). It is tempting to speculate that the GBV-C and GBV-A E2 peptides interfere with HIV entry via competition with HIV–cell fusion, and studies are under way to address this hypothesis.
A limitation of our data is that we removed the C-terminal transmembrane domain of both human and chimpanzee GBV-C E2 protein in our studies. Due to cellular toxicity, we were unable to express the full-length protein in Jurkat cells, even using a tet-on system (data not shown). Nevertheless, taken together, the finding that expression of human and chimpanzee GBV-C E2 protein and the peptide motif of GBV-C, GBV-Ccpz and GBV-A in a CD4+ T-cell line inhibited HIV replication and that the addition of recombinant human GBV-C E2 (Jung et al., 2007) or the synthetic peptide motif to CD4+ T-cells inhibits HIV entry (Xiang et al., 2012) supports the hypothesis that GBV-C E2 protein contributes to the inverse relationship between GBV-C and HIV viral loads observed in human clinical trials (Tillmann et al. 2001; Björkman et al. 2007), and to the beneficial association between GBV-C and survival in HIV-infected humans (reviewed by Mohr & Stapleton, 2009).
GBV-C appears to infect a small proportion of human B- and T-lymphocytes (George et al., 2006); however, no data exist on GBV-Ccpz or GBV-A tropism in vivo. Nevertheless, the finding that GBV-Ccpz E2-expressing cells can reduce infection in bystander MT-2 cells suggests that the HIV inhibitory effect may extend more broadly than infected cells. Furthermore, the mean chimpanzee GBV-Ccpz viral load is greater than 1×108 genome copies ml−1, similar to what is observed for GBV-C infection of humans (Mohr et al., 2011; Rydze et al., 2012). Given that E2 is thought to be part of the virion, a relatively high concentration of the GBV-Ccpz E2 protein is in contact with CD4+ T-cells in vivo. Although the recent ban on chimpanzee research in the USA and Western Europe makes the development of a GBV-Ccpz–HIV model unlikely in developed nations (NIH Webpage, 2012), the data showing that the GBV-A E2 peptide domain also inhibits HIV replication raises the possibility of using GBV-A infection of New World primates as an animal model to examine Pegivirus–SHIV (chimeric simian immunodeficiency virus/HIV-1 virus) interactions. In addition, understanding the structural basis of the GBV-C E2 peptide inhibition of HIV entry may provide novel approaches to inhibiting HIV entry.
Methods
Characterization of the GBV-Ccpz E2 coding sequence.
Serum samples from two chimpanzees previously shown to be chronically infected with GBV-C (Mohr et al., 2011) were obtained at the Texas Biomedical Research Institute in San Antonio, TX. RNA was extracted by Qiagen Mini RNAeasy kit (Qiagen) for RT-PCR and sequence analysis was carried out as previously described (Xiang et al., 2000). The GBV-Ccpz genome region containing the sequence that aligned with the putative complete human GBV-C E2 coding region (1167–2178 based on GenBank accession no. AF121950) was amplified by first generating cDNA with the antisense primer (5′-GCACCGCCTCAGTCACTG-3′; AF121950 nt 5332–5349 based on AF121950; Invitrogen cDNA synthesis kit) followed by nested PCR amplification of two overlapping genome regions. For the upstream amplicon, the following primers were used: outer sense 5′-TACTGGATACTGGAGTATCT-3′; outer antisense 5′-CTCGGTGGTCGACGATCCA-3′; inner sense 5′-TGGCGCCTTCCCTTTGACT-3′; inner antisense 5′-CTAGGACCGCTATGGTGCA-3′. For the downstream amplicon, the following primers were utilized: outer sense: 5′-AGTCAAAGGGAAGGCGCCA-3′; outer antisense 5′-CAGAAGTACGCAGCCAAGCACCA-3′; inner sense 5′-AGATACTCCAGTATCCAGTA-3′; inner antisense 5′-GTCTCCATCCACCAGCGCTCAT-3′. These amplicons were ligated into pCR2.1 and sequenced by the University of Iowa DNA Core Facility using ABI automated sequencing as described previously (Xiang et al., 2006). Subsequently, the putative GBV-Ccpz E2 coding region truncated at the C terminus to remove the predicted transmembrane domain was amplified using the same cDNA as above followed by nested RT-PCR using the following primers: outer and inner sense primer: 5′-AGGATCCCATGGGACCCTTTGTCCCCGGTC-3′, outer antisense 5′-ATGCGGCCGCCAAAGGCGCTGCAAAT-3′, and inner antisenses 5′- AGCGGCCGCCCATGAATGGTCGTCGA-3′. For expressing GBV-Ccpz or GBV-A E2 peptide sequences, synthetic oligonucleotides were purchased (Iowa DNA Technologies) and ligated into the modified pTRE2 vector as previously described for other peptide sequences (Xiang et al., 2006).
E2 sequences from GBV-C isolates obtained from humans and chimpanzees were aligned with published sequences using the clustal w method and evolutionary histories were inferred using the UPGMA method and evolutionary distances were computed using the Maximum Composite Likelihood method using DNAman software (Linnen, Biosoft). The two newly described GBV-Ccpz E2 nucleotide and deduced amino acid sequences (from animals 1855 and Candi; GenBank accession nos JX472278 and JX472279, respectively) were analysed in comparison with the only published GBV-Ccpz E2 sequence (GenBank accession no. AF070476), and representative GBV-C isolates from each human GBV-C genotype (GT): GT 1, GenBank accession no. AB003291; GT 2, AF121950; GT 3, U94695; GT 4, AB018667; GT 5, AY949771; GT 6, AB003292; and GT 7, KY117.
Stable transfection of GBV-C E2 protein and controls in Jurkat cells.
GBV-Ccpz E2 protein from chimpanzee 1855 (GenBank accession no. JX472278) or the 17mer GBV-Ccpz and GBV-A (GenBank accession no. AF023424) E2 peptides were inserted into a modified Tet-off pTRE2-HGY plasmid (Clontech) as previously described (Xiang et al., 2006). This vector has a Kozak sequence, an Ig-kappa secretory leader sequence at the N terminus and a polyhistidine tag in-frame at the C terminus of the E2 insert, followed by stop codons in all three reading frames, and the EMCV IRES which directs translation of GFP. The negative controls included an empty vector control (which expresses GFP) and the human GBV-C E2 sequence in which a frameshift mutation was inserted to prevent translation of the E2 protein (FS) as described previously (Xiang et al., 2006).
Tet-off Jurkat cells (Clontech) were transfected with the GBV-Ccpz E2 plasmids and the control plasmid by Amaxa nucleofection (Lonza). Cells were selected in hygromycin and neomycin G418 (200 µg ml−1 each) medium for 3–4 weeks, and GFP-positive cells were bulk-sorted by FACS (Diva flow selection, University of Iowa Microscopy Core Facility). Chimpanzee and human GBV-C E2 protein expression in Jurkat cells was determined by SDS-PAGE, immunoblot analysis using an anti-His mAb (Penta-His; Qiagen), mouse polyclonal anti-GBV-C E2 serum or rabbit anti-human GBV-C E2 peptide serum as described by Mohr et al. (2010). For the hygromycin-resistant, GFP-positive cell line expressing the GBV-Ccpz 17mer peptide or the control peptide sequence, total cellular RNA was extracted from cells and the peptide sequence was confirmed by RT-PCR and sequence analysis as above.
HIV infection or transduction.
Four strains of HIV CXCR4-tropic (X4 strain) representing clades A, B and D, and two T-20 drug-resistant isolates (T-20) were studied. All were provided by the NIH AIDS Research and Reference Reagent Program (NARRRP) and characteristics are summarized in Table 1 (Mohr et al., 2010). For all infections, 10 ng HIV was applied to 1×106 cells in 24-well culture plates for 3 h at 37 °C, cells were washed three times and maintained in RPMI growth medium (10 % FCS, 1 % penicillin/streptomycin and 1 % glutamine; Gibco) for up to 7 days. Cell culture supernatants were harvested at the indicated times and replication determined by measuring p24 antigen by ELISA. HIV-1 replication was determined by measuring HIV-1 p24 antigen in culture supernatants (Retro-Tek HIV-1 p24 antigen ELISA kits; Zeptometrix). All infections were performed in triplicate and data represent the mean p24 antigen release into the supernatants from the three infections. All triplicate experiments were repeated at least once with consistent results.
Pseudotyped viruses transduction.
Pseudotyped HIV particles were generated in 293T cells using pNL4-3-Luc.R-E- (NIRRRP catalogue number 3417) and either HIV envelope (pHXB2env; NIRRRP catalogue number 1069) or vesicular stomatitis virus glycoprotein (VSVG) envelope (pHEF-VSVG; NIRRRP catalogue number 4693). Jurkat cells (8×104 cells per well) were transduced with the HIV-pseudotyped or VSVG control particles (normalized by HIV p24 antigen content), and luciferase activity was assessed 96 h post-transduction as recommended by the manufacturer using a Luminometer (Promega). As with infections, all transductions were performed in triplicate and all experiments were repeated at least once with consistent results.
HIV entry receptor detection.
Jurkat cells that stably expressed GBV-C E2 sequences or control Jurkat cells were incubated at 4 °C for 30 min with mouse anti-CD4 (PE-conjugated) or anti-CXCR4 (PE-conjugated) mAbs (BD Pharmingen), followed by washing with PBS. Cell-surface CD4 and CXCR4 levels were determined by flow cytometry (FACScan; Becton Dickinson) as previously described (Xiang et al., 2001).
Sequences analysis and statistics.
Sequence analysis was performed using DNAman (Linnen, Biosoft) and mega. Statistics were performed using SigmaStat software V2.03S (Jandel Scientific) and Graphpad Prism (GraphPad version 4.03 software). Comparisons of two samples utilized a Student’s t-test, and comparisons of more than three samples utilized ANOVA.
Acknowledgements
This research work was supported by grants from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development (Merit Review Grants, J. X. and J. T. S.) and the NIH (RO1 AI-58740, J. T. S.). We thank the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, for the following HIV isolates: AZT intermediate isolate (Cat. # 1073) from Dr Douglas Richman; HIV 92UGO29 (Cat. # 1650) from the UNAIDS Network for HIV Isolation and Characterization; xxHIVLAI (Cat. # 2969) from Drs John Mellors and Mr Chaofu Shi; HIVELI (Cat. # 2521) from Dr Jean-Marie Bechet and Dr Luc Montagnier; T-20 resistant isolates (Cat. # 9488 and # 9495) from Trimeris; pNL4-3, Luc.R-E- (Cat. # 3417) from Dr Nathaniel Landau; HIV envelope (pHXB2env; Cat. # 1069) from Dr Kathleen Page and Dr Dan Littman; and VSVG envelope 457-expressing plasmid (pHEF-VSVG; Cat. # 4693) from Dr Lung-Ji Chang. The University of Iowa Flow Cytometry and DNA Core Facilities provided the flow cytometry and DNA sequence analyses.
References
- Adams N. J., Prescott L. E., Jarvis L. M., Lewis J. C. M., McClure M. O., Smith D. B., Simmonds P. (1998). Detection in chimpanzees of a novel flavivirus related to GB virus-C/hepatitis G virus. J Gen Virol 79, 1871–1877 [DOI] [PubMed] [Google Scholar]
- Alter H. J. (1997). G-pers creepers, where’d you get those papers? A reassessment of the literature on the hepatitis G virus. Transfusion 37, 569–572 10.1046/j.1537-2995.1997.37697335149.x [DOI] [PubMed] [Google Scholar]
- Bhattarai N., Stapleton J. T. (2012a). GB virus C: the good boy virus? Trends Microbiol 20, 124–130 10.1016/j.tim.2012.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattarai N., McLinden J. H., Xiang J., Kaufman T. M., Stapleton J. T. (2012b). GB virus C envelope protein E2 inhibits TCR-induced IL-2 production and alters IL-2-signaling pathways. J Immunol 189, 2211–2216 10.4049/jimmunol.1201324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenmeyer L. G., Desai S. M., Muerhoff A. S., Leary T. P., Simons J. N., Montes C. C., Mushahwar I. K. (1998). Isolation of a GB virus-related genome from a chimpanzee. J Med Virol 56, 44–51 [DOI] [PubMed] [Google Scholar]
- Björkman P., Flamholc L., Molnegren V., Marshall A., Güner N., Widell A. (2007). Enhanced and resumed GB virus C replication in HIV-1-infected individuals receiving HAART. AIDS 21, 1641–1643 10.1097/QAD.0b013e32823bc9b7 [DOI] [PubMed] [Google Scholar]
- Blom N., Sicheritz-Pontén T., Gupta R., Gammeltoft S., Brunak S. (2004). Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649 10.1002/pmic.200300771 [DOI] [PubMed] [Google Scholar]
- Bukh J., Kim J. P., Govindarajan S., Apgar C. L., Foung S. K., Wages J., Jr, Yun A. J., Shapiro M., Emerson S. U., Purcell R. H. (1998). Experimental infection of chimpanzees with hepatitis G virus and genetic analysis of the virus. J Infect Dis 177, 855–862 10.1086/515255 [DOI] [PubMed] [Google Scholar]
- Fogeda M., Navas S., Martín J., Casqueiro M., Rodríguez E., Arocena C., Carreño V. (1999). In vitro infection of human peripheral blood mononuclear cells by GB virus C/hepatitis G virus. J Virol 73, 4052–4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fultz P. N., McClure H. M., Swenson R. B., McGrath C. R., Brodie A., Getchell J. P., Jensen F. C., Anderson D. C., Broderson J. R., Francis D. P. (1986). Persistent infection of chimpanzees with human T-lymphotropic virus type III/lymphadenopathy-associated virus: a potential model for acquired immunodeficiency syndrome. J Virol 58, 116–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S. L., Xiang J., Stapleton J. T. (2003). Clinical isolates of GB virus type C vary in their ability to persist and replicate in peripheral blood mononuclear cell cultures. Virology 316, 191–201 10.1016/S0042-6822(03)00585-3 [DOI] [PubMed] [Google Scholar]
- George S. L., Varmaz D., Stapleton J. T. (2006). GB virus C replicates in primary T and B lymphocytes. J Infect Dis 193, 451–454 10.1086/499435 [DOI] [PubMed] [Google Scholar]
- Herrera E., Gomara M. J., Mazzini S., Ragg E., Haro I. (2009). Synthetic peptides of hepatitis G virus (GBV-C/HGV) in the selection of putative peptide inhibitors of the HIV-1 fusion peptide. J Phys Chem B 113, 7383–7391 10.1021/jp900707t [DOI] [PubMed] [Google Scholar]
- ICTV (2012). Executive Committee Approved, Awaiting ICTV ratification. Available at: http://talk.ictvonline.org/search/default.aspx#q=Pegivirus
- Jung S., Eichenmüller M., Donhauser N., Neipel F., Engel A. M., Hess G., Fleckenstein B., Reil H. (2007). HIV entry inhibition by the envelope 2 glycoprotein of GB virus C. AIDS 21, 645–647 10.1097/QAD.0b013e32803277c7 [DOI] [PubMed] [Google Scholar]
- Larios C., Casas J., Alsina M. A., Mestres C., Gómara M. J., Haro I. (2005). Characterization of a putative fusogenic sequence in the E2 hepatitis G virus protein. Arch Biochem Biophys 442, 149–159 10.1016/j.abb.2005.06.027 [DOI] [PubMed] [Google Scholar]
- Maidana Giret M. T., Silva T. M., Sauer M. M., Tomiyama H., Levi J. E., Bassichetto K. C., Nishiya A., Diaz R. S., Sabino E. C. & other authors (2009). GB virus type C infection modulates T-cell activation independently of HIV-1 viral load. AIDS 23, 2277–2287 10.1097/QAD.0b013e32832d7a11 [DOI] [PubMed] [Google Scholar]
- Mohr E. L., Stapleton J. T. (2009). GB virus type C interactions with HIV: the role of envelope glycoproteins. J Viral Hepat 16, 757–768 10.1111/j.1365-2893.2009.01194.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr E. L., Xiang J., McLinden J. H., Kaufman T. M., Chang Q., Montefiori D. C., Klinzman D., Stapleton J. T. (2010). GB virus type C envelope protein E2 elicits antibodies that react with a cellular antigen on HIV-1 particles and neutralize diverse HIV-1 isolates. J Immunol 185, 4496–4505 10.4049/jimmunol.1001980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr E. L., Murthy K. K., McLinden J. H., Xiang J., Stapleton J. T. (2011). The natural history of non-human GB virus C in captive chimpanzees. J Gen Virol 92, 91–100 10.1099/vir.0.026088-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattermann J., Nischalke H. D., Kupfer B., Rockstroh J., Hess L., Sauerbruch T., Spengler U. (2003). Regulation of CC chemokine receptor 5 in hepatitis G virus infection. AIDS 17, 1457–1462 10.1097/00002030-200307040-00006 [DOI] [PubMed] [Google Scholar]
- NIH Webpage (2012). NIH Research Involving Chimpanzees: Notice Number NOT-OD-12–025. Available at: grants.nih.gov/grants/guide/notice-files/NOT-OD-12-025.html
- Polgreen P. M., Xiang J., Chang Q., Stapleton J. T. (2003). GB virus type C/hepatitis G virus: a non-pathogenic flavivirus associated with prolonged survival in HIV-infected individuals. Microbes Infect 5, 1255–1261 10.1016/j.micinf.2003.08.006 [DOI] [PubMed] [Google Scholar]
- Rey D., Vidinic-Moularde J., Meyer P., Schmitt C., Fritsch S., Lang J. M., Stoll-Keller F. (2000). High prevalence of GB virus C/hepatitis G virus RNA and antibodies in patients infected with human immunodeficiency virus type 1. Eur J Clin Microbiol Infect Dis 19, 721–724 10.1007/s100960000352 [DOI] [PubMed] [Google Scholar]
- Rydze R. T., Bhattarai N., Stapleton J. T. (2012). GB virus C infection is associated with a reduced rate of reactivation of latent HIV and protection against activation-induced T-cell death. Antivir Ther 17, 1271–1279 10.3851/IMP2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze-Zander C., Neibecker M., Othman S., Tural C., Clotet B., Blackard J. T., Kupfer B., Luechters G., Chung R. T. & other authors (2010). GB virus C coinfection in advanced HIV type-1 disease is associated with low CCR5 and CXCR4 surface expression on CD4+ T-cells. Antivir Ther 15, 745–752 10.3851/IMP1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons J. N., Pilot-Matias T. J., Leary T. P., Dawson G. J., Desai S. M., Schlauder G. G., Muerhoff A. S., Erker J. C., Buijk S. L., Chalmers M. L. (1995). Identification of two flavivirus-like genomes in the GB hepatitis agent. Proc Natl Acad Sci U S A 92, 3401–3405 10.1073/pnas.92.8.3401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton J. T. (2003). GB virus type C/hepatitis G virus. Semin Liver Dis 23, 137–148 10.1055/s-2003-39943 [DOI] [PubMed] [Google Scholar]
- Stapleton J. T., Foung S., Muerhoff A. S., Bukh J., Simmonds P. (2011). The GB viruses: a review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J Gen Virol 92, 233–246 10.1099/vir.0.027490-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillmann H. L., Heiken H., Knapik-Botor A., Heringlake S., Ockenga J., Wilber J. C., Goergen B., Detmer J., McMorrow M. & other authors (2001). Infection with GB virus C and reduced mortality among HIV-infected patients. N Engl J Med 345, 715–724 10.1056/NEJMoa010398 [DOI] [PubMed] [Google Scholar]
- Xiang J., Wünschmann S., Schmidt W. N., Shao J., Stapleton J. T. (2000). Full-length GB virus C (Hepatitis G virus) RNA transcripts are infectious in primary CD4-positive T cells. J Virol 74, 9125–9133 10.1128/JVI.74.19.9125-9133.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J., Wünschmann S., Diekema D. J., Klinzman D., Patrick K. D., George S. L., Stapleton J. T. (2001). Effect of coinfection with GB virus C on survival among patients with HIV infection. N Engl J Med 345, 707–714 10.1056/NEJMoa003364 [DOI] [PubMed] [Google Scholar]
- Xiang J., George S. L., Wünschmann S., Chang Q., Klinzman D., Stapleton J. T. (2004). Inhibition of HIV-1 replication by GB virus C infection through increases in RANTES, MIP-1α, MIP-1β, and SDF-1. Lancet 363, 2040–2046 10.1016/S0140-6736(04)16453-2 [DOI] [PubMed] [Google Scholar]
- Xiang J., McLinden J. H., Chang Q., Kaufman T. M., Stapleton J. T. (2006). An 85-aa segment of the GB virus type C NS5A phosphoprotein inhibits HIV-1 replication in CD4+ Jurkat T cells. Proc Natl Acad Sci U S A 103, 15570–15575 10.1073/pnas.0604728103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J., McLinden J. H., Chang Q., Jordan E. L., Stapleton J. T. (2008). Characterization of a peptide domain within the GB virus C NS5A phosphoprotein that inhibits HIV replication. PLoS ONE 3, e2580 10.1371/journal.pone.0002580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J., McLinden J. H., Rydze R. A., Chang Q., Kaufman T. M., Klinzman D., Stapleton J. T. (2009). Viruses within the Flaviviridae decrease CD4 expression and inhibit HIV replication in human CD4+ cells. J Immunol 183, 7860–7869 10.4049/jimmunol.0902276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J., McLinden J. H., Kaufman T. M., Mohr E. L., Bhattarai N., Chang Q., Stapleton J. T. (2012). Characterization of a peptide domain within the GB virus C envelope glycoprotein (E2) that inhibits HIV replication. Virology 430, 53–62 10.1016/j.virol.2012.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]