

Abstract
ϕEf11 is a temperate bacteriophage originally isolated by induction from a lysogenic Enterococcus faecalis strain recovered from an infected root canal, and the ϕEf11 prophage is widely disseminated among strains of E. faecalis. Because E. faecalis has emerged as a significant opportunistic human pathogen, we were interested in examining the genes and regulatory sequences predicted to be critical in the establishment/maintenance of lysogeny by ϕEf11 as a first step in the construction of the genome of a virulent, highly lytic phage that could be used in treating serious E. faecalis infections. Passage of ϕEf11 in E. faecalis JH2-2 yielded a variant that produced large, extensively spreading plaques in lawns of indicator cells, and elevated phage titres in broth cultures. Genetic analysis of the cloned virus producing the large plaques revealed that the variant was a recombinant between ϕEf11 and a defective ϕFL1C-like prophage located in the E. faecalis JH2-2 chromosome. The recombinant possessed five ORFs of the defective ϕFL1C-like prophage in place of six ORFs of the ϕEf11 genome. Deletion of the putative lysogeny gene module (ORFs 31–36) and replacement of the putative cro promoter from the recombinant phage genome with a nisin-inducible promoter resulted in no loss of virus infectivity. The genetic construct incorporating all the aforementioned ϕEf11 genomic modifications resulted in the generation of a variant that was incapable of lysogeny and insensitive to repressor, rendering it virulent and highly lytic, with a notably extended host range.
Introduction
ϕEf11 is a temperate bacteriophage that was induced from a lysogenic root canal isolate of Enterococcus faecalis (Stevens et al., 2009). It is a member of the family Siphoviridae, with a long (130 nm) non-contractile tail and a small (41 nm diameter) spherical/icosohedral head. The phage produces small, turbid plaques in lawns of E. faecalis JH2-2. The ϕEf11 DNA has been sequenced and annotated, disclosing a genome of 42 822 bp encoding 65 ORFs (Stevens et al., 2011). Furthermore, our previous studies disclosed that the ϕEf11 DNA restriction pattern produced with certain restriction enzymes, such as NsiI, produced several fragments in submolar amounts (Stevens et al. 2009). This would be expected to occur in the case of a circularly permuted genome due to headful packaging of a concatemeric phage DNA during viral maturation.
The ϕEf11 host species, E. faecalis, and closely related species, such as Enterococcus faecium, have emerged as significant human pathogens, being major aetiological agents of infectious endocarditis, nosocomial infections, burn infections, urinary tract infections, meningitis and surgical wound infections (Lewis & Zervos, 1990; Moellering, 1992; Megran, 1992; Emori & Gaynes, 1993; Jett et al., 1994; Edgeworth et al., 1999; Richards et al., 2000; National Nosocomial Infections Surveillance System, 2004; Biedenbach et al., 2004; Linden, 2007). In terms of oral disease, E. faecalis is the most commonly isolated species from infected root canals of teeth that fail to heal following root canal therapy (Sundqvist et al., 1998; Peciuliene et al., 2000; Pinheiro et al., 2003; Siqueira & Rôças, 2004; Stuart et al., 2006; Zoletti et al., 2006). Complicating management of these infections is the development of resistance among many enterococcal strains against many of the available, previously effective antibiotics, including vancomycin (Havard et al., 1959; Murray & Mederski-Samaroj, 1983; Uttley et al., 1988; Grayson et al., 1991; Bonten et al., 2001; Tenover & McDonald, 2005). Although a modest number of new antibiotics, such as linezolid and daptomycin, have been developed to provide treatment alternatives in cases of infection by organisms that are resistant to all previously available antibiotics, there have been numerous reports of resistance by E. faecalis and E. faecium strains to these antibiotics as well (Eliopoulos et al., 1998; Prystowsky et al., 2001; Gonzales et al., 2001; Herrero et al., 2002; Johnson et al., 2004; Munoz-Price et al., 2005; Kanafani et al., 2007; Hidron et al., 2008; Marshall et al., 2009; Kelesidis et al., 2011; Ross et al., 2011; Ntokou et al., 2012). Therefore, alternative approaches to manage these infections should be explored.
We have been exploring the possibility of engineering variants of phage ϕEf11 that might be effective in controlling these infections. ϕEf11 possesses several characteristics making it a favourable candidate virus to be used in phage therapy: there are no toxin-related genes detected in the ϕEf11 genome, and it encodes several (4–6) genes encoding proteins with lysis-associated functions (Stevens et al., 2011). However, as a temperate virus that is difficult to propagate, wild-type ϕEf11 would certainly not be suitable as a potential therapeutic agent. It would not be expected to infect strains of E. faecalis lysogens due to repressor-mediated superinfection immunity, nor would it be expected to be highly lytic by virtue of its ability to enter into a lysogenic life cycle. Consequently, in its wild-type state, it would have an unacceptably limited host range. Therefore, we sought to determine whether appropriate genetic modifications of ϕEf11 would result in a virulent, highly lytic variant that was insensitive to repression and incapable of lysogeny. These studies shed light on some of the regulatory mechanisms that function in controlling the life cycle of this bacterial virus, and generated a variant phage that had a notably broader host range. Here, we present the identification of the phage virulence determinants, and the development of a virulent version of the temperate E. faecalis bacteriophage ϕEf11.
Methods
Bacterial strains and growth conditions.
TUSoD11 is a lysogenic E. faecalis strain, harbouring a ϕEf11 prophage, which was previously isolated from an infected root canal (Stevens et al., 2009). Following curing, the non-lysogenic variant of this strain was designated E. faecalis TUSoD11 (ΔϕEf11). JH2-2 is a Fusr, Rifr mutant of a clinical E. faecalis isolate (Jacob & Hobbs, 1974) that was generously provided to us by Dr Nathan Shankar. In the course of this study, it was found that this strain harboured a ϕFL1C-type prophage element (Yasmin et al., 2010), indicating that this strain was a lysogen with a defective prophage. Other E. faecalis strains used in this study are listed in Table 1. All strains were grown in brain–heart infusion (BHI) broth (or on BHI agar, with appropriate antibiotics). Escherichia coli One Shot Mach-T1 (Invitrogen) was used in cloning plasmids as described below. Cells were grown in Luria–Bertani (LB) medium supplemented with the appropriate antibiotics. Additional bacterial species used as negative controls in PCR experiments are also listed in Table 1.
Table 1. Bacterial strains used in this study.
| Strain | Characteristics | Source* |
| E. faecalis | ||
| TUSoD11 | Lysogenic root canal isolate harbouring ϕEf11 prophage | 1 |
| JH2-2 | Rifr Fusr, clinical isolate harbouring defective ϕFL1C prophage | 2, 3 |
| OG1RF | Rifr Fusr | 3, 4 |
| MMH594 | Genr | 3 |
| OG1SSp | Strr Spcr | 5 |
| ER3/2s, ER5/1 | Root canal isolates | 6, 7 |
| E1, E2, E3, E4, E5, E6, E7, E8, E10, E11 | Oral isolates | 6, 8 |
| GS1, GS2, GS3, GS4, GS5, GS6, GS7, GS8, GS9, GS10, GS12, GS13, GS14, GS15, GS16, GS17, GS18, GS19, GS21, GS22, GS23, GS24, GS25, GS26, GS27, GS28, GS29, GS30, GS31, GS32, GS33 | Root canal isolates | 9 |
| GS34 | Tongue | 7, 6 |
| OS25 | Oral isolate | 7, 10 |
| AA-OR3, AA-OR4, AA-OR26, AA-OR34 | Oral isolates | 7, 11 |
| AA-T4, AA-T26 | Tongue | 7, 11 |
| V583 | Vanr, clinical isolate | 7, 12 |
| OS16 | Oral isolate | 7, 10 |
| TUSoD1, TUSoD2, TUSoD3 | Lysogenic root canal isolates | 1 |
| TUSoD9, TUSoD10, TUSoD12, TUSoD15, TUSoD17, TUSoD18 | Root canal isolates | 1 |
| Non-enterococcal spp. | ||
| Streptococcus mutans 10449 | Grown in BHI broth | 13 |
| Streptococcus sanguis 43055 | Grown in BHI broth | 13 |
| Fingoldia (Peptostreptococcus) magna (magnus) | Grown in chopped meat broth | 13 |
| Clostridium perfringens 13124 | Grown in modified PY broth | 13 |
| Actinomyces israelii 10049 | Grown in BHI broth | 13 |
| Eubacterium lentum 43033 | Grown in chopped meat broth | 13 |
1, Stevens et al. (2009); 2, Jacob & Hobbs (1974); 3, Dr Nathan Shankar; 4, Dunny et al. (1979); 5, Dr Gary Dunny; 6, Johnson et al. (2006); 7, Dr Christine Sedgley; 8, Sedgley et al. (2004); 9, Sedgley et al. (2005a); 10, Sedgley et al. (2005b); 11, Sedgley et al. (2006); 12, Sahm et al. (1989); 13, ATCC.
Construction of recombinant plasmids.
The nisin promoter (PnisA) cassette containing an erythromycin selection marker (erm) was PCR-amplified using the AccuPrime DNA Taq Polymerase High Fidelity kit (Invitrogen) with primer set PNISaF/PNISR (see Table 2 for primer specifications) from plasmid pMSP3535 (Bryan et al., 2000), a kind gift from Dr B. Buttaro. PCRs were performed in 30 µl reaction mixtures containing 2 µl template DNA, 2 µl (20 pmol) forward primer, 2 µl (20 pmol) reverse primer, 21.5 µl distilled H2O, 2 µl buffer (provided by the manufacturer) and 0.5 µl AccuPrime DNA Taq polymerase. The PCR programme used was: 95 °C for 2 min, followed by 35 cycles of (i) 95 °C for 45 s, (ii) 55 °C for 45 s and (iii) 72 °C for 2 min. This was followed by an additional 5 min extension at 72 °C. Following PCR, the amplicons were detected by agarose gel electrophoresis and ethidium bromide staining. The amplicons generated by this procedure were cloned into pCR8/GW/TOPO vector (Invitrogen) to create pErm-PnisA (Fig. S1, available in Microbiology Online). The two-component nisin sensor system (nisR/nisK) which controls the activation of PnisA by nisin was also amplified from pMSP3535 by PCR, using primer set RKnpF/RKaxR, and cloned into pCR8/GW/TOPO to create pRK. The PnisA fragment plus the erythromycin selection marker was digested from pErm-PnisA with AatII and SphI, and inserted into pRK to create pRK-Erm-PnisA. A fragment (pre31) of 1088 bp from nucleotide coordinates 24 585 to 25 672 of ϕEf11 (upstream of ORF31, the first gene of the putative lysogeny module) and a fragment (post36) of 1090 bp from 28 588 to 29 577 of ϕEf11 (immediately upstream of the putative cro gene, ORF37) were PCR-amplified using primer sets EF31UF/EF31UR and EF37DF/EF37DR, respectively, and cloned into pCR8/GW/TOPO to create pPre31 and pPost36. The post36 fragment was cut out from the pPost 36 with BamHI and SphI and inserted into pRK-Erm-PnisA, to create pPost36-RK-PnisA. The pPre31 was first digested with EcoRI and blunt-ended with the Klenow fragment of DNA polymerase I (Promega), then digested with PstI. Following this, the digested pre31 fragment was cloned into pPost36-RK-PnisA to create the allelic exchange plasmid pΔ31-36 PnisA.
Table 2. PCR primers used in this study.
| Primer | Sequence (5′→3′) | Use |
| EF31UF | GATAGTTCTTGTTTCGACAAATCAC | Amplify upstream of ϕEf11 ORF 31 |
| EF31UR | CTGTCGACGTTCCTGCAGAGCTCTAAATAAATATGGCAAGTA | Amplify upstream of ϕEf11 ORF 31 |
| EF37DF | CTGGATCCATGTGCTATGATTACTCAAAATTAGCAG | Amplify downstream of ϕEf11 ORF 36 |
| EF37DR | CTGCATGCCCTTTACCAGTAATTTTCGGCGT | Amplify downstream of ϕEf11 ORF 36 |
| RKnpF | CTCCATGGTCTCTCCTGCAGATAGAATTCTCATGTTTGACAGCTTATCA | Amplify nisR and nisK |
| RKaxR | CTGCATGCTCTCTCGACGTCGCCAGTTAATAGTTTGCCGAA | Amplify nisR and nisK |
| PNISaF | CTGACGTCACAAAAGCGACTCATAGAATTATTTCCTCC | Amplify Erm-PnisA |
| PNISR | GCTTATCGAAATTAATACGACTCACTATAGG | Amplify Erm-PnisA |
| EF31UUF | AAGAGCACCTCAAATTCCAGT | Detection of ϕEf11 ΔORFs 31–36 (upstream) |
| RK5R | TGATAAGCTGTCAAACATGAGAATTCT | Detection of ϕEf11 ΔORFs 31–36 (upstream) |
| 37DDR | TGTGATTTGCATGTAGACATCTCCT | Detection of ϕEf11 ΔORFs 31–36 (downstream) |
| PNIS3F | TTGTAAAACAGGAGACTCTGCATG | Detection of ϕEf11 ΔORFs 31–36 (downstream) |
| EF31MF | AAGTTGTTTCCGTGTCAACGTGGC | Detection of ϕEf11 ORF 31 deletion |
| EF31MR | GTGTCCATCATGGTCGTTTAGCAG | Detection of ϕEf11 ORF 31 deletion |
| EF36MF | TTATCAGGGTCTGGTGAATGCG | Detection of ϕEf11 ORF 36 deletion |
| EF36MR | GCAACTTATGAGTGAGCGCAA | Detection of ϕEf11 ORF 36 deletion |
| ϕEf11F | GAGAGTGGAAGTGGATTCAATG | Detection of ϕEf11 ORF 43 |
| ϕEf11R | GCACTTTCATCTAAACTCTCG | Detection of ϕEf11 ORF 43 |
| EF44F | ACCAAGATTTGACGCAGAAGTTGCC | Detection of ϕEf11 ORF 44 |
| EF44R | TGGCCATCGTCGTCTTTATCTGCT | Detection of ϕEf11 ORF 44 |
| EF60F | AGACGTTTGGACCGAATAGCTGGT | Detection of ϕEf11 ORF 60 |
| EF60R | TGCGGTAAGCTTCTGCGAATTCAA | Detection of ϕEf11 ORF 60 |
| Fl1A35F | GGGAACTAGCAGTTGAAGAATCGC | Detection of ϕFL1C gp40 |
| Fl1A35R | TTCCTTTGTACTATCTTGATCTCCA | Detection of ϕFL1C gp40 |
| Fl1A37F | GAGCGTTTAGATAAGTCGGATTGG | Detection of ϕFL1C gp44 |
| Fl1A38R | CCAAGTTTCTTTAGCCTGGTCACG | Detection of ϕFL1C gp44 |
Isolation of spontaneous phage ϕEf11/ϕFL1C-like recombinant [ϕEf11(Δ61-1, ϕFL1C40-44)] and the creation of a lysogen harbouring the recombinant prophage.
An exponential phase BHI broth culture of E. faecalis JH2-2 was inoculated with phage ϕEf11. After incubation at 37 °C for 1 h, the culture was centrifuged (17 000 g for 3 min) and the supernatant was filtered (0.45 µm) before being plaque-assayed. After overnight incubation at 37 °C, the plates were examined, and several large, extensively spreading plaques were noted among a background of small, turbid plaques. These large plaques were picked, and the virus in these large plaques was cloned by successive plaque purifications. The genomic DNA from the cloned virus was sequenced by Sanger di-deoxy sequencing reactions as described previously (Stevens et al., 2011).
To create a lysogen harbouring a ϕEf11(Δ61-1, ϕFL1C40-44) prophage, JH2-2 cells from surviving colonies in the centre of the large plaques produced by this virus were cloned and screened for the presence of the recombinant phage genome. This was done by PCR using primers (EF60F/FL1A35R) that recognized ϕEf11 ORF 60 at the 5′ end and ϕFL1C ORF 40 at the 3′ end (see Table 2 for primer specifications). The lysogen harbouring this recombinant prophage was designated E. faecalis JH2-2[ϕEf11(Δ61-1, ϕFL1C40-44); (Fig. 1). In addition, virus spontaneously released from this lysogen was detected by plaque assay, and also confirmed to be recombinant by PCR analysis.
Fig. 1.


Sequence alignment of phages ϕEf11, ϕFL1C and spontaneous recombinant phage [ϕEf11(Δ61-1, ϕFL1C 40-44)] in the region of recombination (ϕEf11 ORFs 60/61-1 and ϕFL1C ORFs 39/40–44). Red indicates ϕEf11 sequences, green indicates ϕFL1C sequences. Sp, Spontaneous recombinant. Genomic coordinates are indicated to the right of each row of sequence. Sites of sequence identity between ϕEf11 and ϕEf11(Δ61-1, ϕFL1C 40-44) are indicated by asterisks. (a) Overview of the regions of ϕEf11 and ϕFL1C that recombined to yield recombinant ϕEf11(Δ61-1, ϕFL1C 40-44). (b) ϕEf11 sequence from 39 307 (within ORF 60) to 451 (within ORF 1), and ϕFL1C sequence from 14 236 (within ORF 39) to 17 451 (within ORF 44). The segment of the ϕEf11 sequence that has been replaced by the ϕFL1C sequence to form the ϕEf11(Δ61-1, ϕFL1C 40-44) recombinant is indicated by Graphic 1. NdeI restriction site in ϕEf11 sequence is indicated.
Deletion of the lysogeny module and replacement of cro promoter with PnisA by allelic exchange.
Cells of E. faecalis lysogen JH2-2[ϕEf11(Δ61-1, ϕFL1C40-44)] were made competent using the procedures described by Shepard & Gilmore (1995). Briefly, the cells were grown in SGM17 medium (37.25 g M17 l−1, 0.5 M sucrose and 8 % glycine) for 48 h at 37 °C. The cells were then harvested by centrifugation, washed twice with EB buffer (0.5 M sucrose and 10 % glycerol), and finally resuspended in EB buffer. Plasmid pΔ31-36 PnisA was linearized with XhoI and then electroporated into the competent JH2-2 lysogens using the Bio-Rad MicroPulser System. Following electroporation, 1 ml SGM17MC medium (SGM17 plus 10 mM MgCl2 and 10 mM CaCl2) was added to the electroporation cuvette, which was then incubated for 2 h. Transformants were selected on BHI agar containing erythromycin (30 µg ml−1). Presumptive transformant colonies were screened for deletion of the lysogeny module genes (ϕEf11 ORFs 31–36) and replacement of Pcro by PnisA by PCR using primers EF31UUF/RK5R, PNIS3F/37DDR, EF31MF/EF31MR and EF36MF/EF36MR. In addition, control of lytic functions in the prophage by PnisA was demonstrated by measuring phage induction in the presence or absence of nisin (40 ng ml−1). The phage recovered from the induced lysogens lacking ORFs 31–36 and Pcro, but containing the PnisA promoter, was designated ϕEf11(vir)PnisA.
Screening for the presence of ϕEf11 prophages in E. faecalis strains.
Primers specific to ϕEf11 were designed from ϕEf11 ORF 43 (GenBank accession number GQ452243). This sequence (ORF 43) of the ϕEf11 genome was chosen as searches of all available databases failed to disclose any homologous sequences to this gene. The forward (ϕEf11F) and reverse (ϕEf11R) primers for amplification of a 165 bp amplicon of this gene are specified in Table 2. Template DNA was prepared as follows: 10 ml broth cultures of each strain to be screened were pelleted by centrifugation, washed in 4 ml wash solution [20 mM Tris/HCl (pH 8.5), 0.85 % NaCl], resuspended in 2 ml lysis buffer [1 % Triton X-100, 20 mM Tris/HCl (pH 8.5), 2 mM EDTA], and heated to 95–100 °C for 10 min. The suspension was then centrifuged and the supernatants were collected and frozen at −80 °C until used in PCR assays (Goncharoff et al., 1993). Extracts from E. faecalis TUSoD11 (lysogenic for ϕEf11) were used as positive controls, and extracts from E. faecalis JH2-2 (non-lysogenic for ϕEf11) and numerous unrelated species (see Table 1) were used as negative controls. Reaction mixtures (total 40 µl) for PCR contained 5 µl template DNA, 5 µl (50 pmol) forward primer, 5 µl (50 pmol) reverse primer, 5 µl distilled H2O and 20 µl 2× Go Taq green PCR master mix (Promega). The PCR programme used was 97 °C for 1 min, followed by 26 cycles of (i) 97 °C for 1 min, (ii) 50 °C for 45 s and (iii) 72 °C for 1 min. This was followed by an additional 4 min at 72 °C. Following PCR, amplification products were detected by agarose (2 %) gel electrophoresis and ethidium bromide staining.
Preparation of cured E. faecalis TUSoD11.
Cells of E. faecalis TUSoD11 were made competent for electroporation as described above. After electroporation with the allelic exchange vector pΔ31-36 PnisA, erythromycin-resistant colonies were screened for homologous recombination-mediated deletion of the lysogeny module genes (ORFs 31–36) in the genome of E. faecalis TUSoD11. Strains exhibiting deletion of ORFs 31–36 were further tested by PCR for the presence of ϕEf11 genes outside of the lysogeny module. In addition to clones containing ϕEf11 genes other than ORFs 31–36, a few rare clones were identified that lacked any of the ϕEf11 genes. Such clones could not be induced, but could now be infected by phage ϕEf11. These cured clones were designated E. faecalis TUSoD11(ΔϕEf11).
Testing adsorption of ϕEf11 and ϕEf11(Δ61-1, ϕFL1C40-44) to lysogenic and non-lysogenic E. faecalis strains.
E. faecalis strains JH2-2, TUSoD11 and the cured strain, TUSoD11 (ΔϕEf11), were grown in BHI medium to exponential phase. Then, 100 µl of ϕEf11 or ϕEf11(Δ61-1, ϕFL1C40-44) preparations were added to 1 ml E. faecalis strains. After incubation at 37 °C for 10 min the mixtures were centrifuged at 17 000 g for 3 min, the supernatants were filtered through 0.45 µm filters and filtrates containing any unadsorbed phage were plaque-assayed, using JH2-2 indicator cells, to determine residual phage titres.
One-step growth curve.
Cells of an exponential phase BHI broth culture (2 ml) of E. faecalis JH2-2 were collected by centrifugation, resuspended in 1 ml BHI broth, and inoculated with 100 µl of a stock culture of either phage ϕEf11, ϕEf11(Δ61-1, ϕFL1C40-44) or ϕEf11 (vir)PnisA. After incubation for 30 min to allow phage adsorption, the cells were recovered by centrifugation, washed three times in BHI broth and finally resuspended in 10 ml BHI broth. Aliquots (500 µl) of the suspension were made, and each was incubated at 37 °C. At various time points, an aliquot was centrifuged to remove the cells, and the supernatant was plaque-assayed, using fresh JH2-2 indicator cells, for phage titre.
Host range determination for ϕEf11, ϕEf11(Δ61-1, ϕFL1C40-44) and ϕEf11(vir)PnisA.
Plaque assays and spot tests were conducted with wild-type phage ϕEf11 and recombinant phages ϕEf11(Δ61-1, ϕFL1C40-44) and ϕEf11(vir)PnisA using the panel of 67 E. faecalis strains listed in Table 1 as indicators. The E. faecalis panel included both lysogenic and non-lysogenic strains. Lytic infection by each phage was detected by plaque assay with each E. faecalis indicator strain.
Results
Isolation of spontaneous ϕEf11/ϕFL1C recombinant
Following repeated propagation and plaque assay of phage ϕEf11 on host strain E. faecalis JH2-2, it became evident that variants of the wild-type virus were being generated. Whereas wild-type ϕEf11 produced small, turbid plaques in lawns of JH2-2, approximately 0.02 % of the plaques appeared as large, extensively spreading, somewhat clearer zones of lysis (Fig. 2). Interestingly, incubation of plaque assays of clones obtained by plaque purification of the virus producing these larger plaques resulted in continued expansion of the plaques to the extent that virtually the entire JH2-2 lawn was lysed (Fig. 2). In contrast, wild-type plaques typically disappeared after extended incubation, presumably due to growth of surviving lysogens within the plaques (Fig. 2). Agarose gel electrophoresis analysis of the NdeI restriction fragments of the DNA from the virus producing these large plaques revealed that it was missing one of the fragments (fragment 6, 2.79 kbp) that was present in the NdeI DNA digestion of the original ϕEf11 isolate (Fig. S2). In addition, it was also noted that another of the NdeI fragments (fragment 2, approx. 9.4 kbp) from the DNA of the virus producing the large plaques had increased in size (compared with NdeI fragment 2 from the original ϕEf11 DNA) by an amount approximately equal to the size of the missing NdeI fragment 6 (Fig. S2). Close inspection of the ϕEf11 NdeI restriction map (Fig. S3) and the ϕEf11 NdeI restriction digest summary (Table S1) revealed that NdeI fragment 6 was composed of the two extreme ends of the genome (fragment coordinates 0–1036 plus 41 068–42 822), and that in a circularly permuted genome, this fragment is immediately adjacent to NdeI fragment 2 (coordinates 33 692–41 068; Fig. S3). Sequencing this region of the genome disclosed that ORFs 60–65 and 1 (coordinates 39 671–42 822 and 1–336) were replaced by ORFs 40–44 (coordinates 14 600–17 336) of E. faecalis phage ϕFL1C (Fig. 1). Furthermore, the NdeI restriction site at coordinate 41 068, which divides NdeI fragment 2 from Ndel fragment 6 in the ϕEf11 DNA, is absent in the ϕFL1C DNA and consequently in the DNA of the recombinant virus (Fig. 1). PCR, restriction fragment analysis and partial sequencing of the recombinant DNA failed to detect any other modifications to the ϕEf11 genome. Consequently, this ϕEf11/ϕFL1C recombinant was designated phage ϕEf11(Δ61-1, ϕFL1C40-44). Because the JH2-2 genome was the only possible source of the ϕFL1C genes, we decided to screen E. faecalis JH2-2 for the ϕFL1C prophage. ϕFL1C (ORFs 40–44)-specific primers (Table 2) were used in PCR with JH2-2 extracts, prepared as described previously. As seen in Fig. S4, ϕFL1C-specific amplicons were generated from the JH2-2 templates and the ϕFL1C-specific primers, confirming the presence of (at least a portion of) a ϕFL1C prophage in the JH2-2 chromosome. PCR, using JH2-2 template DNA and primers specific for regions of the ϕFL1C genome other than ORFs 40–44, failed to produce any amplicons (data not shown).
Fig. 2.

Plaque assay of ϕEf11 wild-type (wt), spontaneous recombinant [(ϕEf11(Δ61-1, ϕFL1C40-44)] and virulent mutant [ϕEf11(vir)PnisA]. (a) wt after incubation for 1 day, (b) wt after incubation for 2 days, (c) spontaneous recombinant after incubation for 1 day, (d) spontaneous recombinant after incubation for 4 days, (e) virulent mutant after incubation for 1 day, (f) virulent mutant after incubation for 4 days.
Deletion of the lysogeny module and replacement of cro promoter in ϕEf11(Δ61-1, ϕFL1C40-44) by allelic exchange
Although the ϕEf11(Δ61-1, ϕFL1C40-44) recombinant exhibited enhanced lytic activity (compared with wild-type virus) as judged by the extensively enlarged plaques it forms in lawns of host cells (Fig. 2), and the elevated titres it achieved in productive infection (Fig. 3), these variants of phage ϕEf11 would still be expected to be subject to repression due to superinfection immunity, and be limited in lytic infection due to the possibility of entering into lysogeny, rather than generating a productive infection. Consequently, we sought to delete all lysogeny-related genes and render regulatory genetic elements insensitive to repressor control. Clones of JH2-2[ϕEf11(Δ61-1, ϕFL1C40-44)] transformed with plasmid pΔ31-36 PnisA were selected on erythromycin-containing media. PCR analysis and sequencing of these erythromycin-resistant JH2-2[ϕEf11(Δ31-36, ΔPCRO,PnisA, erm, nisR/K, Δ61-1, ϕFL1C40-44)] clones demonstrated that they lacked ϕEf11 ORFs 31–36, and the ϕEf11 cro promoter, but contained the nisin promoter (PnisA) and nisR/nisK (Figs 4 and S5). Furthermore, exposure of a population of this lysogenic clone, harbouring a mutant prophage containing the nisin promoter (PnisA) in place of the wild-type cro promoter/operator (PCRO), to nisin (40 ng ml−1) resulted in the induction of phage, yielding a mean (±sd) titre of 6.82×107 p.f.u. ml−1 (±0.31×107). In the absence of nisin, a similar population of these lysogens spontaneously released phage, producing a titre of 5.57×105 p.f.u. ml−1 (±0.31×105). In contrast, phage induction from lysogens {JH2-2[ϕEf11(Δ61-1, ϕFL1C40-44)]} containing a prophage with the wild-type cro promoter/operator did not appear to be affected by the presence of nisin: in the presence of nisin (40 ng ml−1), these cells produced a phage titre of 3.36×105 p.f.u. ml−1 (±0.25×105), whereas the same cells produced a titre of 3.31×105 p.f.u. ml−1 (±0.38×105) in the absence of nisin. These data suggest that productive infection was now under control of the nisin-sensitive promoter (PnisA), albeit this being somewhat leaky.
Fig. 3.
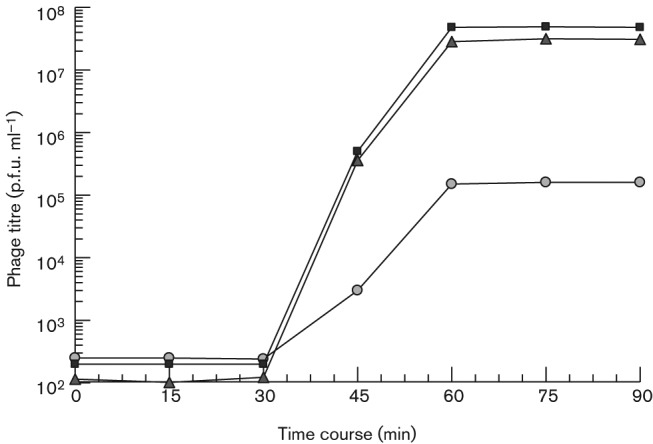
One-step growth curve for phages ϕEf11 (wild-type), ϕEf11(Δ61-1, ϕFL1C40-44) (spontaneous recombinant) and ϕEf11(vir)PnisA (virulent variant). Exponential phase broth cultures of E. faecalis JH2-2 were infected with a phage stock. After adsorption for 30 min, the cells were collected by centrifugation, washed and incubated at 37 °C. At various time points aliquots of the suspension were centrifuged to remove the cells, and the supernatants were plaque assayed for phage titre using JH2-2 indicator cells. (•) ϕEf11 titre (p.f.u. ml−1), (▪) ϕEf11(Δ61-1, ϕFL1C40-44) titre, (▴) ϕEf11(vir)PnisA titre.
Fig. 4.
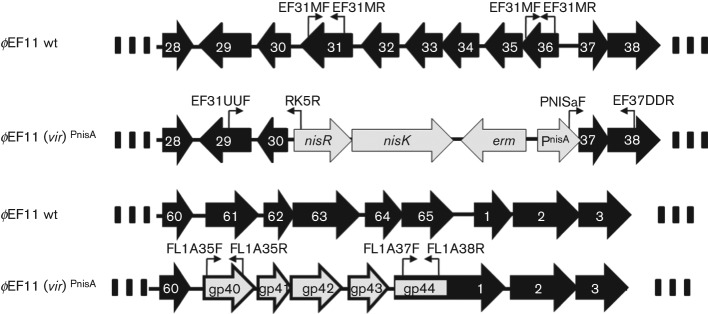
ϕEF11 (wild-type) and ϕEF11(vir)PnisA sequence comparison. Note that in the virulent mutant, ϕEF11(vir)PnisA, ORFs ORFs 31–36 as well as the cro promoter were allelically exchanged for the nisin promoter cassette, and ORFs 61–1 were allelically exchanged with gp40-gp44 of ϕFL1C. Primer binding sites are indicated by  and
and  .
.
The virus obtained, phage [ϕEf11(Δ31-36,ΔPCRO, PnisA, erm,nisR/K,Δ61-1, ϕFL1C40-44)], by nisin induction of the JH2-2[ϕEf11(Δ31-36, ΔPCRO, PnisA, erm, nisR/K, Δ61-1, ϕFL1C40-44)] lysogens produced large, clear plaques (Fig. 2), and was designated ϕEf11(vir)PnisA. As shown below, this derivative of temperate phage ϕEf11 had all the characteristics of a virulent virus.
Isolation of cured E. faecalis TUSoD11
After electroporation of E. faecalis TUSoD11 with the gene exchange vector pΔ31-36 PnisA, erythromycin-resistant colonies were screened by PCR for deletion of ORFs 31–36. Unexpectedly, a few colonies were found with deletions of not only the intended ORF 31–ORF 36 lysogeny module, but also all other phage genes outside this region. These clones may have been generated by the homologous recombination between the gene exchange vector and a permutated and terminally redundant prophage DNA that may have positioned the ORF 30 and ORF 37 regions at either end of the ϕEf11 prophage within the host E. faecalis TUSoD11 chromosome. These E. faecalis clones, lacking any detectable ϕEf11 genes, were designated TUSoD11(ΔϕEf11), and were further tested for phage induction. No phage could be induced from these cells.
Restoration of adsorption of ϕEf11 and ϕEf11(Δ61-1, ϕFL1C40-44) by a cured E. faecalis strain
Although it is not surprising that neither ϕEf11 nor ϕEf11(Δ61-1, ϕFL1C40-44) could produce a viable infection on the lysogenic TUSoD11 strain due to superinfection immunity, it was interesting that incubation of either ϕEf11 or ϕEf11(Δ61-1, ϕFL1C40-44) with a cell suspension of lysogenic E. faecalis strain TUSoD11 failed to result in phage adsorption to the cells. In contrast, cell suspensions of either strain JH2-2 (non-lysogenic with respect to ϕEf11) or TUSoD11(ΔϕEf11), a cured E. faecalis strain, effectively adsorbed both virus strains (Table 3).
Table 3. Phage adsorption by lysogenic and non-lysogenic E. faecalis strains.
Phage suspensions were incubated with each of the indicated E. faecalis strains for 10 min, whereupon the cultures were centrifuged and filtered to remove the cells along with all adsorbed phage. The cell-free filtrates were then assayed for residual phage titre. Values represent the mean of triplicate assays±sd.
| Phage | Phage titre before adsorption | Residual phage titre after adsorption with: | ||
| Lysogen TUSoD11 | Non-lysogen JH2-2 | Non-lysogen cured TUSoD11 | ||
| ϕEf11 | 1.2×105 | 1.17×105±0.16×105 | 3.2×102±0.25×102 | 2.74×102±0.16×102 |
| ϕEf11(Δ61-1, ϕFL1C40-44) | 5.58×107 | 5.23×107±0.23×107 | 4.79×102±0.23×12 | 3.82×102±0.17×102 |
Host range of ϕEf11(vir)PnisA
The ability of ϕEf11(vir)PnisA, in comparison with wt ϕEf11, to generate a productive infection in strains of E. faecalis is shown in Table 4. Whereas wild-type ϕEf11 productively infected only four (6 %) of the 67 E. faecalis strains tested, productive infection occurred in 33 (49 %) of these strains inoculated with phage ϕEf11(vir)PnisA. The panel of E. faecalis strains was also screened by PCR for the presence of a prophage, using ϕEf11-specific primers. Among the strains tested, 14 were found to be ϕEf11 lysogens (data not shown). Of these 14 ϕEf11 lysogens, none was susceptible to wild-type ϕEf11, although four of these lysogenic strains (strains GS2, GS8, GS22 and GS25) could be productively infected by ϕEf11(vir)PnisA. Furthermore, the presence of the ϕEf11 repressor gene (cI, ORF 36) was confirmed in these ϕEf11(vir)PnisA -susceptible lysogenic strains by PCR (data not shown).
Table 4. Host range of E. faecalis phages.
| Phage | ||||
| E. faecalis strain | ϕEf11(wild type) | ϕEf11(Δ61-1, ϕFL1C39-44)(spontaneous recombinant) | ϕEf11(Δ31-36, ΔPCRO, PnisA, Δ61-1,ϕFL1C39-44)(virulent mutant) | |
| OG1RF | – | – | – | |
| ER3/2s | – | – | – | |
| ER5/1 | – | – | + | |
| E1 | + | + | +* | |
| E2† | – | – | – | |
| E3† | – | – | – | |
| E4† | – | – | – | |
| E5† | – | – | – | |
| E6 | – | – | – | |
| E7† | – | – | – | |
| E8 | – | – | + | |
| E10 | – | – | + | |
| E11 | – | – | + | |
| GS1 | – | – | – | |
| GS2† | – | – | + | |
| GS3 | – | – | + | |
| GS4 | – | – | – | |
| GS5 | – | – | – | |
| GS6 | – | – | + | |
| GS7 | – | – | + | |
| GS8† | – | – | + | |
| GS9† | – | – | – | |
| GS10 | – | – | – | |
| GS12 | – | – | – | |
| GS13 | – | – | + | |
| GS14 | – | – | +* | |
| GS15 | – | – | + | |
| GS16 | – | – | + | |
| GS17 | – | – | – | |
| GS18 | – | – | – | |
| GS19 | – | – | + | |
| GS21 | – | – | – | |
| GS22† | – | – | + | |
| GS23† | – | – | – | |
| GS24 | – | – | + | |
| GS25† | – | – | + | |
| GS26 | – | – | + | |
| GS27 | – | – | + | |
| GS28 | – | – | – | |
| GS29† | – | – | – | |
| GS30 | – | – | +* | |
| GS31 | – | – | – | |
| GS32 | – | – | – | |
| GS33† | – | – | – | |
| GS34 | – | – | – | |
| OS25 | – | – | + | |
| AA-OR3 | – | – | + | |
| AA-OR4 | – | – | + | |
| AA-OR26 | – | – | +* | |
| AA-OR34† | – | – | – | |
| AA-T4 | – | – | + | |
| AA-26 | – | – | +* | |
| V583 | – | – | + | |
| OS16 | – | – | + | |
| TUSoD1 | +* | +* | + | |
| TUSoD2 | – | – | – | |
| TUSoD3 | – | – | – | |
| TUSoD9 | – | – | + | |
| TUSoD10 | – | – | – | |
| TUSoD12 | – | – | – | |
| TUSoD15 | – | – | – | |
| TUSoD17 | – | – | – | |
| TUSoD18 | +* | – | – | |
| MMH594 | – | – | – | |
| OG1SSP | – | – | + | |
| DG16 | – | – | – | |
| JH2-2 | + | + | + | |
| Cumulative | 6.0% | 4.5% | 49.3% | |
*Sensitive to phage (spot test).
†Lysogenic E. faecalis strain containing ϕEf11 prophage.
+, Sensitive to phage (plaque assay); –, not sensitive to phage.
Discussion
The outcome between the competing, alternative life cycles of lysogeny and productive infection in temperate bacterial viruses is largely determined by the presence or absence of functional early expression genes, such as those coding for the repressor and the integrase proteins, and the related early gene promoters (Ptashne, 2004). In the case of bacteriophage ϕEf11, a genomic module of six contiguous putative early expression genes with lysogeny-related functions was identified by sequencing and homology comparison with known lysogeny-related genes of other bacterial viruses (Stevens et al., 2011). In the same study, a region of the ϕEf11 genome with marked similarity to the PR and PL early promoter region of the temperate lactococcal bacteriophage TP901-1 (Madsen & Hammer, 1998) was also detected, suggesting a similar regulatory function in ϕEf11. To confirm the predicted functions of these regions of the ϕEf11 genome, and to develop a potentially useful agent for phage therapy, we wished to determine whether deletion or replacement of these sites in phage ϕEf11 would result in a derivative virus with virulent rather than temperate properties. There is substantial precedent for temperate to virulent conversion of phage by modification of these genomic sites (Bailone & Devoret, 1978; Flashman, 1978; Donnelly-Wu et al., 1993; Bruttin & Brüssow, 1996; Ford et al., 1998).
Our previous analysis of the phage ϕEf11 genome concluded that a module of six contiguous genes (ORFs 31–36) was responsible for functions related to the establishment of lysogeny (Stevens et al., 2011). This module included a putative integrase (ORF 31) and a putative cI repressor (ORF 36). In the present investigation, we found that these six genes are completely dispensable for lytic cycle function, as deletion of these genes did not prevent productive infection by the virus. Infection of lawns of host cells by the mutant virus lacking these genes produced clear plaques. Furthermore, we were unable to recover surviving (presumptive lysogenic) cells from the plaques produced by the mutant virus lacking ORFs 31–36. Therefore, these data suggest that the predicted lysogeny-related functions of these genes were correct, and that by deleting these genes from the viral genome, we have generated a ϕEf11 mutant that is incapable of lysogeny. It should be noted, however, that complementation studies to confirm the function of these (putative lysogeny-related) genes could not be carried out because the ϕEf11(vir)PnisA genome includes a nisin promoter, in place of the wild-type cro promoter, which is not sensitive to repression by the cI gene product (see below).
Similarly, we previously identified a stem–loop structure surrounded by PL and PR promoter sequences in the ϕEf11 genome lying between a putative cI repressor gene and a putative cro gene, which was highly similar to the PR/PL promoter/operator region of Lactococcus bacteriophage TP901-1 (Stevens et al., 2011; Madsen & Hammer 1998). In deleting this sequence, and replacing it with a nisin-inducible promoter, we have generated a virus that was capable of productively infecting E. faecalis (ϕEf11) lysogens, in the presence of the ϕEf11 cI repressor protein. Therefore, these data support our computationally based prediction that this region of the ϕEf11 genome (i.e. between ORFs 36 and 37) is a regulatory sequence that determines the outcome of phage infection between lysogeny and a productive infective cycle. Thus, it appears that the ϕEf11(vir)pnisA variant resulting from replacing the wild-type PR/PL promoter/operator sequence with a nisin-inducible promoter, PnisA, is indeed insensitive to cI repression.
Surprisingly, spontaneous recombinational replacement of five genes (ORFs 61–65) of the DNA replication/modification module and one gene (ORF 1/terminase A) of the packaging module by five genes (ORFs 40–44) of E. faecalis phage ϕFL1C also had an effect on the virulence properties of the virus. While this genetic recombination had no effect upon host range, it did markedly alter the lytic properties observed during infection of either broth cultures or soft agar overlay lawns of susceptible host cells. Broth cultures rapidly and more thoroughly cleared, after infection by the recombinant phage ϕEf11(Δ61-1, ϕFL1C40-44), as compared with infection by the wild-type ϕEf11 virus. Similarly, plaques produced by the recombinant phage ϕEf11(Δ61-1, ϕFL1C40-44) appeared as large, extensively spreading lytic zones with a clearer centre, compared with those formed by the wild-type ϕEf11 virus. As the predicted function of the replaced genes involved either DNA replication/modification (ORFs 61–65), or packaging (ORF 1), we hypothesize that the replacement (ϕFL1C) genes contributed to a more robust, more productive lytic infection by increasing the efficiency of either phage DNA synthesis or packaging, or both.
The results of one-step growth experiments for wild-type ϕEf11 and recombinant ϕEf11(Δ61-1, ϕFL1C40-44) phages appear to bear out the above hypothesis that recombination of ϕEf11 with the ϕFL1C genes results in a greatly (>100-fold) enhanced production of progeny virus (Fig. 3). As both the wild-type (ϕEf11) and the recombinant ϕEf11(Δ61-1, ϕFL1C40-44) phages are temperate, and we do not know what proportion of infected cells undergoes productive infection and what proportion becomes lysogens, we cannot accurately determine a burst size for either strain (although the latent period of both strains, approx. 30 min, appears to be similar). That is, it is not possible to determine from these data whether the difference in titre observed between the two phage strains is due to a greater burst size produced by the recombinant or a higher proportion of cells in the population that undergoes lytic infection (or both). Nevertheless, these data do demonstrate that a much greater titre of virus is produced in E. faecalis populations infected by the ϕEf11(Δ61-1, ϕFL1C40-44) recombinant as compared with the wild-type virus.
In addition, the source of the ϕFL1C genes (i.e., the E. faecalis JH2-2 chromosome) was unexpected, as previous studies reported that this E. faecalis strain was susceptible to ϕFL1C infection, and in fact could form ϕFL1C lysogens following ϕFL1C infection, suggesting that this strain did not initially harbour a ϕFL1C prophage (Yasmin et al., 2010). This conundrum was solved when we used PCR to attempt to detect other regions of the ϕFL1C genome in JH2-2. No other regions of the ϕFL1C genome could be detected in JH2-2, suggesting that the ϕFL1C sequence that we detected was part of a defective (incomplete) prophage, or was the only ϕFL1C-like portion of a complete prophage. Under such circumstances, it would not be surprising for JH2-2 cells (lacking the ϕFL1C immunity functions) to be susceptible to ϕFL1C infection, while being the source of a limited number of ϕFL1C genes.
The lack of adsorption of either the wild-type or the spontaneous recombinant phage ϕEf11(Δ61-1, ϕFL1C40-44) by lysogenic E. faecalis strain TUSoD11 suggests either that this lysogen lacked the phage receptor, or that the phage receptor had been modified, rendering it incapable of binding the phage ligand. The fact that curing TUSoD11 rendered it capable of adsorbing ϕEf11 supports the notion that lysogeny either altered or eliminated the phage receptor on the cell surface. Prophage-mediated modification of phage receptors has been long known and well documented for several other phage/host systems (Uetake et al., 1958; Holloway & Cooper, 1962; Losick & Robbins, 1967; Castillo & Bartell, 1974; Gemski et al., 1975; Kuzio & Kropinski, 1983; Tomás & Kay, 1984). Enzymes specified by these phages catalyse modifications of phage receptor sites (e.g. O-acetylation of the lipopolysaccharide side chains, changes in the bonding between the lipopolysaccharide trisaccharide units from α1→4 to β1→4) on the host cell, resulting in the inability of the cell to adsorb additional phage. Although the previous examples of phage receptor modification involve Gram-negative bacteria, it is possible that a similar phenomenon may occur in E. faecalis as the phage receptors in most Gram-positive, low G+C bacteria are cell surface polysaccharides (Vidaver & Brock, 1966; Douglas & Wolin, 1971; Cleary et al., 1977; Yokokura 1977; Keogh & Pettingill, 1983; Valyasevi et al., 1990; Schäfer et al., 1991). If this is the case, then evolutionarily speaking, it may be that originally E. faecalis TUSoD11 was non-lysogenic and possessed cell-surface phage receptors. Upon infection and lysogenization by a temperate phage ϕEf11 at some time in the past, the cell’s phage receptors were lost or modified, resulting in a non-phage-adsorbable cell surface. However, once cured of the phage, the cell surface changed back to its original form that could again adsorb the phage and support virus infection. However, note that we found some lysogenic E. faecalis strains to be sensitive to ϕEf11(vir)PnisA, indicating that they must be able to adsorb the virus. Why some lysogens (e.g. E. faecalis GS8) should be able to adsorb the phage while others (e.g. TUSoD11) do not remains to be determined.
In summary, we have developed a virulent variant of temperate E. faecalis phage ϕEf11 that is incapable of lysogeny, is insensitive to cI repression, is highly lytic and has a greatly extended host range compared with the wild-type virus. In the course of constructing this virus we have increased our understanding of the genome of phage ϕEf11 by confirming the predicted function of several of the genomic loci. These include regions responsible for establishing and maintaining lysogeny. In addition, we have determined that incorporating allelic alternatives in a region of the genome responsible for DNA replication/modification and packaging results in enhancement of productive infection in the host cell population, thereby capturing phage mozaicism happening in real-time.
We recognize that further genetic modifications will be necessary to develop a phage that may be clinically useful in managing enterococcal infections. Replacing the repressor-sensitive cro promoter with the repressor-insensitive, nisin-inducible promoter system to drive phage lytic infection functions proved to be a very effective and useful strategy in making genetic modifications in the virus. However, because the inducing agent, nisin, is a toxic protein, this approach would not be suitable in an agent designed for therapeutic application. Consequently, we are exploring alternative, constitutive promoters to replace the nisin-inducible promoter in variants of the ϕEf11(vir)PnisA strain. Similarly, we used antibiotic (erythromycin) resistance to select transformant lysogen clones containing prophages with the desired genotype. It would be necessary to delete this determinant prior to using this agent clinically. Finally, while the virulent variant phage that we have constructed possesses an eminently greater host range than the wild-type virus, there are still E. faecalis strains that do not appear to be sensitive to this virus. We are exploring additional strategies aimed at further expanding the host range of the ϕEf11(vir)PnisA virulent variant that we have constructed.
Acknowledgements
This work was supported by the National Institute of Dental & Craniofacial Research Public Health Service grant 1R15DE021016-01.
Footnotes
One supplementary table and five supplementary figures are available with the online version of this paper.
References
- Bailone A., Devoret R. (1978). Isolation of ultravirulent mutants of phage λ. Virology 84, 547–550. 10.1016/0042-6822(78)90273-8 [DOI] [PubMed] [Google Scholar]
- Biedenbach D. J., Moet G. J., Jones R. N. (2004). Occurrence and antimicrobial resistance pattern comparisons among bloodstream infection isolates from the SENTRY Antimicrobial Surveillance Program (1997–2002). Diagn Microbiol Infect Dis 50, 59–69. 10.1016/j.diagmicrobio.2004.05.003 [DOI] [PubMed] [Google Scholar]
- Bonten M. J. M., Willems R., Weinstein R. A. (2001). Vancomycin-resistant enterococci: why are they here, and where do they come from? Lancet Infect Dis 1, 314–325. 10.1016/S1473-3099(01)00145-1 [DOI] [PubMed] [Google Scholar]
- Bruttin A., Brüssow H. (1996). Site-specific spontaneous deletions in three genome regions of a temperate Streptococcus thermophilus phage. Virology 219, 96–104. 10.1006/viro.1996.0226 [DOI] [PubMed] [Google Scholar]
- Bryan E. M., Bae T., Kleerebezem M., Dunny G. M. (2000). Improved vectors for nisin-controlled expression in gram-positive bacteria. Plasmid 44, 183–190. 10.1006/plas.2000.1484 [DOI] [PubMed] [Google Scholar]
- Castillo F. J., Bartell P. F. (1974). Studies on the bacteriophage 2 receptors of Pseudomonas aeruginosa. J Virol 14, 904–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary P. P., Wannamaker L. W., Fisher M., Laible N. (1977). Studies of the receptor for phage A25 in group A streptococci: the role of peptidoglycan in reversible adsorption. J Exp Med 145, 578–593. 10.1084/jem.145.3.578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly-Wu M. K., Jacobs W. R., Jr, Hatfull G. F. (1993). Superinfection immunity of mycobacteriophage L5: applications for genetic transformation of mycobacteria. Mol Microbiol 7, 407–417. 10.1111/j.1365-2958.1993.tb01132.x [DOI] [PubMed] [Google Scholar]
- Douglas L. J., Wolin M. J. (1971). Cell wall polymers and phage lysis of Lactobacillus plantarum. Biochemistry 10, 1551–1555. 10.1021/bi00785a007 [DOI] [PubMed] [Google Scholar]
- Dunny G. M., Craig R. A., Carron R. L., Clewell D. B. (1979). Plasmid transfer in Streptococcus faecalis: production of multiple sex pheromones by recipients. Plasmid 2, 454–465. 10.1016/0147-619X(79)90029-5 [DOI] [PubMed] [Google Scholar]
- Edgeworth J. D., Treacher D. F., Eykyn S. J. (1999). A 25-year study of nosocomial bacteremia in an adult intensive care unit. Crit Care Med 27, 1421–1428. 10.1097/00003246-199908000-00002 [DOI] [PubMed] [Google Scholar]
- Eliopoulos G. M., Wennersten C. B., Gold H. S., Schülin T., Souli M., Farris M. G., Cerwinka S., Nadler H. L., Dowzicky M. & other authors (1998). Characterization of vancomycin-resistant Enterococcus faecium isolates from the United States and their susceptibility in vitro to dalfopristin-quinupristin. Antimicrob Agents Chemother 42, 1088–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emori T. G., Gaynes R. P. (1993). An overview of nosocomial infections, including the role of the microbiology laboratory. Clin Microbiol Rev 6, 428–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flashman S. M. (1978). Mutational analysis of the operators of bacteriophage lambda. Mol Gen Genet 166, 61–73. 10.1007/BF00379730 [DOI] [PubMed] [Google Scholar]
- Ford M. E., Sarkis G. J., Belanger A. E., Hendrix R. W., Hatfull G. F. (1998). Genome structure of mycobacteriophage D29: implications for phage evolution. J Mol Biol 279, 143–164. 10.1006/jmbi.1997.1610 [DOI] [PubMed] [Google Scholar]
- Gemski P., Jr, Koeltzow D. E., Formal S. B. (1975). Phage conversion of Shigella flexneri group antigens. Infect Immun 11, 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharoff P., Figurski D. H., Stevens R. H., Fine D. H. (1993). Identification of Actinobacillus actinomycetemcomitans: polymerase chain reaction amplification of lktA-specific sequences. Oral Microbiol Immunol 8, 105–110. 10.1111/j.1399-302X.1993.tb00554.x [DOI] [PubMed] [Google Scholar]
- Gonzales R. D., Schreckenberger P. C., Graham M. B., Kelkar S., DenBesten K., Quinn J. P. (2001). Infections due to vancomycin-resistant Enterococcus faecium resistant to linezolid. Lancet 357, 1179. 10.1016/S0140-6736(00)04376-2 [DOI] [PubMed] [Google Scholar]
- Grayson M. L., Eliopoulos G. M., Wennersten C. B., Ruoff K. L., De Girolami P. C., Ferraro M. J., Moellering R. C., Jr (1991). Increasing resistance to beta-lactam antibiotics among clinical isolates of Enterococcus faecium: a 22-year review at one institution. Antimicrob Agents Chemother 35, 2180–2184. 10.1128/AAC.35.11.2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havard C. W., Garrod L. P., Waterworth P. M. (1959). Deaf or dead? A case of subacute bacterial endocarditis treated with penicillin and neomycin. BMJ 1, 688–689. 10.1136/bmj.1.5123.688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero I. A., Issa N. C., Patel R. (2002). Nosocomial spread of linezolid-resistant, vancomycin-resistant Enterococcus faecium. N Engl J Med 346, 867–869. 10.1056/NEJM200203143461121 [DOI] [PubMed] [Google Scholar]
- Hidron A. I., Schuetz A. N., Nolte F. S., Gould C. V., Osborn M. K. (2008). Daptomycin resistance in Enterococcus faecalis prosthetic valve endocarditis. J Antimicrob Chemother 61, 1394–1396. 10.1093/jac/dkn105 [DOI] [PubMed] [Google Scholar]
- Holloway B. W., Cooper G. N. (1962). Lysogenic conversion in Pseudomonas aeruginosa. J Bacteriol 84, 1321–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A. E., Hobbs S. J. (1974). Conjugal transfer of plasmid-borne multiple antibiotic resistance in Streptococcus faecalis var. zymogenes. J Bacteriol 117, 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jett B. D., Huycke M. M., Gilmore M. S. (1994). Virulence of enterococci. Clin Microbiol Rev 7, 462–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A. P., Mushtaq S., Warner M., Livermore D. M. (2004). Activity of daptomycin against multi-resistant Gram-positive bacteria including enterococci and Staphylococcus aureus resistant to linezolid. Int J Antimicrob Agents 24, 315–319. 10.1016/j.ijantimicag.2004.04.006 [DOI] [PubMed] [Google Scholar]
- Johnson E. M., Flannagan S. E., Sedgley C. M. (2006). Coaggregation interactions between oral and endodontic Enterococcus faecalis and bacterial species isolated from persistent apical periodontitis. J Endod 32, 946–950. 10.1016/j.joen.2006.03.023 [DOI] [PubMed] [Google Scholar]
- Kanafani Z. A., Federspiel J. J., Fowler V. G., Jr (2007). Infective endocarditis caused by daptomycin-resistant Enterococcus faecalis: a case report. Scand J Infect Dis 39, 75–77. 10.1080/00365540600786465 [DOI] [PubMed] [Google Scholar]
- Kelesidis T., Humphries R., Uslan D. Z., Pegues D. A. (2011). Daptomycin nonsusceptible enterococci: an emerging challenge for clinicians. Clin Infect Dis 52, 228–234. 10.1093/cid/ciq113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keogh B. P., Pettingill G. (1983). Adsorption of bacteriophage eb7 on Streptococcus cremoris EB7. Appl Environ Microbiol 45, 1946–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzio J., Kropinski A. M. (1983). O-antigen conversion in Pseudomonas aeruginosa PAO1 by bacteriophage D3. J Bacteriol 155, 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis C. M., Zervos M. J. (1990). Clinical manifestations of enterococcal infection. Eur J Clin Microbiol Infect Dis 9, 111–117. 10.1007/BF01963635 [DOI] [PubMed] [Google Scholar]
- Linden P. K. (2007). Optimizing therapy for vancomycin-resistant enterococci (VRE). Semin Respir Crit Care Med 28, 632–645. 10.1055/s-2007-996410 [DOI] [PubMed] [Google Scholar]
- Losick R., Robbins P. W. (1967). Mechanism of ϵ-15 conversion studies with a bacterial mutant. J Mol Biol 30, 445–455. 10.1016/0022-2836(67)90361-0 [DOI] [PubMed] [Google Scholar]
- Madsen P. L., Hammer K. (1998). Temporal transcription of the lactococcal temperate phage TP901-1 and DNA sequence of the early promoter region. Microbiology 144, 2203–2215. 10.1099/00221287-144-8-2203 [DOI] [PubMed] [Google Scholar]
- Marshall B. M., Ochieng D. G., Levy S. B. (2009). Commensals: underappreciated reservoir of antibiotic resistance. Microbe 4, 231–238. [Google Scholar]
- Megran D. W. (1992). Enterococcal endocarditis. Clin Infect Dis 15, 63–71. 10.1093/clinids/15.1.63 [DOI] [PubMed] [Google Scholar]
- Moellering R. C., Jr (1992). Emergence of Enterococcus as a significant pathogen. Clin Infect Dis 14, 1173–1176. 10.1093/clinids/14.6.1173 [DOI] [PubMed] [Google Scholar]
- Munoz-Price L. S., Lolans K., Quinn J. P. (2005). Emergence of resistance to daptomycin during treatment of vancomycin-resistant Enterococcus faecalis infection. Clin Infect Dis 41, 565–566. 10.1086/432121 [DOI] [PubMed] [Google Scholar]
- Murray B. E., Mederski-Samaroj B. (1983). Transferable β-lactamase. A new mechanism for in vitro penicillin resistance in Streptococcus faecalis. J Clin Invest 72, 1168–1171. 10.1172/JCI111042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Nosocomial Infections Surveillance System (2004). National Nosocomial Infections Surveillance (NNIS) System Report, data summary from January 1992 through June 2004, issued October 2004. Am J Infect Control 32, 470–485. 10.1016/j.ajic.2004.10.001 [DOI] [PubMed] [Google Scholar]
- Ntokou E., Stathopoulos C., Kirsto I., Dimitroulia E., Labrou M., Vasdeki A., Makris D., Zakynthinos E., Tsakris A., Pournaras S. (2012). Intensive care unit dissemination of multiple clones of linezolid-resistant Enterococcus faecalis and Enterococcus faecium. J Antimicrob Chemother 67, 1819–1823. [DOI] [PubMed] [Google Scholar]
- Peciuliene V., Balciuniene I., Eriksen H. M., Haapasalo M. (2000). Isolation of Enterococcus faecalis in previously root-filled canals in a Lithuanian population. J Endod 26, 593–595. 10.1097/00004770-200010000-00004 [DOI] [PubMed] [Google Scholar]
- Pinheiro E. T., Gomes B. P. F. A., Ferraz C. C. R., Sousa E. L. R., Teixeira F. B., Souza-Filho F. J. (2003). Microorganisms from canals of root-filled teeth with periapical lesions. Int Endod J 36, 1–11. 10.1046/j.1365-2591.2003.00603.x [DOI] [PubMed] [Google Scholar]
- Prystowsky J., Siddiqui F., Chosay J., Shinabarger D. L., Millichap J., Peterson L. R., Noskin G. A. (2001). Resistance to linezolid: characterization of mutations in rRNA and comparison of their occurrences in vancomycin-resistant enterococci. Antimicrob Agents Chemother 45, 2154–2156. 10.1128/AAC.45.7.2154-2156.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M. (2004). A Genetic Switch, 3rd edn Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Richards M. J., Edwards J. R., Culver D. H., Gaynes R. P. (2000). Nosocomial infections in combined medical–surgical intensive care units in the United States. Infect Control Hosp Epidemiol 21, 510–515. 10.1086/501795 [DOI] [PubMed] [Google Scholar]
- Ross J. E., Farrell D. J., Mendes R. E., Sader H. S., Jones R. N. (2011). Eight-year (2002–2009) summary of the linezolid (Zyvox® Annual Appraisal of Potency and Spectrum; ZAAPS) program in European countries. J Chemother 23, 71–76. [DOI] [PubMed] [Google Scholar]
- Sahm D. F., Kissinger J., Gilmore M. S., Murray P. R., Mulder R., Solliday J., Clarke B. (1989). In vitro susceptibility studies of vancomycin-resistant Enterococcus faecalis. Antimicrob Agents Chemother 33, 1588–1591. 10.1128/AAC.33.9.1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A., Geis A., Neve H., Teuber M. (1991). Bacteriophage receptors of Lactobacillus lactis subsp. ‘diacetylactis’ F7/2 and Lactococcus lactis subsp. cremoris Wg2–1. FEMS Microbiol Lett 78, 69–74. [DOI] [PubMed] [Google Scholar]
- Sedgley C. M., Lennan S. L., Clewell D. B. (2004). Prevalence, phenotype and genotype of oral enterococci. Oral Microbiol Immunol 19, 95–101. 10.1111/j.0902-0055.2004.00122.x [DOI] [PubMed] [Google Scholar]
- Sedgley C. M., Molander A., Flannagan S. E., Nagel A. C., Appelbe O. K., Clewell D. B., Dahlén G. (2005a). Virulence, phenotype and genotype characteristics of endodontic Enterococcus spp. Oral Microbiol Immunol 20, 10–19. 10.1111/j.1399-302X.2004.00180.x [DOI] [PubMed] [Google Scholar]
- Sedgley C. M., Nagel A. C., Shelburne C. E., Clewell D. B., Appelbe O., Molander A. (2005b). Quantitative real-time PCR detection of oral Enterococcus faecalis in humans. Arch Oral Biol 50, 575–583. 10.1016/j.archoralbio.2004.10.017 [DOI] [PubMed] [Google Scholar]
- Sedgley C. M., Buck G., Appelbe O. K. (2006). Prevalence of Enterococcus faecalis at multiple oral sites in endodontic patients using culture and PCR. J Endod 32, 104–109. 10.1016/j.joen.2005.10.022 [DOI] [PubMed] [Google Scholar]
- Shepard B. D., Gilmore M. S. (1995). Electroporation and efficient transformation of Enterococcus faecalis grown in high concentrations of glycine. Methods Mol Biol 47, 217–226. [DOI] [PubMed] [Google Scholar]
- Siqueira J. F., Jr, Rôças I. N. (2004). Polymerase chain reaction-based analysis of microorganisms associated with failed endodontic treatment. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 97, 85–94. 10.1016/S1079-2104(03)00353-6 [DOI] [PubMed] [Google Scholar]
- Stevens R. H., Porras O. D., Delisle A. L. (2009). Bacteriophages induced from lysogenic root canal isolates of Enterococcus faecalis. Oral Microbiol Immunol 24, 278–284. 10.1111/j.1399-302X.2009.00506.x [DOI] [PubMed] [Google Scholar]
- Stevens R. H., Ektefaie M. R., Fouts D. E. (2011). The annotated complete DNA sequence of Enterococcus faecalis bacteriophage φEf11 and its comparison with all available phage and predicted prophage genomes. FEMS Microbiol Lett 317, 9–26. 10.1111/j.1574-6968.2010.02203.x [DOI] [PubMed] [Google Scholar]
- Stuart C. H., Schwartz S. A., Beeson T. J., Owatz C. B. (2006). Enterococcus faecalis: its role in root canal treatment failure and current concepts in retreatment. J Endod 32, 93–98. 10.1016/j.joen.2005.10.049 [DOI] [PubMed] [Google Scholar]
- Sundqvist G., Figdor D., Persson S., Sjögren U. (1998). Microbiologic analysis of teeth with failed endodontic treatment and the outcome of conservative re-treatment. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 85, 86–93. 10.1016/S1079-2104(98)90404-8 [DOI] [PubMed] [Google Scholar]
- Tenover F. C., McDonald L. C. (2005). Vancomycin-resistant staphylococci and enterococci: epidemiology and control. Curr Opin Infect Dis 18, 300–305. 10.1097/01.qco.0000171923.62699.0c [DOI] [PubMed] [Google Scholar]
- Tomás J. M., Kay W. W. (1984). Effect of bacteriophage P1 lysogeny on lipopolysaccharide composition and the lambda receptor of Escherichia coli. J Bacteriol 159, 1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetake H., Luria S. E., Burrous J. W. (1958). Conversion of somatic antigens in Salmonella by phage infection leading to lysis or lysogeny. Virology 5, 68–91. 10.1016/0042-6822(58)90006-0 [DOI] [PubMed] [Google Scholar]
- Uttley A. H. C., Collins C. H., Naidoo J., George R. C. (1988). Vancomycin-resistant enterococci. Lancet i, 57–58. 10.1016/S0140-6736(88)91037-9 [DOI] [PubMed] [Google Scholar]
- Valyasevi R., Sandine W. E., Geller B. L. (1990). The bacteriophage kh receptor of Lactococcus lactis subsp. cremoris KH is the rhamnose of the extracellular wall polysaccharide. Appl Environ Microbiol 56, 1882–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidaver A. K., Brock T. D. (1966). Purification and properties of a bacteriophage receptor material from Streptococcus faecium. Biochim Biophys Acta 121, 298–314. 10.1016/0304-4165(66)90119-X [DOI] [PubMed] [Google Scholar]
- Yasmin A., Kenny J. G., Shankar J., Darby A. C., Hall N., Edwards C., Horsburgh M. J. (2010). Comparative genomics and transduction potential of Enterococcus faecalis temperate bacteriophages. J Bacteriol 192, 1122–1130. 10.1128/JB.01293-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokokura T. (1977). Phage receptor material in Lactobacillus casei. J Gen Microbiol 100, 139–145. [DOI] [PubMed] [Google Scholar]
- Zoletti G. O., Siqueira J. F., Jr, Santos K. R. N. (2006). Identification of Enterococcus faecalis in root-filled teeth with or without periradicular lesions by culture-dependent and -independent approaches. J Endod 32, 722–726. 10.1016/j.joen.2006.02.001 [DOI] [PubMed] [Google Scholar]