

Abstract
The capsids of ssRNA phages comprise a single copy of an ~45 kDa maturation protein that serves to recognize the conjugative pilus as receptor, to protect the ends of the viral RNA and also to escort the genomic RNA into the host cytoplasm. In the Alloleviviridae, represented by the canonical phage Qβ, the maturation protein A2 also causes lysis. This is achieved by inhibiting the activity of MurA, which catalyses the first committed step of murein biosynthesis. Previously, it was shown that Qβ virions, with a single copy of A2, inhibit MurA activity. This led to a model for lysis timing in which, during phage infection, A2 is not active as a MurA inhibitor until assembled into virion particles, thus preventing premature lysis before a sufficient yield of viable progeny has accumulated. Here we report that MurA inactivates purified Qβ particles, casting doubt on the notion that A2 must assemble into particles prior to MurA inhibition. Furthermore, quantification of A2 protein induced from a plasmid indicated that lysis is entrained when the amount of the lysis protein is approximately equimolar to that of cellular MurA. Qβ por mutants, isolated as suppressors that overcome a murArat mutation that reduces the affinity of MurA for A2, were shown to be missense mutations in A2 that increase the translation of the maturation protein. Because of the increased production of A2, the por mutants have an attenuated infection cycle and reduced burst size, indicating that a delicate balance between assembled and unassembled A2 levels regulates lysis timing.
Introduction
Three lytic phages with single-strand nucleic acid genomes – Alloleviviridae (ssRNA; Qβ), Leviviridae (ssRNA; MS2) and Microviridae (ssDNA; ϕX174) – are known to effect lysis by expression of a single gene without muralytic activity (Bernhardt et al., 2002). Of these, two cause lysis by inhibiting an essential enzyme in the murein precursor pathway: Qβ A2 inhibits MurA (Bernhardt et al., 2001) and ϕX174 E inhibits MraY (Bernhardt et al., 2000). In both cases, lysis occurs when the host cell attempts to septate in the absence of murein synthesis. The third, MS2 L, effects lysis by an unknown mechanism.
Qβ has long been one of the paradigm experimental systems for studying viral RNA-dependent RNA polymerase and RNA evolution, in part because of its simplicity (Blumenthal & Carmichael, 1979; Domingo et al., 1978; Domingo & Holland, 1997; Drake, 1993). The Qβ genome consists of only three genes, encoding four proteins: maturation or A2, coat, read-through coat or A1 and replicase (Fig. 1a). A single copy of A2 is mounted on the Qβ virion, comprising a T = 3 capsid containing ~165 copies of coat, ~15 copies of A1 and the 4.2 kb ssRNA (van Duin & Tsareva, 2006). Among the three simple phage systems, the Qβ lysis system is unique in that the lysis protein, A2, has other functions: it protects the genomic RNA from RNase degradation (Weber et al., 1975), mediates specific adsorption to the F-pilus and chaperones the genome into the host cytoplasm (Kozak & Nathans, 1971). As the other functions of A2 require binding to Qβ RNA, the phage particle and the host F-pilus, an assortment of potential regulatory modes are possible in the infected cell. A2 expression is tightly regulated by extensive RNA secondary structure surrounding its translational initiation region (Beekwilder et al., 1996; Groeneveld et al., 1995) (Fig. 1b), and in vitro translation experiments indicate that A2 is produced only from nascent transcripts (Robertson & Lodish, 1970; Staples et al., 1971).
Fig. 1.
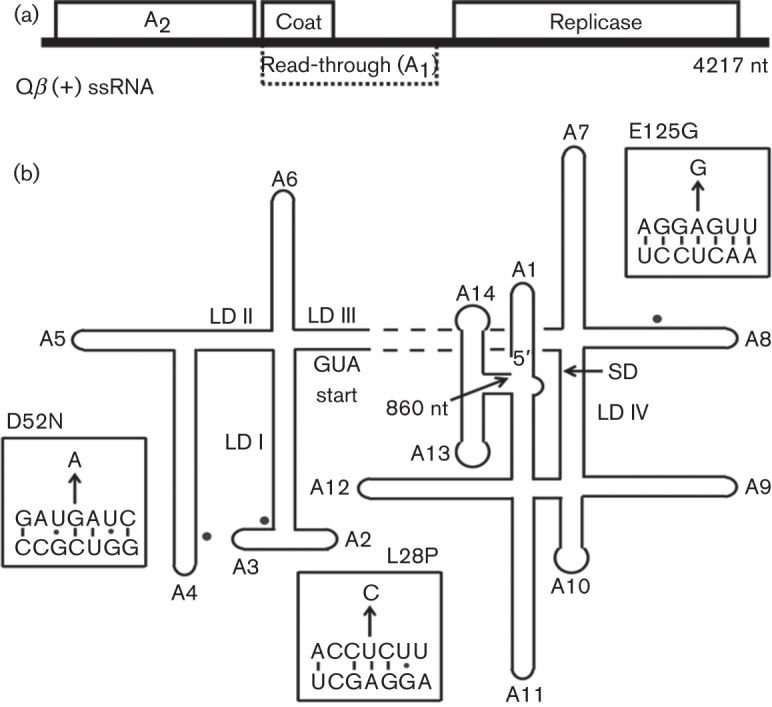
Qβ genome. (a) Qβ is a (+) ssRNA phage with a 4.2 kb genome. Replication of the chromosome is dependent on encoded replicase and four additional host proteins. The Qβ capsid is composed of three gene products: coat, the major virion structural protein; A1, a minor component translated from read-through of the leaky coat UGA stop codon; and a single copy of A2. (b) RNA secondary structure of Qβ 5′-end (nucleotides 1–860). Stem-loops and long-distance interactions (LD) are drawn and labelled according to Beekwilder et al. (1996). Single nucleotide substitutions of Qβ por mutants are depicted with filled circles, with specific sequence substitutions shown in boxes (arrow). SD, Shine–Dalgarno.
Winter & Gold (1983) revealed another function for this protein by showing that induction of A2 cloned on a medium copy plasmid was necessary and sufficient to induce host lysis. Nearly 20 years later, Bernhardt et al. (2001) discovered that induction of a plasmid-borne cDNA copy of A2 caused cell-wall synthesis to cease at ~20 min prior to lysis, indicating that A2 was acting as a ‘protein antibiotic’ targeting murein biosynthesis. To address the mechanism of Qβ lysis, survivors of A2 induction were screened for insensitivity to viral infection. Two independent rat (resistance to A2) mutants were mapped to murA, which encodes UDP-N-acetylglucosamine enolpyruvyltransferase, the enzyme catalysing the committed step for the biosynthetic pathway of murein precursors. Both rat alleles proved to have a single missense change, Leu138Gln (L138Q). Biochemical studies of A2 were impeded due to insolubility of the protein apart from the capsid (Bernhardt et al., 2001, 2002). However, MurA activity in a crude lysate was shown to be inhibited by intact Qβ particles. This led to a model for regulation of lysis in Qβ infections in which A2 does not become inhibitory until fully assembled into the capsid (Hatfull, 2001).
Here we report experiments to test this model, in which we have quantified both MurA and A2 during the Qβ infection cycle and determined the critical concentration of A2 required for inhibition of MurA. In addition, the characteristics of Qβ mutants that escape the lysis block imposed by the MurA rat mutations are analysed. The results are discussed in terms of a new model for regulation of lysis in Qβ infections.
Methods
Bacterial strains and growth conditions.
Escherichia coli strains that were used in this study are listed in Table 1. XL1-Blue was used for all plasmid constructions. XL1-Bluerat cells were used for phage infections. HfrH and HfrH lacZ : : Tn5 served as lawns for bacteriophage plating and gradient induction for protein quantification, respectively. ER2738 was used for phage propagation and protein expression. Details of strain and plasmid construction are included in the Supplementary Information, available with the online version of this paper. Primers used for plasmid construction are listed in Table S1. E. coli strains were grown under aerobic conditions at 37 °C in standard Luria–Bertani (LB) medium supplemented with 100 µg ampicillin ml−1, 40 µg kanamycin ml−1 or 10 µg tetracycline ml−1 when appropriate. For Qβ infections, this medium was supplemented with 2 mM CaCl2.
Table 1. Strains and plasmids used in this study.
| Strain or plasmid | Genotype and relevant features | Source or reference |
| E. coli strain | ||
| XL1-Blue | recA endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′ proAB lacZΔM15 : : Tn10] | Stratagene |
| XL1-Bluerat | recA endA1 gyrA96 thi-1 hsdR17 supE44 relA1 murA1 lac [F′ proAB lacZΔM15 : : Tn10], murA1 contains the murArat1 mutation corresponding to an amino acid substitution of L138Q | Bernhardt et al. (2001) |
| HfrH | λ- fhuA : : Tn10 lacIq relA1 spoT1 thi-1 | Laboratory stock |
| HfrHrat | λ- fhuA : : Tn10 lacIq relA1 murA1 spoT1 thi-1 | This study |
| HfrH lacZ : : Tn5 | λ− lacIq1 lacZ : : Tn5 relA1 spoT1 thi-1 | Laboratory stock |
| HfrH ΔmurA : : kan | λ- fhuA : : Tn10 lacIq relA1 ΔmurA : : kan spoT1 thi-1 | This study |
| pZE12-murABs | ΔmurA : : kan transduced from strain ZK1745 | Brown et al. (1995) |
| ER2738 | (F′proA+B+ lacIq Δ(lacZ)M15 zzf : : Tn10) fhuA2 glnV Δ(lac-proAB) thi-1 Δ(hsdS-mcrB)5 | New England Biolabs |
| Plasmid | ||
| pZE12-A2 | Qβ A2 cloned under the PLlacO-1 promoter, which is IPTG-inducible | This study |
| pZE12-murA | E. coli murA cloned under the PLlacO-1 promoter, which is IPTG-inducible | Bernhardt et al. (2001) |
| pZE12-murAHis | An oligo-histidine tag (G2H6G2) was adjoined to the C terminus of murA | Reed et al. (2012) |
| pZE12-murABs | Bacillus subtilis murAA cloned under the PLlacO-1 promoter, which is IPTG-inducible | This study |
Qβ purification and titre.
Qβ particles used in in vitro inactivation experiments were purified via caesium chloride gradient centrifugation as described by Strauss & Sinsheimer (1963). Phage for infections was prepared from plate lysates. Approximately 105 p.f.u. of Qβ was included in a bacterial soft agar overlay. Plates were incubated at 37 °C for 16 h. The overlay was scraped into a tube with SM medium (Sambrook et al., 1989) and incubated for 1 h. The phage stock was centrifuged at 2800 g for 20 min and the supernatant was filtered (0.45 µm).
To measure released p.f.u. accumulation, 1 ml infected culture was pelleted, and the supernatant was titrated. Serial dilutions of samples were prepared and 100 µl aliquots were included in a soft agar overlay with a 200 μl aliquot of HfrH grown to mid-exponential phase. After incubation, as above, plaques were counted.
Phage inactivation assay.
Approximately 4×1011 p.f.u. of Qβ was mixed with 1 μg purified MurAHis protein, as previously described (Reed et al., 2012). A Qβ-only control as well as a sample including 1 μg BSA (Sigma-Aldrich) was prepared in parallel. Samples were brought to 20 μl with buffer (0.1 M Tris, pH 8.0) and incubated at 37 °C for 1 h. Samples were then brought to 1 ml with SM medium. Tenfold serial dilutions were prepared and 100 μl of various dilutions was included in the bacterial overlay for plating comparison. Plates were incubated at 37 °C for 6–8 h and efficiency of plating was assessed. RNase A (Sigma, 1.25 ng μl−1) was also included in the inactivation assay with purified particles and MurA.
Determination of A2 affinity for MurA.
Percentage viability of Qβ (~0.3 μM) was measured after 30 min incubation with increasing amounts of MurA. Determination of Ki was done by plotting viability versus MurA concentration with the graphing software KaleidaGraph (Synergy Software) and fitting to the following equation (Riley-Lovingshimer & Reinhart, 2001):
| V = (Vmax[MurA]+VoKm</sub>+Vo[MurA])/(Km+[MurA]) | (1) |
where Vo is the percentage viable when [MurA] = 0. Vmax is m1, Vo is m2 and the Ki (Km) is m3 (units of μM).
MurA quantification.
Quantification of MurA was performed by standard SDS-PAGE and immunoblotting procedures (Gründling et al., 2000). HfrH cellular samples were precipitated by incubating on ice for 30 min with 10 % TCA (trichloroacetic acid); pellets were washed with acetone, resuspended by boiling for 5 min in SDS-PAGE sample loading buffer [50 mM Tris/HCl, 2 % SDS, 5 % glycerol, 100 mM β-mercaptoethanol (pH 6.8)] and analysed on an 8 % Tris-Tricine gel. For quantification, a standard of purified MurAHis was prepared. Blots were probed with α-MurA raised against the synthetic peptide, CHGKRPKAVNVRTAP (GenScript), at 1 : 3000 dilution and goat-anti-rabbit secondary antibody (Thermo Fisher Scientific) at a 1 : 3000 dilution. The SuperSignal West Femto developer kit (Thermo Fisher Scientific) was used for chemiluminescence detection. A murA knockout in which the chromosomal murA was replaced with a kanamycin cassette and complemented with the heterologous murA from Bacillus subtilis (HfrH murA : : kan pZE12-murABs) was used as the negative control for MurA. Strain construction is described in the Supplementary Information.
A2 induction and quantification.
The critical concentration for A2-mediated lysis was determined by modulating A2 expression with graded inductions of HfrH lacZ : : Tn5 pZE12-A2 at final IPTG concentrations of 0, 12.5, 25, 50, 100 and 1000 μM. This strain is phenotypically LacY− due to polarity of the Tn5 insertion into lacZ; the permease is not expressed, so induction of the lac promoter is linearly dependent on inducer concentration (Jensen et al., 1993; Siegele & Hu, 1997). Cell growth was monitored as a function of absorbance (A550) versus time. At various time points, samples were taken for TCA precipitation as described above. SDS-PAGE and immunoblotting was performed, as described above, with a 1 : 10 000 dilution of α-A2 raised against a synthetic peptide, PKLPRGLRFGA (Bethyl Laboratories), and 1 : 3000 dilution of goat-anti-rabbit 2° antibody (Thermo Fisher Scientific). An A2 standard was obtained by purifying inclusion bodies from cells expressing A2 protein. The inclusion bodies were processed as described by Palmer & Wingfield (2001) and dissolved in 10 % SDS. The concentration of the purified A2 protein was determined by amino acid analysis, performed by the Protein Chemistry Laboratory (Texas A&M University).
Quantification of A2 during an infection was performed as described above, with samples of XL1-Blue cells infected with an input m.o.i. of 1 Qβ p.f.u. per cell. The amount of A2 per cell was determined by measuring the total amount of A2 ml−1, dividing by the total number of cells ml−1, and multiplying by 1.6 to correct for the expected number of uninfected cells based on Poisson statistics.
Results
MurA inactivates Qβ particles
We were initially interested in attempting to purify a Qβ-MurA complex. However, when we incubated purified Qβ (~0.3 μM) with purified MurA (1 μM), we were surprised to find that the virions were quantitatively inactivated (Fig. 2a, b). In contrast, identical incubations with BSA had no effect on plating efficiency (Fig. 2c). Occlusion of virion particles from adsorbing to the F-pilus by MurA was ruled out because the virion-MurA complex is diluted 5×105-fold before inclusion in the overlay. We exploited this inactivation reaction to determine that the apparent affinity of virion-associated A2 for MurA was ~40 nM (Fig. 3). The inactivation reaction was further examined by fractionation on a sucrose step gradient. Analysis of the samples showed that a fraction of the genomic RNA became RNase-sensitive in the Qβ sample that was incubated with MurA but not with the virion-only control (Table S2 and Fig. S1). A similar RNase sensitivity has been reported for ssRNA particles that lacked maturation protein (Boedtker & Gesteland, 1975); here, however, the A2 protein is still present. Thus, the simplest interpretation is that MurA binding causes a conformational change of the maturation protein that inactivates the virion for adsorption to the F-pilus and allows access of RNase to the viral RNA.
Fig. 2.

Qβ particles are inactivated by incubation with MurA in vitro. Plating of purified Qβ particles (a) in an HfrH overlay. Incubation of Qβ with MurA (b) or BSA (c) was performed prior to plating.
Fig. 3.
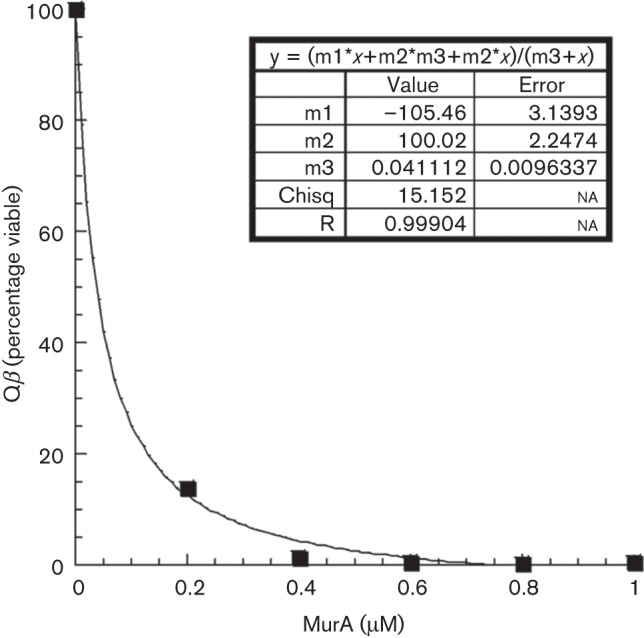
Determination of Ki for Qβ inactivation by MurA. Percentage viability of Qβ (~0.3 μM) was measured after 30 min incubation with increasing amounts of MurA. The Ki was determined by plotting viability versus MurA concentration and fitting to equation (1) using the graphical program KaleidaGraph.
Quantification of MurA and A2 in vivo
The finding that MurA can inactivate the virions called into question the previous model that the titration of extant MurA levels by assembled virions accounted for the temporal regulation of lysis in Qβ infections. To begin addressing the question of how A2-mediated lysis is regulated in vivo, it was necessary to quantify both MurA and A2. The endogenous level of MurA was determined by quantitative Western blotting, using purified MurA as a standard. The result, 390±30 molecules per cell (mean±sd of three blots, Fig. S2), is in agreement with another determination of 410 MurA molecules per cell by MS profiling (Ishihama et al., 2008). In view of the affinity measured above (40 nM), at this level of MurA (~400 nM) A2 should bind stoichiometrically. Accordingly, we estimated the level of A2 necessary for lysis by performing a gradient induction of an IPTG-inducible plasmid clone. Lysis was observed at 40 min at 100 μM IPTG (Fig. 4). At this level of induction, ~350 and ~500 A2 molecules accumulate per cell at 15 and 30 min, respectively (Table 2). As it has been reported previously that there is delay of 20 min after the cessation of cell-wall synthesis before lysis is observed (Bernhardt et al., 2001), the lysis time determined here, 40 min, indicates that quantitative inhibition of MurA was achieved at ~20 min, when ~400 A2 molecules would be expected. Lysis occurs even earlier at higher levels of inducer (Fig. 4).
Fig. 4.
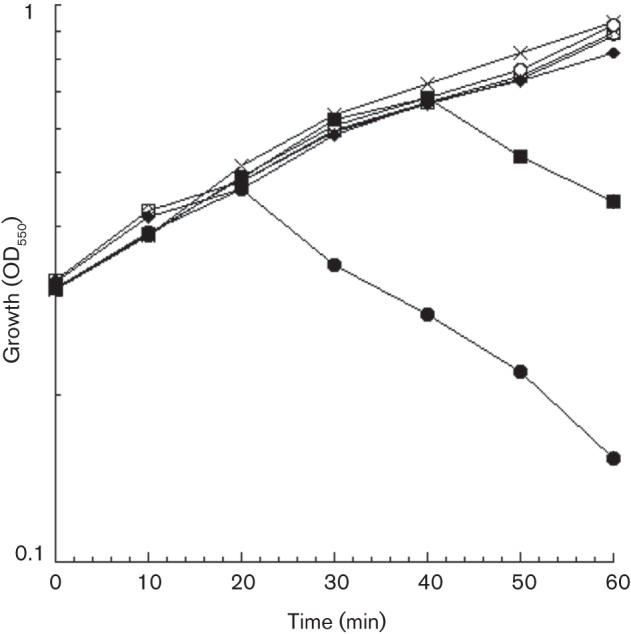
Plasmid-borne A2 induces cellular lysis. A2-mediated lysis was assessed by monitoring growth (OD550) of HfrH lacZ : : Tn5 cultures induced with various concentrations of IPTG: pZE12-luc control (1 mM, ×); pZE12-A2 (uninduced, ▵; 12.5 μM, □; 25 μM, ○; 50 μM, ⧫; 100 μM, ▪; 1 mM, •).
Table 2. Critical concentration of A2 to induce host lysis.
nd, Not determined.
| Level of induction (μM IPTG)* | Lysis | A2 molecules per cell | Time of analysis (min) |
| 0 | − | nd | nd |
| 12.5 | − | nd | nd |
| 25 | − | 2.2×102 | 60 |
| 50 | − | 2.3×102 | 60 |
| 100 | + | 3.6×102 | 15 |
| 100 | + | 4.9×102 | 30 |
| 1000 | + | 1.4×103 | 15 |
pZE12-A2.
Quantification of A2 accumulation during infection
The context of the viral infection cycle might change the stoichiometry of A2 and MurA, especially considering that in this framework A2 has multiple roles. To address this issue, A2 was quantified during a Qβ infection of the male strain XL1-Blue. The total A2 concentration was found to exceed that of MurA by 30 min after infection (Table 3 and Fig. 5a), so, as noted above, lysis could be expected at ~50 min. However, culture-wide lysis, monitored both by turbidity and by release of Qβ virions (Fig. 5a, b), was not observed until 80 min, suggesting that cell-wall synthesis was not blocked until ~60 min, when the level of A2 (1.2×103 molecules) is threefold higher than the level of MurA. This suggests that during the Qβ infection cycle, a significant fraction of A2 is sequestered from binding to MurA.
Table 3. Qβ A2 quantification.
nd, Not determined.
| Time (min) | XL1-Blue | |||
| Qβ A2WT | Qβ A2L28P | Qβ A2D52N | Qβ A2E125G | |
| 15 | nd | nd | nd | nd |
| 30 | 5.1×102 | 1.0×103 | 1.7×103 | 1.3×103 |
| 45 | 7.5×102 | nd | nd | nd |
| 60 | 1.2×103 | nd | nd | nd |
| 75 | 2.9×103 | nd | nd | nd |
Fig. 5.

Qβ and Qβ por phenotype of infected cells. (a) Infection of XL1-Blue cells with Qβ. Reduction in cellular mass accumulation is observed starting at 30 min with a maximum at 80 min with phage input m.o.i. of 1. A2 accumulation (◊) was quantified at intervals during infection. The dashed line represents the number of MurA molecules in XL1-Blue. (b) Qβ has a higher released p.f.u. titre after a single infection cycle than Qβ por. (c) A2 accumulation during infection of XL1-Blue. Loading was normalized to total volume. Samples were taken at the time points listed (min). (d) Qβ por mutants have an earlier lysis phenotype in XL1-Blue-infected cells. Qβ infection (a) reproduced with Qβ por infections. Qβ-only control (×), Qβ A2WT (•), Qβ A2L28P (⧫), Qβ A2D52N (▪) and Qβ A2E125G (▾).
The por mutants produce A2 more rapidly and cause early lysis
Further evidence addressing the relationship between the rate of A2 production and lysis timing was obtained from analysis of Qβ por (plates on rat) mutants (Bernhardt et al., 2001). These mutants were isolated as rare plaque-formers on the murArat host (Fig. S3). Eight independent mutants were obtained (Table S3); sequence analysis revealed that each had mutations in the 5′ end of the A2 gene that corresponds to the N terminus of the protein. The simplest interpretation was that missense changes in A2 could suppress the defect in the inhibition of the MurAL138Q mutant and implied that the N-terminal domain of A2 carried the lytic determinant. This would be consistent with the observation that the principal differences between the lytic maturation proteins of Qβ and other Alloleviviridae and the non-lytic maturation proteins of the Leviviridae are in the N-terminal region. However, clones of the A2por alleles, although fully lytic in the parental host, failed to support inducible lysis in the murArat background (Fig. S4). Moreover, Western blot analysis of samples taken from infections with the por mutants clearly showed that A2 was accumulating more rapidly than in the parental phage infection (Fig. 5c). These results indicated that the ability of por mutants to support plaque formation on the murArat background is due to a higher rate of synthesis of A2 in vivo, rather than an altered ability to inhibit MurAL138Q. Inspection of the secondary structure map of the A2 mRNA shows that each of the mutations alters a base-pairing element that is predicted to be involved in repressing translational initiation (Fig. 1b). We conclude that the por mutations derepress the translational control of A2.
Based on the previous results with the inducible cDNA clones of A2 (Fig. 4), we expected that the por mutants would show earlier lysis in infections of wild-type cells. We found that this is indeed the case (Fig. 5d), with lysis onset occurring 30–45 min earlier than with wild-type Qβ. With all three por alleles, the level of total A2 production by 30 min was equal to or greater than that achieved by the wild-type phage at 60 min, accounting for the earlier lysis (Table 3 and Fig. 5c). The early lysis also reduces the yield of progeny (Fig. 5b), demonstrating that the normal regulation of the lysis protein is important for the fitness of Qβ.
Discussion
The discovery that two of the paradigm experimental phage systems, the ssDNA phage ϕX174 and the ssRNA phage Qβ, both achieved host lysis by specifically inhibiting steps in the murein precursor biosynthesis pathway was interesting from the perspective of phage-based design of ‘protein antibiotics’ (Bernhardt et al., 2001). In addition, these findings also raised the question of how these phages regulated the timing of lysis, and thus the length and fecundity of their infection cycles. The fact that the lytic protein in Qβ, A2, also served as an essential component of the virion led to the proposal of the simplest possible regulatory scheme that would ensure the production of a certain level of virions; that is, the virions themselves, each carrying one copy of A2, would accumulate until they titrated out the level of cytoplasmic MurA, the target of A2 (Hatfull, 2001). However, here we have shown that MurA actually inactivates the Qβ virion in vitro (Fig. 2), apparently causing conformational changes in A2 that make the genomic RNA susceptible to exogenous RNase (Fig. S1). Alternative schemes might be based on either a low affinity of virion-associated A2 for MurA, so that only a small fraction of virions would be inactivated, or that A2 is produced in a large excess over MurA, so that attrition of infectious virions by contact with MurA would be negligible. Here we have demonstrated that neither of these conditions applies. First, based on the in vitro inactivation experiments, the level of MurA in the cell is an order of magnitude higher than needed to saturate A2 (400 nM MurA compared with 40 nM for the apparent Kd) (Fig. 3). Moreover, graded induction of a cloned A2 gene indicates that cell-wall biosynthesis is blocked when the level of A2 is equimolar with cellular MurA (Fig. 4). Finally, during the infection cycle, A2 is produced in an amount (1.2×103 molecules at the interpolated time of inhibition of cell-wall biosynthesis; Table 3, 60 min) only threefold higher than MurA (~400 molecules).
These considerations suggest that regulation of lysis timing in Qβ infections involves another viral-specific component. Qβ is an extremely simple virus, with only four proteins, and so the candidates for the regulatory component are limited. Here we propose that the regulatory molecule is the Qβ genomic RNA. This notion is based on several lines of evidence. First, in vitro gene expression experiments have suggested that initiation of translation of the maturation protein cistron occurs only in nascent RNA molecules that lack inhibitory secondary structures (Robertson & Lodish, 1970; Staples et al., 1971). More recently, it has been proposed that Qβ virion assembly starts with circularization of the viral RNA by a single molecule of A2 binding both the 5′ and the 3′ ends of the RNA (Dykeman et al., 2011; Shiba & Suzuki, 1981). Based on these observations and the data presented here, we propose a model in which A2 is synthesized from nascent Qβ RNAs in slight stoichiometric excess. The majority of A2 molecules are bound to the viral RNA and are thus committed to assembly, whereas a small fraction are free and able to form the inhibitory complex with MurA. In support of this perspective, we note that the por mutants, selected for their ability to overcome the rat phenotype of murAL138Q, produce A2 at two- to three-fold higher levels than the parental phage, presumably due to the disruption of local RNA secondary structures. This is necessary, because the L138Q mutation in MurA reduces the affinity for A2 (Reed et al., 2012). As a consequence, lysis occurs much earlier in the Qβ por infections and the average yield of virions is drastically curtailed (Fig. 5b, d). This model predicts that A2 bound to the viral RNA in the context of the virion assembly pathway is insensitive to MurA. Preliminary experiments looking for protection of the virion by purified Qβ genomic RNA have been unsuccessful (data not shown), suggesting that RNA structures specific to the assembly pathway may be involved. It should be pointed out that this type of system offers the evolutionary capability of changing the lysis timing of the Qβ infection cycle. Presumably, many nucleotide positions in the A2 cistron can be altered to effect marginal changes in A2 production, which would then have graded effects on the timing of lysis. Regardless, this model presents a conceptual framework for further analysis of Qβ, which, with its genetic simplicity, is an attractive paradigm system for attempting to achieve quantitative and predictive understanding of a viral infection cycle (Tsukada et al., 2009).
Acknowledgements
We thank the Young laboratory members, past and present, for their helpful criticisms and suggestions. We also would like to thank Michelle Lovingshimer for help with Ki calculations. This work was supported by Public Health Service grant GM27099 to R. Y., the Robert A. Welch Foundation and the Program for Membrane Structure and Function, a Program of Excellence grant from the Office of the Vice President for Research at Texas A&M University.
Footnotes
Four supplementary figures, three supplementary tables and supplementary methods are available with the online version of this paper.
References
- Beekwilder J., Nieuwenhuizen R., Poot R., van Duin J. (1996). Secondary structure model for the first three domains of Qβ RNA. Control of A-protein synthesis. J Mol Biol 256, 8–19. [DOI] [PubMed] [Google Scholar]
- Bernhardt T. G., Roof W. D., Young R. (2000). Genetic evidence that the bacteriophage ϕ X174 lysis protein inhibits cell wall synthesis. Proc Natl Acad Sci U S A 97, 4297–4302. 10.1073/pnas.97.8.4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt T. G., Wang I. N., Struck D. K., Young R. (2001). A protein antibiotic in the phage Qβ virion: diversity in lysis targets. Science 292, 2326–2329. 10.1126/science.1058289 [DOI] [PubMed] [Google Scholar]
- Bernhardt T. G., Wang I. N., Struck D. K., Young R. (2002). Breaking free: “protein antibiotics” and phage lysis. Res Microbiol 153, 493–501. 10.1016/S0923-2508(02)01330-X [DOI] [PubMed] [Google Scholar]
- Blumenthal T., Carmichael G. G. (1979). RNA replication: function and structure of Qβ-replicase. Annu Rev Biochem 48, 525–548. 10.1146/annurev.bi.48.070179.002521 [DOI] [PubMed] [Google Scholar]
- Boedtker H., Gesteland R. F. (1975). Physical properties. In RNA Phages, pp. 1–28. Edited by Zinder N. D. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Brown E. D., Vivas E. I., Walsh C. T., Kolter R. (1995). MurA (MurZ), the enzyme that catalyzes the first committed step in peptidoglycan biosynthesis, is essential in Escherichia coli. J Bacteriol 177, 4194–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E., Holland J. J. (1997). RNA virus mutations and fitness for survival. Annu Rev Microbiol 51, 151–178. 10.1146/annurev.micro.51.1.151 [DOI] [PubMed] [Google Scholar]
- Domingo E., Sabo D., Taniguchi T., Weissmann C. (1978). Nucleotide sequence heterogeneity of an RNA phage population. Cell 13, 735–744. 10.1016/0092-8674(78)90223-4 [DOI] [PubMed] [Google Scholar]
- Drake J. W. (1993). Rates of spontaneous mutation among RNA viruses. Proc Natl Acad Sci U S A 90, 4171–4175. 10.1073/pnas.90.9.4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykeman E. C., Grayson N. E., Toropova K., Ranson N. A., Stockley P. G., Twarock R. (2011). Simple rules for efficient assembly predict the layout of a packaged viral RNA. J Mol Biol 408, 399–407. 10.1016/j.jmb.2011.02.039 [DOI] [PubMed] [Google Scholar]
- Groeneveld H., Thimon K., van Duin J. (1995). Translational control of maturation-protein synthesis in phage MS2: a role for the kinetics of RNA folding? RNA 1, 79–88. [PMC free article] [PubMed] [Google Scholar]
- Gründling A., Bläsi U., Young R. (2000). Biochemical and genetic evidence for three transmembrane domains in the class I holin, λ S. J Biol Chem 275, 769–776. 10.1074/jbc.275.2.769 [DOI] [PubMed] [Google Scholar]
- Hatfull G. F. (2001). Microbiology. The great escape. Science 292, 2263–2264. 10.1126/science.1062957 [DOI] [PubMed] [Google Scholar]
- Ishihama Y., Schmidt T., Rappsilber J., Mann M., Hartl F. U., Kerner M. J., Frishman D. (2008). Protein abundance profiling of the Escherichia coli cytosol. BMC Genomics 9, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen P. R., Westerhoff H. V., Michelsen O. (1993). The use of lac-type promoters in control analysis. Eur J Biochem 211, 181–191. [DOI] [PubMed] [Google Scholar]
- Kozak M., Nathans D. (1971). Fate of maturation protein during infection by coliphage MS2. Nat New Biol 234, 209–211. [DOI] [PubMed] [Google Scholar]
- Palmer I., Wingfield P. T. (2001). Preparation and extraction of insoluble (inclusion-body) proteins from Escherichia coli. Current Protocols in Protein Science 70, 6.3.1–6.3.20. 10.1002/0471140864.ps0603s00 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed C. A., Langlais C., Kuznetsov V., Young R. (2012). Inhibitory mechanism of the Qβ lysis protein A2. Mol Microbiol 86, 836–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley-Lovingshimer M. R., Reinhart G. D. (2001). Equilibrium binding studies of a tryptophan-shifted mutant of phosphofructokinase from Bacillus stearothermophilus. Biochemistry 40, 3002–3008. [DOI] [PubMed] [Google Scholar]
- Robertson H. D., Lodish H. F. (1970). Messenger characteristics of nascent bacteriophage RNA. Proc Natl Acad Sci U S A 67, 710–716. 10.1073/pnas.67.2.710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E. F., Maniatis T., Sambrook J., Fritsch E. F., Maniatis T. (1989). Molecular Cloning: a Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Shiba T., Suzuki Y. (1981). Localization of A protein in the RNA-A protein complex of RNA phage MS2. Biochim Biophys Acta 654, 249–255. 10.1016/0005-2787(81)90179-9 [DOI] [PubMed] [Google Scholar]
- Siegele D. A., Hu J. C. (1997). Gene expression from plasmids containing the araBAD promoter at subsaturating inducer concentrations represents mixed populations. Proc Natl Acad Sci U S A 94, 8168–8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples D. H., Hindley J., Billeter M. A., Weissmann C. (1971). Localization of Q-β maturation cistron ribosome binding site. Nat New Biol 234, 202–204. [DOI] [PubMed] [Google Scholar]
- Strauss J. H., Jr, Sinsheimer R. L. (1963). Purification and properties of bacteriophage MS2 and of its ribonucleic acid. J Mol Biol 7, 43–54. 10.1016/S0022-2836(63)80017-0 [DOI] [PubMed] [Google Scholar]
- Tsukada K., Okazaki M., Kita H., Inokuchi Y., Urabe I., Yomo T. (2009). Quantitative analysis of the bacteriophage Qβ infection cycle. Biochim Biophys Acta 1790, 65–70. 10.1016/j.bbagen.2008.08.007 [DOI] [PubMed] [Google Scholar]
- van Duin J., Tsareva N. (2006). Single-stranded RNA phages. In The Bacteriophages, pp. 175–196. Edited by Calendar R. Oxford: Oxford University Press. [Google Scholar]
- Weber K., Konigsberg W., Zinder N. D. (1975). Proteins of the RNA phages. In RNA Phages, pp. 51–84. Edited by Zinder N. D. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Winter R. B., Gold L. (1983). Overproduction of bacteriophage Q β maturation (A2) protein leads to cell lysis. Cell 33, 877–885. 10.1016/0092-8674(83)90030-2 [DOI] [PubMed] [Google Scholar]