

Abstract
The M1 protein is a major structural protein that has multiple functions in various steps within the life cycle of the influenza A virus (IAV). However, little is currently known about the role of M1 in IAV replication in vivo and the associated pathogenesis. In this study, six isogenic H1N1 WSN33 viruses, constructed to express unique M1 proteins derived from various strains, subtypes or WSN33 itself, were tested to determine in vitro and in vivo functional exchangeability of M1 proteins in the replication and pathogenesis of the WSN33 virus. Despite five chimeric M1 viruses replicating to levels similar to those of the parental WSN33 virus in cell cultures, all M1 chimeras exhibited improved replication and enhanced virulence in mice when compared with the WSN33 virus. Interestingly, M1 proteins derived from swine viruses caused more severe clinical diseases than those from human or quail. These data indicate that the M1 protein is an important determinant of viral replication and pathogenic properties in mice, although the functions of M1 observed in vivo are not adequately reflected in simple infections of cultured cells. Chimeric M1 viruses that are variable in their clinical manifestations described here will aid future understanding of the role of M1 in IAV pathogenesis.
Introduction
The M1 protein of the influenza A virus (IAV) is the most abundant protein of the virion and forms a bridge-like membrane matrix that structurally and functionally connects the internal viral ribonucleoprotein (vRNP) core and external viral envelope proteins (Nayak et al., 2009). The M1 protein is instrumental in orchestrating the virus life cycle. This orchestration is believed to be achieved through M1-mediated interactions with other viral proteins, viral RNA, cellular proteins and lipid (Akarsu et al., 2003; Ali et al., 2000; Avalos et al., 1997; Bui et al., 2000; Chen et al., 2008; Demirov et al., 2012; Elster et al., 1997; Enami et al., 1994; Hirayama et al., 2004; Huang et al., 2009; Liu et al., 2009; Noton et al., 2007; Pal et al., 2011; Reinhardt & Wolff, 2000; Roberts et al., 1998; Ruigrok et al., 2000; Watanabe et al., 2006; Wu et al., 2011; Zhao et al., 1998; Zhirnov & Klenk, 1997). Despite mechanisms that are not yet fully understood, it is generally believed that during virus entry M1 must undergo low pH-induced structural transition in the endosomal compartment probably from high-order and stable oligomers to unstable M1 monomers (Bui et al., 1996; Calder et al., 2010; Fontana et al., 2012; Zhirnov, 1992). Disassociation of the M1 protein shell facilitates release of vRNPs to the cytoplasm so they can enter the nucleus and initiate viral genome transcription and replication (Bui et al., 1996; Martin & Helenius, 1991b). The newly synthesized M1 protein migrates to the nucleus and subsequently binds to vRNPs through the nuclear export protein, which directs nucleocytoplasmic transport of vRNP complexes (Akarsu et al., 2003; Bucher et al., 1989; Elton et al., 2001; Neumann et al., 2000; O’Neill et al., 1998; Yasuda et al., 1993). In addition, M1 present in the cytoplasm also interacts with vRNPs via viral RNA or nucleoprotein (Baudin et al., 2001; Elster et al., 1997; Noton et al., 2007; Wakefield & Brownlee, 1989; Watanabe et al., 1996; Ye et al., 1999). These interactions are found to inhibit viral transcription (Perez & Donis, 1998; Watanabe et al., 1996). This latter event converts IAV replication from viral genome transcription to virus assembly and budding. The role of M1 in the final stage of the virus life cycle is currently unclear, as recent studies have demonstrated that M1 is not required for budding of IAV virus-like particles (Chen et al., 2007; Wang et al., 2010). Despite this fact, the M1 protein plays an indisputable role in determining viral morphology. Its unique topology in the virion implicates M1 as a key player in coordinating discrete steps during the assembly and budding of IAV (Calder et al., 2010; Elleman & Barclay, 2004; Roberts et al., 1998).
In addition to its structural and regulatory roles, the M1 protein has also been shown to be involved in host adaptation, as well as contributing to the virulence of IAVs (Buckler-White et al., 1986; Fan et al., 2009; Furuse et al., 2009; Lee et al., 2001; McCullers et al., 2005; Murphy et al., 1989; Reid et al., 2002; Ward, 1995, 1996, 1997). For example, an earlier study reported that M1 contributed to restrict the replication of an avian IAV in monkeys (Buckler-White et al., 1986). A cluster of mutations, centring largely on the C-terminal domain of M1, have been implicated as determinants for advantageous growth in mice and subsequent acquisition of virulence (Brown et al., 2001; Govorkova et al., 2000). Perhaps the most significant finding that links M1 with viral pathogenesis was discovered during the study of the influenza B virus, where McCullers et al. (2005) demonstrated that a single amino acid change in the C-terminal domain of M1 conferred mouse adaptation and virulence of influenza B virus. Recent observations that the M segment from pandemic H1N1 (pH1N1) promotes efficient spread of reassortant viruses in animal models, including pig and guinea pig, seem to further support the essential role of M1 in the replication, transmission and pathogenesis of influenza viruses (Chou et al., 2011; Ducatez et al., 2011; Ma et al., 2012).
The M1 protein contains 252 aa and is the most conserved protein found in IAV. Despite their conserved nature, the M1 proteins of IAVs exhibit up to 20 % amino acid sequence variation (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html). Little is known about the role of M1-associated sequence variation in IAV replication and virulence. In this study, we constructed six isogenic recombinant influenza H1N1 WSN33 viruses that differed only in their M1 proteins, which were derived from various strains of different subtypes or originated from WSN33 itself. The chimeric M1 viruses were examined in detail in terms of replication kinetics in cell cultures as well as replication and virulence properties in a mouse model. Results from our experiments showed that despite similar levels of replication in vitro, the chimeric viruses exhibited different patterns of replication and virulence in mice. Thus, these observations suggest in vivo functions of different M1 proteins that are not adequately reflected in simple infections of culture cells, and provide compelling evidence that the M1 protein of IAV is a determinant of viral replication and virulence properties in experimentally infected mice.
Results
Generation of wild-type (WT), recombinant M-segment and chimeric M1 WSN33 viruses
Two groups of viruses, in addition to WT WSN33, were generated to address the impact on the replication of IAV, resulting from functional replacement of different M1 proteins. The first group involved substitutions of the WSN33 M segment with its counterparts from the following viruses: influenza A/Swine/Iowa/15/30 (H1N1), A/Swine/Missouri/4296424/06 (H2N3), A/Swine/Texas/4199-2/98 (H3N2), A/California/04/09 (H1N1) and A/Quail/Hong Kong/G1/97 (H9N2). The second group consisted of six isogenic WSN viruses, differing only in their M1 proteins, which were derived from the viruses in the first group. To generate the second group of viruses, a PCR-ligation-PCR technique was used to introduce M1 coding sequences from other viruses to the parental WSN33 M segment in a way that did not alter its M2 coding sequence (Fig. 1) (Chen et al., 2001; Li et al., 2002). Stocks of these viruses were generated by reverse genetics and titrated in Madin–Darby canine kidney (MDCK) cells by plaque assay as described in Methods.
Fig. 1.

Construction of chimeric M segments. (a) Genomic structure of M segment of influenza A/WSN/33 virus (adapted and modified from Palese & Shaw, 2007). Numbering of the nucleotide positions (and in b) is based on the M1 coding sequence of WSN33 virus. Nucleotide positions 1 and 759 correspond, respectively, to the first and last nucleotides coding for M1 ORF. Nucleotides at positions 26 and 715 are splicing donor and acceptor sites, respectively, to generate M2 mRNA (intron is indicated by the V-shaped line). Nucleotide position 982 is the last coding nucleotide for M2 protein. (b) The overlapped coding sequences of M1 and M2 and the splicing site of M2. Nucleotides 715–759 are used to encode the amino acids for both M1 and M2, through a RNA splicing mechanism (Palese & Shaw, 2007). The most C-terminal 13 aa of M1 encoded by nucleotides 716–759 (bold) and the most N-terminal 9 aa of M2 encoded by nucleotides 1–26 and 715 (underlined) are identical among the six different M segments used in this study. The nucleotide at position 715 and upstream of the WSN33 M segment were replaced with its counterpart from each of these five viruses: A/swine/Iowa/15/30 (H1N1), A/Swine/Missouri/4296424/06 (H2N3), A/Swine/Texas/4199-2/98 (H3N2), A/California/04/09 (H1N1) and A/Quail/Hong Kong/G1/97 (H9N2) using the standard PCR and cloning technique, respectively, which does not alter the M2 amino acids of WSN33.
In vitro characterization of recombinant M-segment and chimeric M1 WSN33 viruses
Fig. 2 summarizes the growth kinetics experiments of WT and mutant WSN33 viruses. In general, all recombinant M-segment and chimeric M1 viruses replicated in a similar fashion as the WT rWSN in infected MDCK cells with the notable exception of rWSN-HK97M1. rWSN-HK97M1 displayed a 36 h lag relative to WT and other M1 chimeric viruses and appeared not to be able to reach a WT-like peak level of virus replication observed during the time-course of the experiments. Despite this difference, our results collectively suggested that the WSN33 M segment or M1 protein can be functionally replaced by its counterparts from other IAVs in vitro. Considering that the focus of this study was on the M1 protein, we further characterized in vivo replication and virulence properties of WT WSN33 and chimeric M1 viruses.
Fig. 2.
Replication kinetics of WT rWSN33, recombinant M-segment and chimeric M1 viruses in MDCK cells. The cultured MDCK cells were infected with either WT rWSN33 or the indicated recombinant M-segment (a) or chimeric M1 (b) viruses using an m.o.i. of 0.001. Virus replication following infection of MDCK cells was monitored every 12 h over a period of 3 days by measuring virus infectivity (p.f.u. ml−1) in the culture supernatant. The data shown represents the means±sd of two independent experiments.
In vivo functional exchangeability of M1 proteins in influenza WSN33 virus replication
We used a mouse model to study replication and pathogenicity of isogenic WSN33 viruses differing only in M1 proteins. Six groups, each consisting of 13 mice, were inoculated intranasally with 5×103 p.f.u. of rWSN, rWSN-IA30M1, rWSN-HK97M1, rWSN-TX98M1, rWSN-MO06M1 and rWSN-CA09M1. In the control group, 13 mice were inoculated with virus-free minimal essential medium (MEM) by the same intranasal route. We selected this particular infection dose because we speculated that a low viral inoculum in mice could maximize an animal’s sensitivity to virus replication and the associated virulence, allowing slight changes in M1 protein to be detected by changes in virulence in mice.
rWSN-infected mice did not show clear clinical symptoms and only displayed a slight weight loss starting at day 7 post-infection (p.i.); however, all the mice regained the weight by day 11 p.i. (Fig. 3a). None of mice in this group succumbed to infection over a period of 14 days p.i. Modest levels of disease may have been caused by a relative low dose of virus inoculum (i.e. 5×103). Inoculation of mice with similar doses of WSN33 virus resulting in neither detectable weight loss nor death has been previously reported in a recent study (Garcia et al., 2010). In marked contrast, all the mice in our study inoculated with M1 chimeras exhibited substantial weight loss starting at day 4 p.i., and visible clinical symptoms including ruffled fur, lethargy, anorexia and dyspnoea were observed. Despite being significantly pathogenic in mice, the five M1 chimeric viruses behaved differently in conferring virulence in the mice, measured by survival rate (Fig. 3b). Among these, rWSN-IA30M1 and rWSN-TX98M1 appeared to be more virulent, as all the mice inoculated with these two viruses succumbed to infection or reached the experimental end point by days 10 and 12 p.i., respectively. rWSN-MO06M1 also exhibited relatively high virulence as it caused approximately 80 % mortality of mice. Two chimeric rWSN-HK97M1 and rWSN-CA09M1 viruses seemed to possess a moderate virulence as approximately 40–55 % of infected mice survived their infections over the 14 day observation period. It is interesting to note that the mice study did not reproduce an attenuated phenotype of rWSN-HK97M1 as demonstrated in MDCK cells (Fig. 2b) (compared with the WT WSN33). Despite an unknown mechanism, we suspect that the rWSN-HK97M1 protein may not interact with its cellular targets in MDCK cells as efficiently as those in mice, which are required for optimal virus replication. Taken together, these data demonstrate an M1-dependent virulence phenomenon in mice, highlighting the important role of the M1 protein in IAV pathogenesis.
Fig. 3.
Body weight changes (a) and survival rates (b) in mice infected with the WT rWSN and chimeric M1 viruses. Mice were infected intranasally with 5×103 p.f.u. of the viruses indicated or mock infected with 50 µl MEM. Body weights were monitored daily up to 14 days p.i. Mice were euthanized after losing 25 % of their initial body weight.
The severity of influenza disease has been correlated closely with the levels of virus replication in experimentally infected mice. Therefore, we closely monitored the levels of virus replication in mouse lungs by TCID50 ml−1 analysis in MDCK cells. Three mice from each group were sacrificed on both days 3 and 5 p.i., in this study. Notably, all mice inoculated with the five chimeric M1 viruses had significantly higher lung viral titres than those infected with the WSN33 virus by approximately 1 or 2 logs at both time points (Fig. 4a). This finding suggests a reasonable association between the level of virus replication and disease severity in a mouse model of IAV infection. However, we did not observe a strict correlation between these two parameters, particularly among mice inoculated with M1 chimeric viruses. For example, mean viral titre values in mice inoculated with rWSN-MO06M1 were found to be 1 log higher when compared with rWSN-IA30M1 on day 3 p.i.; however, rWSN-MO06M1 caused less mortality than rWSN-IA30M1 and similar discordance was also observed in other virus groups.
Fig. 4.
Viral titres of homogenized lung tissues (a) and microscopic lung lesions (b) of mice infected with the WT rWSN and chimeric M1 viruses on 3 and 5 days p.i. The values are means±sem from each group (* P<0.05; ** P<0.01; *** P<0.001).
To further evaluate the pathogenesis of isogenic WSN33 viruses differing only in the M1 protein, we performed histopathological examination of infected mice lungs. The WT rWSN and chimeric M1 viruses induced pneumonia in infected mouse lungs (Figs 4b and 5). As shown in Fig. 4(b), the severity of lung lesions determined by mean histological lesion scores as described previously (Liu et al., 2011) increased with time, suggesting progressive disease development due to virus replication. Overall, mice inoculated with M1 chimeric viruses developed histological lesion scores higher than that of the WT WSN33 inoculated group. Significant differences in histological lung lesions were observed between the WT rWSN and three M1 chimeric viruses (rWSN-CA09M1, rWSN-MO06M1 and rWSN-HK97M1) on day 3 p.i., and between the rWSN and three M1 chimeric (rWSN-CA09M1, rWSN-IA30M1 and rWSN-HK97M1) viruses on day 5 p.i. (Fig. 4b). As noted in virus load analysis, there was no absolute correlation between lung lesion score and virulence among mice infected with M1 chimeras. For example, the rWSN-HK97M1 induced higher lung lesions in mouse lungs rather than rWSN-TX98M1, but it caused less mortality than the latter. Taken together, all these results demonstrate that M1 chimeric viruses gain better replication fitness and are more virulent in mice as compared with the WT WSN33.
Fig. 5.
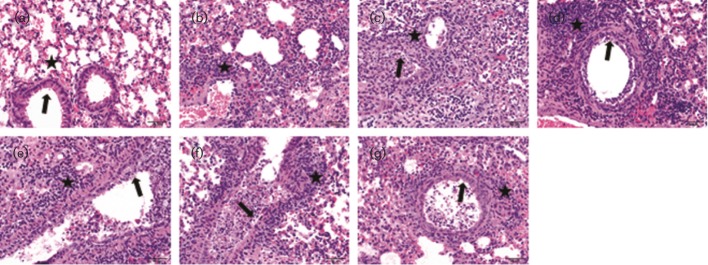
Haematoxylin and eosin staining of microscopic lung sections from mice infected with WT rWSN and M1 chimeric viruses on 5 days p.i. (a) Control: the bronchioles are lined by normal cuboidal epithelium (arrow) and the alveoli are clear (asterisk) in control mice. (b) WT rWSN33 virus: mild bronchiolar epithelial necrosis was seen and the alveolar interstitium is infiltrated by moderate numbers of lymphocytes and neutrophils (asterisk). Chimeric M1 viruses including (c) rWSN-CA09M1, (d) rWSN-IA30M1, (e) rWSN-MO06M1, (f) rWSN-TX98M1 and (g) rWSN-HK97M1: moderate bronchiolar epithelial degeneration and necrosis is visible (arrow) with sloughing of necrotic cells in the lumen. The peribronchiolar and interstitial areas are expanded by moderate numbers of lymphocytes and neutrophils (asterisk). Bars, 50 µm.
Analysis of viral protein expression in chimeric and WT WSN33 viruses
In order to further determine whether chimeric viruses behave similarly to WT WSN33 virus in biological and biochemical properties, we next characterized these viruses for M1, haemagglutinin (HA) and neuraminidase (NA) protein expression. We infected MDCK cells with 1 m.o.i. of WT rWSN and M1 chimeras, respectively, and purified virions from culture supernatants collected 12 and 24 h p.i. Virions were analysed for M1 protein expression, HA activity and NA function. We chose 1 m.o.i. inoculum over 0.001 m.o.i. used for generation of growth kinetics data because the latter could not allow us to detect viral protein expression at 12 and 24 h following virus infection.
For M1 protein detection and quantification by Western blot assay, the expression level of WT rWSN M1 protein was arbitrarily set at 100 % and the M1 level for each chimeric virus was normalized by comparison with WT rWSN (Fig. 6a, b). At 12 h p.i., all chimeras except for rWSN-HK97M1 exhibited nearly identical amounts of virion-associated M1 as that observed in WT rWSN. The rWSN-HK97M1 had slightly less M1 protein, being about 20 % that of the WT rWSN. The M1 reduction in virions of rWSN-HK97M1 seemed to correlate with reduced accumulation of intracellular M1 protein. This trend continued at 24 h p.i. At this time point, the amount of M1 protein in the rWSN-HK97M1 virions was increased and exhibited only about 10 % reduction relative to the WT rWSN.
Fig. 6.

Analysis of M1 and NA proteins. (a, b) M1 protein expression levels in infected MDCK cells and virions harvested at 12 and 24 h p.i. M1 protein bands (resolved by SDS-PAGE and Western blot assay) were analysed by ImageJ software (http://rsbweb.nih.gov/ij/) to quantify the percentage of M1 protein of respective chimeric virus relative to that of WT rWSN virus. The expression level of WT rWSN M1 protein was arbitrarily set at 100 % and the M1 level for each chimeric virus was normalized by comparison with WT rWSN. Data shown are means±sd for three independent experiments. (c) Virion-associated NA activity. MDCK monolayers were infected with chimeric and WT rWSN viruses at a dose of 1 m.o.i. Supernatants were collected at 12 and 24 h p.i., and clarified with low-speed centrifugation. Clarified supernatants were reacted with substrate 2′-(4-methylumbelliferyl)-α-N-acetylneuraminic acid sodium salt hydrate (MUNANA), and relative fluorescence units (RFU) were measured. Data shown are means±sd for three independent experiments.
We used a traditional haemagglutination assay to quantify the amount of HA protein in virions and also employed a fluorescence-based enzymic assay to measure the function of NA protein. As demonstrated in Table 1 and Fig. 6(c), all chimeric viruses exhibited WT-like HA and NA activities at two time points with the exception of rWSN-HK97M1. This virus expressed NA protein very similar to WT rWSN but showed reduced HA titres: 1 log2 and 2 log2 lower relative to WT rWSN at 12 and 24 h p.i.
Table 1. HA titres of clarified supernatants (log2).
MDCK monolayers were infected with chimeric and WT viruses at a dose of 1 m.o.i., supernatants were collected at 12 and 24 h p.i. and clarified with low-speed centrifugation (2000 r.p.m. for 10 min). The HA titre of clarified supernatants were measured using 0.5 % turkey red blood cells. Data shown are means±sd for three independent experiments.
| Virus | Time p.i. (h) | |
| 12 | 24 | |
| rWSN-IA30M1 | 5.67±0.58 | 6.67±0.58 |
| rWSN-CA09M1 | 5.67±0.58 | 6.67±0.58 |
| rWSN-MO06M1 | 6.00±0.00 | 7.67±0.58 |
| rWSN-TX98M1 | 6.00±0.00 | 7.67±0.58 |
| rWSN-HK97M1 | 4.67±0.58 | 5.67±0.58 |
| rWSN | 6.0±0.0 | 7.67±0.58 |
Results of our experiments demonstrated that all M1 chimeric viruses are similar to WT rWSN with regards to the expression of M1, NA and HA proteins, which is in good agreement with our in vitro cell-culture-based replication experiment. In replication kinetics we noted that rWSN-HK97M1 virus was slightly attenuated in comparison with WT rWSN and other chimeric viruses and this moderate attenuation is probably due to the reduction of M1 and HA proteins in virus particles.
Discussion
Studies described here reveal a novel M1-dependent replication and virulence phenomenon of influenza A WSN33 H1N1 virus in a mammalian host. Thus, the results of our experiments suggest that the M1 protein is an important determinant for viral replication and pathogenic properties in vivo. These findings maintain a theme established by previous studies concerning a role of the M1 protein in IAV replication and pathogenesis (Buckler-White et al., 1986; Fan et al., 2009; McCullers et al., 2005; Ward, 1995, 1997). The differences observed between the in vitro and in vivo phenotypes of the M1 chimeric viruses used in this study are interesting, and emphasize the importance of evaluating protein function under in vivo conditions, as in vitro cell culture assays may not detect critical in vivo properties of influenza viral proteins such as M1.
The M1 protein of IAV is a primary determinant for viral morphogenesis (Calder et al., 2010; Elleman & Barclay, 2004; Roberts et al., 1998). It should be noted that we have not performed electron microscopy studies to define the morphology of these viruses, mainly due to our observation that these recombinant viruses replicated similarly to the WT virus in MDCK cells. It is likely that these viruses have a similar morphology. However, it is still possible that the described viruses may exhibit different morphologies including viral budding efficiency. Notable differences in viral morphogenesis contributing to viral replication might not be measurable in cell culture but can be detected and amplified in mice. Future study towards this aspect should help to define the molecular mechanism of M1-associated virulence.
It is believed that higher levels of viral replication are associated with more severe influenza disease. Results of our experiments in this study supported this established concept. For example, mice inoculated with chimeric M1 viruses showed more severe clinical symptoms compared with mice inoculated with the WSN33 virus. Additionally, the mice infected with the chimeric viruses produced virus at a level about 1 log higher than the mice infected with the WSN33 virus. However, our data also suggested that the relationship between disease severity and viral load is not absolute in all cases. For example, mice infected by M1 chimeric viruses had similar viral loads (measured at days 3 and 5 p.i.) but resulted in variable disease progression. Studying the contribution of cytokines, chemokines, and infiltration of immune cells, in addition to virus load, to clinical disease progression in the near term may provide some insights to the observed discordance.
The M1 protein of IAV has three distinctive domains (N, M and C) and certain functions have been assigned to each individual domain (Akarsu et al., 2003; Bui et al., 2000; Burleigh et al., 2005; Cao et al., 2012; Das et al., 2012; Demirov et al., 2012; Elleman & Barclay, 2004; Elster et al., 1997; Harris et al., 2001; Hui et al., 2006; Liu & Ye, 2005; Martin & Helenius, 1991a; Okada et al., 2003; Perez & Donis, 1998; Ye et al., 1995). Through comparative analysis of the six M1 amino acid sequences used in this study (Fig. 7), we found that these M1 proteins show up to an 11 % sequence divergence. We also noticed that unique variations between WSN33 M1 and other M1 proteins occur only at five positions, 41, 115, 126, 137 and 204 (Fig. 7). M1 position 41, located in the M1 N domain, has been linked to a role in determining virion morphology (Elleman & Barclay, 2004; Roberts et al., 1998). Early studies have shown that an A to V change at this position resulted in a loss in filamentous phenotype (Elleman & Barclay, 2004). It remains unclear whether the other four positions are involved in virus replication and virulence, as none of these residues are located in a known functional domain of the M1 protein. Nevertheless, large-scale phylogenetic analyses of M1 sequences of IAVs from different subtypes or different hosts suggest that these positions, particularly 115, 126 and 137 located in the M1 M domain, might be involved in host adaptation and virulence (Furuse et al., 2009; Lam et al., 2008; Reid et al., 2002). Addressing the impact of amino acid variations at these positions in the M1 protein on the observed disease enhancement poses great potential as a focus point in future study. In addition to these five positions, we also observed sequence variations in other locations among five M1 proteins introduced to WSN33 virus (data not shown). Investigating these mutations in the future is also important because it will help to elucidate the genetic basis of clinical disease progression rates in mice as observed among chimeric M1 viruses.
Fig. 7.

Alignment of the complete amino acid sequences of M1 proteins used in this study. Identical residues (*), highly conserved residues (:), and less conserved residues (.) are indicated. Note that amino acid variations at M1 positions 41, 115, 126, 137 and 204 only occur in WSN33 M1, not in the other five M1, and are marked by bold highlighting.
The M1 protein of IAV and M1-like proteins present in other enveloped RNA viruses are able to interact with both the internal core protein complex and the external viral envelope proteins due to their unique topology in the virion (Nayak et al., 2009; Rossman & Lamb, 2011). In this regard, the primary role of these proteins is to act as a bridge to structurally and functionally coalesce various components into budding virions through mechanisms that are largely unknown to date (Nayak et al., 2009; Rossman & Lamb, 2011). It has been clearly shown that the matrix protein of vesicular stomatitis virus (VSV) also plays a key role in evading the host innate immune response (Faul et al., 2009). The ability of VSV M protein to inhibit the host alpha interferon/beta interferon response is genetically separable from its role in virus structure and assembly (Black et al., 1993). An interesting thought for the future would be to design similar experiments to address whether the M1 protein of IAV can inhibit host innate immunity. If this is found to be true, could amino acid variations at the five aforementioned positions of the M1 protein impact M1’s ability in overcoming host innate immune response that may result in different clinical outcomes? Future elucidation of these questions will provide insights into the pathobiology of IAV M1 protein.
Methods
Cells, virus and animals.
MDCK and 293T human embryonic kidney cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10 % FBS at 37 °C with 5 % CO2. Influenza virus A/WSN/33 (H1N1) was generated by using the reverse genetic system that contains eight plasmids (pHW181-PB2, pHW182-PB1, pHW183-PA, pHW184-HA, pHW185-NP, pHW186-NA, pHW187-M and pHW188-NS), which was kindly provided by E. Hoffmann, St Jude Children’s Research Hospital. Virus was propagated in MDCK cells at an m.o.i. of 0.001. Six-week-old female BALB/c mice were purchased from Charles River Farms.
RT-PCR and sequencing.
Viral RNA was extracted from collected cell-culture supernatants with the QIAamp viral RNA kit (Qiagen). Purified RNA was treated with DNase I (Ambion) at 37 °C for 30 min for complete removal of potential plasmid DNA contamination followed by treatment with DNase inactivation reagent according to the manufacturer’s instructions. Following purification and treatment, RNA was reverse-transcribed and its corresponding cDNA amplified with primers using the OneStep RT-PCR kit (Invitrogen) and the amplification products were used for sequencing.
Plasmid constructions.
To generate recombinant M-segment viruses in the backbone of the WSN33 virus, the M segments from IAVs were cloned into the plasmid pHW2000 as described previously (Hoffmann et al., 2000a, b). These viruses included: A/swine/Iowa/15/30 (H1N1), A/Swine/Missouri/4296424/06 (H2N3), A/Swine/Texas/4199-2/98 (H3N2), A/California/04/09 (H1N1) and A/Quail/Hong Kong/G1/97 (H9N2). The resulting plasmids were named pHW-IA30M, pHW-TX98M, pHW-MO06M, pHW-HK97M and pHW-CA09M. To generate chimeric M1 segments, we used standard PCR and cloning methods. We generated M1 substitution constructs by replacing the native M1 coding sequence in the WSN33 M segment with each of the five sequences encoding M1 from other IAVs as listed above. The coding sequence for the WSN33 M2 was not altered in these chimeric M segments, which were designated pHW-IA30M1+WSN-M2, pHW-HK97M1+WSN-M2, pHW-TX98M1+WSN-M2, pHW-MO06M1+WSN-M2 and pHW-CA09M1+WSN-M2, respectively. Primers and protocols for generation of these constructs will be provided upon request.
Production of infectious IAVs by reverse genetics.
WT A/WSN/33, recombinant M-segment and chimeric M1 viruses were produced by the reverse genetic technique as described previously (Hoffmann et al., 2000b). Briefly, to rescue the WT WSN virus, 293T and MDCK cells were co-cultured at approximately 70 % confluence in 12-well plates and transfected with a mixture of eight plasmids (pHW181-PB2, pHW182-PB1, pHW183-PA, pHW184-HA, pHW185-NP, pHW186-NA, pHW187-M and pHW188-NS), which were prepared in Trans-LT1 (Mirus) and Opti-MEM I medium according to the manufacturer’s instructions. Recombinant M-segment and chimeric M1 viruses were generated in the same way but substituting the pHW187-M with the plasmid containing the M or chimeric M1 segment as mentioned in the plasmid constructions’ section. The mixtures were incubated at room temperature for 45 min before they were added to the cells. After 6 h culture, transfection mixtures were replaced with Opti-MEM I medium followed by an additional 18 h incubation at 37 °C. Then, Opti-MEM I medium containing tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin (Sigma-Aldrich) at a concentration of 1 µg ml−1 was added to each well. At 72 h post-transfection, viral supernatants were collected, centrifuged at 2000 r.p.m. (Sorvall Legend Mach 1.6R, rotor 75003348) for 10 min, aliquoted and stored at −80 °C until further analysis. The resulting viruses were the WT (rWSN), recombinant M (rWSN-IA30M, rWSN-HK97M, rWSN-TX98M, rWSN-MO06M and rWSN-CA09M) and chimeric M1 (rWSN-IA30M1, rWSN-HK97M1, rWSN-TX98M1, rWSN-MO06M1 and rWSN-CA09M1) viruses and were confirmed by sequencing.
Replication kinetics and plaque assay.
Studies on viral replication kinetics were performed on a monolayer of MDCK cells using an inoculum of 0.001 m.o.i. Viral samples were collected at 0, 24, 48 and 72 h p.i., respectively. After centrifugation at 2000 r.p.m. (Sorvall Legend Mach 1.6R, rotor 75003348) for 10 min to remove cellular debris, samples were stored at −80 °C until they were titrated by plaque assay.
The conventional plaque assay was performed to measure viral infectivity as described previously (Burleigh et al., 2005). Confluent MDCK cells grown in 12-well plates were infected with 0.5 ml of 10-fold series dilutions of viral samples. After 1 h incubation at 37 °C, unabsorbed viruses were removed by washing with PBS, and infected monolayers were overlaid with 1.5 ml of overlay medium containing 2× DMEM supplemented with 0.3 % BSA, 2 µg TPCK-trypsin ml−1, and 1.8 % agar. After 3 days incubation at 37 °C, the overlay was removed and cells were fixed and stained with 0.1 % crystal violet (Acros; dissolved in 10 % formaldehyde solution) for 30 min at room temperature. The plates were gently rinsed with water to remove excess stain followed by air-drying for 10 min at room temperature. Titres (p.f.u.) were calculated by the counting the number of plaques and multiplying this value by the reciprocal of the dilution factor.
SDS-PAGE and Western blot analysis.
MDCK cells were inoculated with 1 m.o.i. of rWSN-IA30M1, rWSN-CA09M1, rWSN-MO06M1, rWSN-TX98M1, rWSN-HK97M1 and rWSN, respectively. Viral samples were collected at 12 and 24 h p.i. After brief centrifugation at 2000 r.p.m. (Sorvall Legend Mach 1.6R, rotor 75003348) for 10 min to remove cellular debris, clarified supernatants were taken out for haemagglutination and NA activity assays. The remaining samples were filtered and concentrated by 40-fold through ultracentrifugation (30 000 r.p.m. for 2 h; Beckman L-60, rotor SW41). Cell lysates from infected MDCK cells (12 and 24 h p.i.) were prepared with cell lysis buffer.
Concentrated virions and cell lysates were separated by 12 % SDS-PAGE and transferred to a nitrocellulose membrane. After blocking, the membranes were incubated with mouse anti-influenza A matrix protein (AbD Serotec) followed by hybridization with goat anti-mouse IgG–HRP (Sigma). The protein bands were visualized on film using an ECL system (Pierce). The M1 bands were analysed by ImageJ software (http://rsbweb.nih.gov/ij/) to quantify the percentage of M1 protein expression level of respective chimeric virus relative to WT rWSN.
HA and NA activity assay.
NA activity was measured by using the standard fluorescent assay. The clarified supernatant and fluorogenic substrate MUNANA (Sigma) were added to a 96-well plate, mixed and incubated at 37 °C for 1 h. After stopping the reaction by adding 50 mM glycine buffer, fluorescence was measured in a fluorometer with an excitation wavelength of 355 nm and an emission wavelength of 460 nm, and relative fluorescent units (RFUs) were recorded. A micro-haemagglutination assay (WHO. Manual for the laboratory diagnosis and virological surveillance of influenza. 2011, http://www.who.int/influenza/resources/document/manual diagnosis surveillance influenza/en/index.html/ was used to detect the HA titre of viruses with 0.5 % turkey red blood cells.
Mouse study.
Animal research was conducted in a Biosafety Level 2 animal facility under the guidance of Kansas State University’s Institutional Animal Care and Use Committee. Six-week-old female BALB/c mice (Charles River Laboratories) were randomly divided into six groups (13 mice per group). Mice were anaesthetized with isoflurane USP (Phoenix Pharmaceutical) and intranasally inoculated with a 50 µl volume containing either virus-free MEM (i.e. negative control) or with 5×103 p.f.u. of each of six isogenic influenza H1N1 WSN33 viruses differing only in the M1 proteins: rWSN (with native M1), rWSN-IA30M1, rWSN-HK97M1, rWSN-TX98M1, rWSN-MO06M1 and rWSN-CA09M1. Mice were monitored daily for weight loss, survival and general health status for a period of 14 days. After the onset of disease, general health status was recorded twice daily. Mice with a weight loss of more than 25 % of the initial body weight prior to virus infection were euthanized and recorded as dead. Three mice from each group were euthanized at day 3 and 5 p.i., respectively, and all other mice were euthanized at day 14 p.i. During necropsy, the right lung was collected and stored at −80 °C for later analysis, and the left lung was fixed in 10 % formalin for histopathologic assessment (Liu et al., 2011). Haematoxylin and eosin staining was performed as the routine procedure (Sheehan & Hrapchak, 1980). Virus detection was performed in a 10 % tissue homogenate in PBS that had been homogenized twice for 1 min in a Mini BeadBeater-8 (Biospec Products). The homogenate was centrifuged at 640 g for 5 min, and the supernatant was transferred to 1.5 ml reaction tubes for virus isolation and titration in MDCK cells as described previously (Richt et al., 2003).
Statistical analysis.
Virus titres, weight loss, microscopic lung lesion scores and mortality were analysed by using analysis of variance (ANOVA) in GraphPad Prism version 5.0 (GraphPad software); a P value of 0.05 or less was considered significant. Those response variables shown to have a significant effect by treatment group were subjected to comparisons for all pairs by using the Tukey–Kramer test. Pair-wise mean comparisons between inoculated and control groups were made using the Student’s t-test.
Acknowledgements
We thank Haixia Liu, Darlene Sheffer, Jingqun Ma, Jingjiao Ma and Jolene Carlson for helping with the animal study. This work was partially supported by South Dakota State University AES 3AH-203, South Dakota 2010 Biological Control and Analysis by Applied Photonics (BCAAP) Fund and NIH 5K02AI076125-02 award to F. L.; and by the European Commission FP7-GA258084 and Kansas State University Start-up SRO001 to W. J. M.; and by NIH under contract numbers HHSN266200700005C to J. A. R.
References
- Akarsu H., Burmeister W. P., Petosa C., Petit I., Müller C. W., Ruigrok R. W., Baudin F. (2003). Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2). EMBO J 22, 4646–4655 10.1093/emboj/cdg449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A., Avalos R. T., Ponimaskin E., Nayak D. P. (2000). Influenza virus assembly: effect of influenza virus glycoproteins on the membrane association of M1 protein. J Virol 74, 8709–8719 10.1128/JVI.74.18.8709-8719.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos R. T., Yu Z., Nayak D. P. (1997). Association of influenza virus NP and M1 proteins with cellular cytoskeletal elements in influenza virus-infected cells. J Virol 71, 2947–2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin F., Petit I., Weissenhorn W., Ruigrok R. W. (2001). In vitro dissection of the membrane and RNP binding activities of influenza virus M1 protein. Virology 281, 102–108 10.1006/viro.2000.0804 [DOI] [PubMed] [Google Scholar]
- Black B. L., Rhodes R. B., McKenzie M., Lyles D. S. (1993). The role of vesicular stomatitis virus matrix protein in inhibition of host-directed gene expression is genetically separable from its function in virus assembly. J Virol 67, 4814–4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E. G., Liu H., Kit L. C., Baird S., Nesrallah M. (2001). Pattern of mutation in the genome of influenza A virus on adaptation to increased virulence in the mouse lung: identification of functional themes. Proc Natl Acad Sci U S A 98, 6883–6888 10.1073/pnas.111165798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher D., Popple S., Baer M., Mikhail A., Gong Y. F., Whitaker C., Paoletti E., Judd A. (1989). M protein (M1) of influenza virus: antigenic analysis and intracellular localization with monoclonal antibodies. J Virol 63, 3622–3633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler-White A. J., Naeve C. W., Murphy B. R. (1986). Characterization of a gene coding for M proteins which is involved in host range restriction of an avian influenza A virus in monkeys. J Virol 57, 697–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui M., Whittaker G., Helenius A. (1996). Effect of M1 protein and low pH on nuclear transport of influenza virus ribonucleoproteins. J Virol 70, 8391–8401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui M., Wills E. G., Helenius A., Whittaker G. R. (2000). Role of the influenza virus M1 protein in nuclear export of viral ribonucleoproteins. J Virol 74, 1781–1786 10.1128/JVI.74.4.1781-1786.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burleigh L. M., Calder L. J., Skehel J. J., Steinhauer D. A. (2005). Influenza a viruses with mutations in the m1 helix six domain display a wide variety of morphological phenotypes. J Virol 79, 1262–1270 10.1128/JVI.79.2.1262-1270.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calder L. J., Wasilewski S., Berriman J. A., Rosenthal P. B. (2010). Structural organization of a filamentous influenza A virus. Proc Natl Acad Sci U S A 107, 10685–10690 10.1073/pnas.1002123107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S., Liu X., Yu M., Li J., Jia X., Bi Y., Sun L., Gao G. F., Liu W. (2012). A nuclear export signal in the matrix protein of Influenza A virus is required for efficient virus replication. J Virol 86, 4883–4891 10.1128/JVI.06586-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Li F., Montelaro R. C. (2001). Functional roles of equine infectious anemia virus Gag p9 in viral budding and infection. J Virol 75, 9762–9770 10.1128/JVI.75.20.9762-9770.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B. J., Leser G. P., Morita E., Lamb R. A. (2007). Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J Virol 81, 7111–7123 10.1128/JVI.00361-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B. J., Leser G. P., Jackson D., Lamb R. A. (2008). The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J Virol 82, 10059–10070 10.1128/JVI.01184-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou Y. Y., Albrecht R. A., Pica N., Lowen A. C., Richt J. A., García-Sastre A., Palese P., Hai R. (2011). The M segment of the 2009 new pandemic H1N1 influenza virus is critical for its high transmission efficiency in the guinea pig model. J Virol 85, 11235–11241 10.1128/JVI.05794-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S. C., Watanabe S., Hatta M., Noda T., Neumann G., Ozawa M., Kawaoka Y. (2012). The highly conserved arginine residues at positions 76 through 78 of influenza A virus matrix protein M1 play an important role in viral replication by affecting the intracellular localization of M1. J Virol 86, 1522–1530 10.1128/JVI.06230-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirov D., Gabriel G., Schneider C., Hohenberg H., Ludwig S. (2012). Interaction of influenza A virus matrix protein with RACK1 is required for virus release. Cell Microbiol 14, 774–789 10.1111/j.1462-5822.2012.01759.x [DOI] [PubMed] [Google Scholar]
- Ducatez M. F., Hause B., Stigger-Rosser E., Darnell D., Corzo C., Juleen K., Simonson R., Brockwell-Staats C., Rubrum A. & other authors (2011). Multiple reassortment between pandemic (H1N1) 2009 and endemic influenza viruses in pigs, United States. Emerg Infect Dis 17, 1624–1629 10.3201/eid1709.110338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elleman C. J., Barclay W. S. (2004). The M1 matrix protein controls the filamentous phenotype of influenza A virus. Virology 321, 144–153 10.1016/j.virol.2003.12.009 [DOI] [PubMed] [Google Scholar]
- Elster C., Larsen K., Gagnon J., Ruigrok R. W., Baudin F. (1997). Influenza virus M1 protein binds to RNA through its nuclear localization signal. J Gen Virol 78, 1589–1596 [DOI] [PubMed] [Google Scholar]
- Elton D., Simpson-Holley M., Archer K., Medcalf L., Hallam R., McCauley J., Digard P. (2001). Interaction of the influenza virus nucleoprotein with the cellular CRM1-mediated nuclear export pathway. J Virol 75, 408–419 10.1128/JVI.75.1.408-419.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enami K., Sato T. A., Nakada S., Enami M. (1994). Influenza virus NS1 protein stimulates translation of the M1 protein. J Virol 68, 1432–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan S., Deng G., Song J., Tian G., Suo Y., Jiang Y., Guan Y., Bu Z., Kawaoka Y., Chen H. (2009). Two amino acid residues in the matrix protein M1 contribute to the virulence difference of H5N1 avian influenza viruses in mice. Virology 384, 28–32 10.1016/j.virol.2008.11.044 [DOI] [PubMed] [Google Scholar]
- Faul E. J., Lyles D. S., Schnell M. J. (2009). Interferon response and viral evasion by members of the family Rhabdoviridae. Viruses 1, 832–851 10.3390/v1030832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana J., Cardone G., Heymann J. B., Winkler D. C., Steven A. C. (2012). Structural changes in Influenza virus at low pH characterized by cryo-electron tomography. J Virol 86, 2919–2929 10.1128/JVI.06698-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse Y., Suzuki A., Kamigaki T., Oshitani H. (2009). Evolution of the M gene of the influenza A virus in different host species: large-scale sequence analysis. Virol J 6, 67 10.1186/1743-422X-6-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia C. C., Russo R. C., Guabiraba R., Fagundes C. T., Polidoro R. B., Tavares L. P., Salgado A. P., Cassali G. D., Sousa L. P. & other authors (2010). Platelet-activating factor receptor plays a role in lung injury and death caused by Influenza A in mice. PLoS Pathog 6, e1001171 10.1371/journal.ppat.1001171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govorkova E. A., Gambaryan A. S., Claas E. C., Smirnov Y. A. (2000). Amino acid changes in the hemagglutinin and matrix proteins of influenza A (H2) viruses adapted to mice. Acta Virol 44, 241–248 [PubMed] [Google Scholar]
- Harris A., Forouhar F., Qiu S., Sha B., Luo M. (2001). The crystal structure of the influenza matrix protein M1 at neutral pH: M1-M1 protein interfaces can rotate in the oligomeric structures of M1. Virology 289, 34–44 10.1006/viro.2001.1119 [DOI] [PubMed] [Google Scholar]
- Hirayama E., Atagi H., Hiraki A., Kim J. (2004). Heat shock protein 70 is related to thermal inhibition of nuclear export of the influenza virus ribonucleoprotein complex. J Virol 78, 1263–1270 10.1128/JVI.78.3.1263-1270.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann E., Neumann G., Hobom G., Webster R. G., Kawaoka Y. (2000a). “Ambisense” approach for the generation of influenza A virus: vRNA and mRNA synthesis from one template. Virology 267, 310–317 10.1006/viro.1999.0140 [DOI] [PubMed] [Google Scholar]
- Hoffmann E., Neumann G., Kawaoka Y., Hobom G., Webster R. G. (2000b). A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A 97, 6108–6113 10.1073/pnas.100133697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., Chen J., Wang H., Sun B., Wang H., Zhang Z., Zhang X., Chen Z. (2009). Influenza A virus matrix protein 1 interacts with hTFIIIC102-s, a short isoform of the polypeptide 3 subunit of human general transcription factor IIIC. Arch Virol 154, 1101–1110 10.1007/s00705-009-0416-7 [DOI] [PubMed] [Google Scholar]
- Hui E. K., Smee D. F., Wong M. H., Nayak D. P. (2006). Mutations in influenza virus M1 CCHH, the putative zinc finger motif, cause attenuation in mice and protect mice against lethal influenza virus infection. J Virol 80, 5697–5707 10.1128/JVI.02729-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam T. T., Hon C. C., Pybus O. G., Kosakovsky Pond S. L., Wong R. T., Yip C. W., Zeng F., Leung F. C. (2008). Evolutionary and transmission dynamics of reassortant H5N1 influenza virus in Indonesia. PLoS Pathog 4, e1000130 10.1371/journal.ppat.1000130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. H., Youn J. W., Kim H. J., Seong B. L. (2001). Identification and characterization of mutations in the high growth vaccine strain of influenza virus. Arch Virol 146, 369–377 10.1007/s007050170181 [DOI] [PubMed] [Google Scholar]
- Li F., Chen C., Puffer B. A., Montelaro R. C. (2002). Functional replacement and positional dependence of homologous and heterologous L domains in equine infectious anemia virus replication. J Virol 76, 1569–1577 10.1128/JVI.76.4.1569-1577.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Qiao C., Marjuki H., Bawa B., Ma J., Guillossou S., Webby R. J., Richt J. A., Ma W. (2011). Combination of PB2 271A and SR polymorphism at positions 590/591 is critical for viral replication and virulence of swine influenza virus in cultured cells and in vivo. J Virol 86, 1233–1237 10.1128/JVI.79.3.1918-1923.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Sun L., Yu M., Wang Z., Xu C., Xue Q., Zhang K., Ye X., Kitamura Y., Liu W. (2009). Cyclophilin A interacts with influenza A virus M1 protein and impairs the early stage of the viral replication. Cell Microbiol 11, 730–741 10.1111/j.1462-5822.2009.01286.x [DOI] [PubMed] [Google Scholar]
- Ma W., Liu Q., Bawa B., Qiao C., Qi W., Shen H., Chen Y., Ma J., Li X. & other authors (2012). The neuraminidase and matrix genes of the 2009 pandemic influenza H1N1 virus cooperate functionally to facilitate efficient replication and transmissibility in pigs. J Gen Virol 93, 1261–1268 10.1099/vir.0.040535-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K., Helenius A. (1991a). Nuclear transport of influenza virus ribonucleoproteins: the viral matrix protein (M1) promotes export and inhibits import. Cell 67, 117–130 10.1016/0092-8674(91)90576-K [DOI] [PubMed] [Google Scholar]
- Martin K., Helenius A. (1991b). Transport of incoming influenza virus nucleocapsids into the nucleus. J Virol 65, 232–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers J. A., Hoffmann E., Huber V. C., Nickerson A. D. (2005). A single amino acid change in the C-terminal domain of the matrix protein M1 of influenza B virus confers mouse adaptation and virulence. Virology 336, 318–326 10.1016/j.virol.2005.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy B. R., Buckler-White A. J., London W. T., Snyder M. H. (1989). Characterization of the M protein and nucleoprotein genes of an avian influenza A virus which are involved in host range restriction in monkeys. Vaccine 7, 557–561 10.1016/0264-410X(89)90283-1 [DOI] [PubMed] [Google Scholar]
- Nayak D. P., Balogun R. A., Yamada H., Zhou Z. H., Barman S. (2009). Influenza virus morphogenesis and budding. Virus Res 143, 147–161 10.1016/j.virusres.2009.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann G., Hughes M. T., Kawaoka Y. (2000). Influenza A virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J 19, 6751–6758 10.1093/emboj/19.24.6751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noton S. L., Medcalf E., Fisher D., Mullin A. E., Elton D., Digard P. (2007). Identification of the domains of the influenza A virus M1 matrix protein required for NP binding, oligomerization and incorporation into virions. J Gen Virol 88, 2280–2290 10.1099/vir.0.82809-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill R. E., Talon J., Palese P. (1998). The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J 17, 288–296 10.1093/emboj/17.1.288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada A., Miura T., Takeuchi H. (2003). Zinc- and pH-dependent conformational transition in a putative interdomain linker region of the influenza virus matrix protein M1. Biochemistry 42, 1978–1984 10.1021/bi027176t [DOI] [PubMed] [Google Scholar]
- Pal S., Santos A., Rosas J. M., Ortiz-Guzman J., Rosas-Acosta G. (2011). Influenza A virus interacts extensively with the cellular SUMOylation system during infection. Virus Res 158, 12–27 10.1016/j.virusres.2011.02.017 [DOI] [PubMed] [Google Scholar]
- Palese P., Shaw M. (2007). Orthomyxoviridae: the viruses and their replication. In Field’s Virology, 5th edn, pp. 1647–1689 Edited by Fields B. N., Knipe D. M., Howley P. M. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins [Google Scholar]
- Perez D. R., Donis R. O. (1998). The matrix 1 protein of influenza A virus inhibits the transcriptase activity of a model influenza reporter genome in vivo. Virology 249, 52–61 10.1006/viro.1998.9318 [DOI] [PubMed] [Google Scholar]
- Reid A. H., Fanning T. G., Janczewski T. A., McCall S., Taubenberger J. K. (2002). Characterization of the 1918 “Spanish” influenza virus matrix gene segment. J Virol 76, 10717–10723 10.1128/JVI.76.21.10717-10723.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt J., Wolff T. (2000). The influenza A virus M1 protein interacts with the cellular receptor of activated C kinase (RACK) 1 and can be phosphorylated by protein kinase C. Vet Microbiol 74, 87–100 10.1016/S0378-1135(00)00169-3 [DOI] [PubMed] [Google Scholar]
- Richt J. A., Lager K. M., Janke B. H., Woods R. D., Webster R. G., Webby R. J. (2003). Pathogenic and antigenic properties of phylogenetically distinct reassortant H3N2 swine influenza viruses cocirculating in the United States. J Clin Microbiol 41, 3198–3205 10.1177/104063870401600501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts P. C., Lamb R. A., Compans R. W. (1998). The M1 and M2 proteins of influenza A virus are important determinants in filamentous particle formation. Virology 240, 127–137 10.1006/viro.1997.8916 [DOI] [PubMed] [Google Scholar]
- Rossman J. S., Lamb R. A. (2011). Influenza virus assembly and budding. Virology 411, 229–236 10.1016/j.virol.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruigrok R. W., Barge A., Durrer P., Brunner J., Ma K., Whittaker G. R. (2000). Membrane interaction of influenza virus M1 protein. Virology 267, 289–298 10.1006/viro.1999.0134 [DOI] [PubMed] [Google Scholar]
- Sheehan D. C., Hrapchak B. B. (1980). Nuclear and cytoplasmic staining. In Theory and Practice of Histotechnology, pp. 137–159 Edited by Sheehan, D. C., Hrapchak, B. B., Columbus, O. H.: Battelle Press. 10.1016/j.virol.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield L., Brownlee G. G. (1989). RNA-binding properties of influenza A virus matrix protein M1. Nucleic Acids Res 17, 8569–8580 10.1093/nar/17.21.8569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Harmon A., Jin J., Francis D. H., Christopher-Hennings J., Nelson E., Montelaro R. C., Li F. (2010). The lack of an inherent membrane targeting signal is responsible for the failure of the matrix (M1) protein of influenza A virus to bud into virus-like particles. J Virol 84, 4673–4681 10.1128/JVI.02306-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A. C. (1995). Specific changes in the M1 protein during adaptation of influenza virus to mouse. Arch Virol 140, 383–389 10.1007/BF01309872 [DOI] [PubMed] [Google Scholar]
- Ward A. C. (1996). Neurovirulence of influenza A virus. J Neurovirol 2, 139–151 10.3109/13550289609146876 [DOI] [PubMed] [Google Scholar]
- Ward A. C. (1997). Virulence of influenza A virus for mouse lung. Virus Genes 14, 187–194 10.1023/A:1007979709403 [DOI] [PubMed] [Google Scholar]
- Watanabe K., Handa H., Mizumoto K., Nagata K. (1996). Mechanism for inhibition of influenza virus RNA polymerase activity by matrix protein. J Virol 70, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K., Fuse T., Asano I., Tsukahara F., Maru Y., Nagata K., Kitazato K., Kobayashi N. (2006). Identification of Hsc70 as an influenza virus matrix protein (M1) binding factor involved in the virus life cycle. FEBS Lett 580, 5785–5790 10.1016/j.febslet.2006.09.040 [DOI] [PubMed] [Google Scholar]
- Wu C. Y., Jeng K. S., Lai M. M. (2011). The SUMOylation of matrix protein M1 modulates the assembly and morphogenesis of influenza A virus. J Virol 85, 6618–6628 10.1128/JVI.02401-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda J., Nakada S., Kato A., Toyoda T., Ishihama A. (1993). Molecular assembly of influenza virus: association of the NS2 protein with virion matrix. Virology 196, 249–255 10.1006/viro.1993.1473 [DOI] [PubMed] [Google Scholar]
- Ye Z., Robinson D., Wagner R. R. (1995). Nucleus-targeting domain of the matrix protein (M1) of influenza virus. J Virol 69, 1964–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z., Liu T., Offringa D. P., McInnis J., Levandowski R. A. (1999). Association of influenza virus matrix protein with ribonucleoproteins. J Virol 73, 7467–7473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Ekström M., Garoff H. (1998). The M1 and NP proteins of influenza A virus form homo- but not heterooligomeric complexes when coexpressed in BHK-21 cells. J Gen Virol 79, 2435–2446 [DOI] [PubMed] [Google Scholar]
- Zhirnov O. P. (1992). Isolation of matrix protein M1 from influenza viruses by acid-dependent extraction with nonionic detergent. Virology 186, 324–330 10.1016/0042-6822(92)90090-C [DOI] [PubMed] [Google Scholar]
- Zhirnov O. P., Klenk H. D. (1997). Histones as a target for influenza virus matrix protein M1. Virology 235, 302–310 10.1006/viro.1997.8700 [DOI] [PubMed] [Google Scholar]