

Abstract
Extracellular human immunodeficiency virus type type 1 (HIV-1) viral protein R (Vpr) is a pleiotropic protein accomplishing several functions within the viral life cycle. While Vpr has been described extensively as an intracellular protein, very little is known about its role as an extracellular protein. In fact, HIV-1 Vpr has been detected in the blood, serum, and cerebrospinal fluid of HIV-1-infected patients, with concentrations increasingly higher in late-stage disease. To determine the role exogenous Vpr plays in HIV-associated central nervous system dysfunction, primary human fetal astrocytes were exposed to recombinant Vpr and a time- and dose-dependent decrease was demonstrated in two fundamental intracellular metabolites (ATP and glutathione (GSH)). Additionally, exposure to exogenous Vpr led to increased caspase activity and secretion of proinflammatory cytokines IL-6 and IL-8 and chemoattractants, monocyte chemotactic protein-1 and migration inhibition factor. Extracellular Vpr also dampened the glycolytic pathway through impairment of GAPDH activity, causing a decline in the levels of ATP. The reduction in intracellular ATP increased reactive oxygen species buildup, decreasing GSH concentrations, which affected several genes in the oxidative stress pathway. In addition, exposure of the SK-N-SH neuroblastoma cell line to conditioned medium from exogenous Vpr-treated astrocytes decreased synthesis of GSH, leading to their apoptosis. These observations point to a role that Vpr plays in altering astrocytic metabolism and indirectly affecting neuronal survival. We propose a model that may explain some of the neurological damage and therefore neurocognitive impairment observed during the course of HIV-1 disease.
Keywords: adenosine triphosphate, astrocytic-neuronal network, glutathione, glyceraldehyde 3-phosphate dehydrogenase, HIV-1 viral protein R
Introduction
Oxidative stress (OS) is the condition wherein accumulation of reactive oxygen species (ROS) impairs intracellular protection mechanisms implicated in several pathological processes, including a number of neurodegenerative disorders (Baruchel and Wainberg, 1992). Glutathione (GSH), a tripeptide composed of glycine, glutamate, and cysteine, plays a critical role in the removal of ROS. Within the central nervous system (CNS), astrocytes are the most critical cell type in regulating homeostasis and disposal of neurotoxic molecules (Shinya et al, 2004). By importing extracellular cysteine and glucose, astrocytes synthesize GSH and lactate (respectively) through aerobic glycolysis (Pellerin and Magistretti, 1994); they then secrete GSH and lactate, which can be taken up by neurons, thus protecting them from OS. Exposure to HIV-1 increases ROS production in neuroglial cell cultures, leading to apoptosis (Pollicita et al, 2009), while excessive ROS accumulation within astrocytes may impair glutamate uptake (Sorg et al, 1997; Trotti et al, 1996), with subsequent extracellular glutamate accumulation and neuronal demise (Klegeris et al, 1997; Trotti et al, 1996). HIV-1-infected individuals are often at increased risk for OS due to impaired antioxidant defense mechanisms (Perl and Banki, 2000; Treitinger et al, 2000) and decreased levels of GSH (Baruchel and Wainberg, 1992). Imbalance between reduced glutathione (GSH) and oxidized glutathione (GSSG) triggers intracellular redox-sensitive pathways, ultimately causing DNA damage and correlating positively with CD4 T-cell count and disease progression (Herzenberg et al, 1997). Other HIV-1 proteins have been found to induce OS (Lassiter et al, 2009). Tat induces ROS accumulation through Nrf2 (Zhang et al, 2009) and inducible nitric oxide synthase signaling in astroglia (Liu et al, 2002), activation of macrophages and microglial cells, as well as T-cell infiltration in the CNS (Zhou et al, 2007), while gp120 sensitizes neurons to hydrogen peroxide-induced OS (Agrawal et al, 2009). HIV-1 viral protein R (Vpr) was found to induce OS through the hypoxia-inducible factor pathway in microglia (Deshmane et al, 2009), and we recently demonstrated Vpr as a causative agent of OS in an astroglioma cell line through depletion of ATP and GSH reservoirs (Ferrucci et al, 2012). ROS-induced signaling leads to increased HIV-1 gene transcription (Perl and Banki, 2000), which reseeds viral infection.
Vpr has been shown to interact with the adenine nucleotide translocator (ANT) on the inner mitochondrial membrane (Jacotot et al, 2001; Jacotot et al, 2000), where amino acids Arg73 and Arg80 in Vpr are indispensable (Sabbah et al, 2006). Although these observations were determined using recombinant proteins or cell-free isolated mitochondria, there is no evidence that exogenous Vpr interacts directly with ANT, although recently Vpr was shown to transduce cells (Greiner et al, 2011; Sherman et al, 2002). Upon transduction, Vpr may bind to ANT, thus hampering the passage of newly synthesized ATP molecules from the mitochondria, which in turn decreases the ATP-dependent concentration of GSH. Conversely, decreases in the levels of ATP and GSH could be the result of uncoupled signaling events, similar to the action of the chemotherapeutic agent doxorubicin. Through impairment of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in the glycolytic pathway (Cui et al, 2006), decreased ATP induces excessive accumulation of ROS, thus reducing GSH storage. To test these hypotheses, the mode of action of Vpr was examined using a Vpr mutant protein with reduced binding affinity to ANT (Vpr R73,80A). Both parental and mutant Vpr caused reduction in the levels of ATP and GSH and equally affected GAPDH activity in human astrocytes, suggesting action through an ANT-independent signaling pathway. Additionally, increased secretion of proinflammatory cytokines (IL-6 and IL-8), monocyte chemotactic protein-1 (MCP-1), and migration inhibitor factor (MIF) and decreased levels of plasminogen activator inhibitor-1 (PAI-1) were observed. Removal of Vpr or Vpr R73,80A from the extracellular medium did not rescue astrocytic levels of either metabolite. Treatment of neurons with conditioned medium from Vpr- or Vpr R73,80A-exposed astrocytes reduced neuronal ability to synthesize GSH and led to neuronal apoptosis. These studies define a unique role for extracellular Vpr in altering astrocytic function, which impacts neuronal survival and might be associated with some of the pathological processes in during the course of HIV-1 disease.
Materials and Methods
Cell maintenance
NAC, BSO, L-GSH, and doxorubicin hydrochloride were obtained from Sigma-Aldrich (St. Louis, MO), while GSH beads for Vpr purification were from GE Healthcare (Waukesha, WI). Whole cell lysates were prepared in 0.5 × RIPA buffer (Thermo Fisher Scientific, Waltham, MA), with the addition of a protease and phosphatase inhibitor cocktail (Calbiochem; EMD Millipore, Billerica, MA); protein concentrations were calculated using the Bradford protein assay (Thermo Fisher Scientific). The pcDNA3.1 vector (Invitrogen, Carlsbad, CA) was used to clone the 6 histidine (6His)-hemagglutinin (HA)-Vpr plasmid. CPT and the caspase inhibitor Ac-DEVD-CHO were purchased from BioVision (Milpitas, CA) and Promega (Madison, WI), respectively. Total cellular RNA was prepared as described by the manufacturer (USB Products; Affymetrix, Santa Clara, CA). Primary HFAs were purchased from ScienCell Research Laboratories (Carlsbad, CA) and maintained in astrocyte medium containing 1% astrocyte growth supplement, 2% fetal bovine serum (FBS), and 1% penicillin/streptomycin as described by the manufacturer. All experiments using HFAs were performed with cells passaged no more than twice as the astrocytic intracellular marker glial fibrillary acidic protein (GFAP) was gradually lost. The SK-N-SH neuroblastoma cell line (ATCC) was maintained in 1× Eagle’s minimal essential medium supplemented with 10% FBS, 1 mM sodium pyruvate, and 1.5-g/l sodium bicarbonate. All cells were maintained at 37°C in 5% CO2 at 90% relative humidity.
Plasmid construction and site-directed mutagenesis
To clone Vpr, the noninfectious HIV-1 pNL4-3R+E− molecular clone was used (obtained through the National Institutes of Health AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, provided by Dr. Nathaniel Landau) (Connor et al, 1995; He et al, 1995)). The 6His-HA-Vpr and GST-6His-HA-Vpr plasmids were previously described (Ferrucci et al, 2012). The Vpr mutant plasmid (GST-6His-HA-Vpr R73,80A), wherein two arginine residues at positions 73 and 80 were changed to alanine, was obtained through two subsequent site-directed mutagenesis reactions from GST-6His-HA-Vpr plasmid using the QuickChange Site-Directed Mutagenesis procedure described by the manufacturer (Stratagene, La Jolla, CA). Initially, the Vpr R73A variant (arginine to alanine change at position 73) was generated using the two reverse complement primers 5′–GCTGTTTATCCATTTCGCAATTGGGTGTCGAC and 5′–GTCGACACCCAATTGCGAAATGGATAAACAGC (the two nucleotides yielding the amino acid mutation are underlined). Parental plasmid DNA was digested by incubation with 10 U of Dpn I for 1 h at 37°C, and the obtained PCR-amplified plasmid was transformed by heat shock into XL1-Blue-competent cells (Stratagene) to yield GST-6His-HA-Vpr R73A plasmid. This plasmid was then used to generate the double mutant GST-6His-HA-Vpr R73,80A with identical conditions as indicated above using the two reverse complement primers 5′–GGTGTCGACATAGCGCAATAGGCGTTACTC and 5′–GAGTAACGCCTATTGCGCTATGTCGACACC (mutated nucleotides underlined). All plasmid DNAs were confirmed by sequencing (Genewiz, South Plainfield, NJ).
Western immunoblot analyses
Harvested cells were washed twice in 1×PBS, collected by low-speed centrifugation and lysed by three freeze-thaw cycles. Lysed samples were cleared of nucleic acids and supernatant was collected, from which the protein concentration was assessed. Equal amounts of protein for all samples were resolved on a 10% SDS-PAGE gel and transferred to a 0.45-μm Immobilon-P PVDF membrane, followed by blocking with 5% (w/v) nonfat dry milk in 1×PBS (1 h at room temperature). Membranes were incubated overnight at 4°C with primary antibody; specific protein bands were detected using an HRP-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA). Spotted antigen-antibody complexes were visualized using a chemiluminescent detection procedure (Pierce, Rockford, IL). Densitometry was performed with an AlphaEase FC software package (Alpha Innotech, Randburg, South Africa). The following antibodies were used: GFAP, caspase-3 (Cell Signaling, Danvers, MA); β-actin (Sigma-Aldrich); and GAPDH (GenScripts, Nanjing, Jiangsu, Japan).
Purification of recombinant Vpr
Purification of Vpr or its mutant was performed as previously described (Ferrucci et al, 2012). Briefly, GST-6His-HA-Vpr or the Vpr R73,80A was induced by addition of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Cultures were then pelleted by centrifugation and lysed in NETN lysis buffer, Tris pH 8.0 (20 mM), NaCl (100 mM), EDTA (1 mM), NP-40 (0.5%), phenylmethylsulfonyl fluoride (1 mM), and protease inhibitor cocktail (1:100). The sonicated lysate was purified by affinity chromatography with GSH-sepharose beads. The retained protein was washed and released from the column in elution buffer (30 mM reduced GSH in 50 mM Tris·HCl buffer, pH 8). Endotoxin was removed from eluted proteins with the ToxinEraser Endotoxin Removal procedure (GenScript). The GST tag was removed by cleavage with 10 U/mg thrombin for 2 h at room temperature. An additional affinity chromatography step was performed using a His SpinTrap nickel column (GE Healthcare) and protein dialyzed overnight at 4°C in 1×PBS. All preparations employed in our study presented residual endotoxin concentration lower than the detectable levels (< 0.001 EU/ml, as measured by the ToxinSensor Chromogenic LAL Endotoxin Assay procedure (Genscript)) and final protein concentration was measured at an absorbance of 280 nm.
ATP, GSH, and GAPDH activity assays
For all experiments involving measurement of intracellular concentrations of ATP and GSH, we used the CellTiter-Glo Luminescent Cell Viability and GSH-Glo Glutathione Assay (Promega), respectively, as previously described (Ferrucci et al, 2012). Reduced GSH levels in SK-N-SH neuroblastoma cells were determined similarly, with the following exceptions: cells were grown directly in single wells of a 96-well opaque plate, treated with conditioned medium for the indicated time, and lysed in 1×GSH-Glo reagent (Promega). For determination of the levels of reduced over oxidized glutathione (GSH/GSSG), a similar approach was followed using the GSH/GSSG-Glo Glutathione Assay (Promega) as described by the manufacturer. GSH/GSSG ratios were calculated from the formula GSH = tGSH − GSSG/2, then being 2•tGSH/GSSG − 1.
To measure GAPDH intracellular activity, a fluorimetric assay procedure (KDalert GAPDH Assay; Ambion, Foster City, CA) was used as described by the manufacturer. All untreated and treated cell populations were harvested and lysed in 100-μl KDalert lysis buffer. Ten-microliter amounts of lysate were dispensed in triplicate in single wells of a 96-well opaque plate with black walls. After addition of 90 μl of master mix solution, the readings were taken using a FluoroMax-3 with a Spex Micromax 384 MicroPlate Reader (Horiba Scientific, Irvine, CA) set at excitation and emission wavelengths of 560 and 590 nm, respectively. All readings were subtracted from the background value (lysis buffer alone). Graphs were plotted as percentage over untreated cells ± standard errors.
Human cytokine array analyses
Supernatant from untreated or Vpr-treated primary HFAs was collected at 48 h and used in the detection of differentially secreted cytokines utilizing the Proteome Profiler Array (R&D Systems, Minneapolis, MN). Supernatants (500 μl) from each of the two samples were incubated with a cytokine detection antibody cocktail and dispensed on top of each membrane. After an overnight incubation at 4°C, cytokine-antibody complexes were detected with a streptavidin-HRP conjugated cocktail. Each cytokine, spotted in duplicate, was visualized by a chemiluminescent detection method (Pierce). To quantitate the brightness of each spot, densitometry was performed as described above, where each pair of cytokines was normalized to the untreated integrated density value, and the ratio of fold over untreated values was calculated. The average of each pair was plotted with its standard error; column bars with a positive or negative fold ratio represent cytokines up- or downregulated in extracellular Vpr-treated samples compared with untreated samples, respectively.
Caspase detection assay
Caspase-3 and -7 activities were measured using the luminescent Caspase-Glo 3/7 assay (Promega), based on a luminogenic substrate containing the tetrapeptide sequence DEVD, targeted by the two caspases. Primary HFAs were plated at a density of 2000 cells in individual wells of a 96-well opaque tissue-culture plate and either left untreated or exposed to different compounds. As a positive control for induction of caspase-3 and -7, HFAs were exposed for 48 h to 2 μM CPT. HFAs were also treated alone or in combination with recombinant Vpr, anti-Vpr antibody (kind gift of Dr. Kopp, NIH, Bethesda, MD), NAC, and the caspase inhibitor Ac-DEVD-CHO (Promega). NAC was applied to cells 24 h prior to harvesting. Forty-eight hours after exposure, cell culture medium was removed, and HFAs were carefully washed in 1×PBS, lysed in situ, and incubated at room temperature in the dark for 20 min. Luminescent signals were recorded using a Glomax luminometer (Promega) and subjected to background correction (no cells). Augmented caspase activity was detected as an enhanced luminescent signal. The data were plotted as fold over untreated cells.
Isolation of RNA and qRT-PCR microarray
Total cellular RNA was isolated from primary HFAs (untreated or exposed to rVpr for 48 h) using the PrepEase RNA spin procedure as described by the manufacturer (USB, Affymetrix) and used to study the genes of the OS pathway that were differentially regulated. Five micrograms of total RNA was subjected to reverse transcription using the RT2 First Strand procedure described by the manufacturer (SABiosciences, Frederick, MD) to yield complementary DNA. qRT-PCR was performed on complementary DNA samples using RT SYBR Green/ROX qPCR Master Mix (SABioscience). The reaction mixtures (25 μl) were dispensed in individual wells of a 96-well plate prespotted with primers of genes belonging to the OS and antioxidant defense pathway (SABioscience). Cycling conditions were set for 40 cycles, each consisting of 15 s at 95°C and 1 min at 60°C on an ABI 7300 Thermocycler (Applied Biosystems, Foster City, CA). For each gene, a threshold cycle was calculated with the 7300 Thermocycler built-in software. Augmented mRNA levels in Vpr-treated samples were analyzed as fold over untreated (calibrator) and expressed in a linear scale. Using a software package (SABioscience), genes with the highest fold expression between untreated and treated were listed and plotted. For those genes with the highest fold values, protein levels were compared between untreated and treated samples by western immunoblotting as previously described.
In situ TUNEL assay
To detect programmed cell death by terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) assay, the In Situ Cell Death Detection procedure was used as described by the manufacturer (Roche, Nutley, NJ). Briefly, adherent SK-N-SH neuroblastoma cells were cultured on coverslips as described previously and exposed overnight to conditioned medium from astrocytes either left untreated or treated with rVpr. Cells were then washed twice with 1×PBS prior to fixation in 4% paraformaldehyde for 20 min at room temperature. Permeabilization of plasma membranes was performed with 0.2% Triton X-100 in 1×PBS for 5 min at room temperature. Cells were then blocked in 5% goat serum, 1% bovine serum albumin in 1×PBS for 1 h at room temperature and incubated with the TUNEL mixture (1:10 of Enzyme Solution to Label Solution, both Roche proprietary solutions) at 37°C in the dark for 1 h. SK-N-SH neuroblastoma cells were subsequently incubated in 300 nM DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen)-containing 1×PBS for 2 min at room temperature to stain nuclei. Cells were washed three times with 1×PBS between each step. Coverslips were then rinsed in ddH2O, mounted on microscope slides with ProLong antifade reagent (Invitrogen), and immediately imaged with a deconvolution microscope (Olympus, Center Valley, PA). For all cell types, a minimum of 10 fields at ×20 magnification were analyzed. The images reported are a representative field of the entire cell population. All fields of view were reported as two separate channels (DAPI and fluorescein isothiocyanate [FITC]) representing nuclei and TUNEL+ cells, respectively, until merged in a third image, wherein apoptotic cells displayed nuclei with an overlapped blue and green staining. As a positive control, SK-N-SH neuroblastoma cells were treated with 200 μM CPT to induce programmed cell death.
Statistical analysis
The results were analyzed statistically by a two-tailed Student t-test. Differences between groups were considered significant if p values <0.05 were obtained.
Results
Kinetic analysis of extracellular Vpr-induced decreases in the levels of ATP and GSH
Since the first report of HIV-1 Vpr as an extracellular soluble protein (Levy et al, 1994), evidence has accumulated demonstrating the damaging effects this protein confers on both neurons and astrocytes (Cheng et al, 2007; Jones et al, 2007; Kitayama et al, 2008; Noorbakhsh et al, 2010; Regis et al, 2010; Sabbah and Roques, 2005). HIV-1 Vpr has been detected in both the serum and cerebrospinal fluid (CSF) of HIV-1-infected patients and this protein has been shown to increase in quantity with disease progression (Levy et al, 1994; Levy et al, 1995). Additional studies, have also shown that Vpr was found as a free extracellular protein in the serum of patients infected with HIV-1 (Hoshino et al, 2007; Jones et al, 2007). However, only the concentration of extracellular Vpr in the plasma of HIV-1-infected patients has been measured and was estimated to be 5–10 ng/ml (Hoshino et al, 2007). The concentration of extracellular Vpr has never been reported for CSF. We recently reported that treatment of the astroglioma U-87 MG cell line with extracellular recombinant HIV-1 Vpr protein induced cellular deprivation of ATP and GSH, OS by increased accumulation of ROS, and a decreased ratio of reduced over oxidized glutathione upon (Ferrucci et al, 2012). In the same study, primary human fetal astrocytes (HFAs) exposed to exogenous recombinant Vpr (rVpr) showed a significant decrease in the levels of ATP and GSH in a time- and rVpr dose-dependent manner, confirming results within the astroglioma U-87 MG cell line. To further examine the effects rVpr exerts on astrocytes, primary HFAs were exposed to different concentrations of rVpr, displaying a time- and dose-dependent decline in the GSH/GSSG ratio (reduced/oxidized glutathione), indicative of excessive oxidation (Table I).
Table I.
Extracellular Vpr induces a time- and dose-dependent decline in GSH/GSSG ratiosa.
| Treatment | 24 Hours | 48 Hours |
|---|---|---|
| Untreated | 53.4 ± 3.8 | 55.6 ± 2.5 |
| Vpr 1 μg/ml | 18.6 ± 2.8b | 7.0 ± 2.4b |
| Vpr 2 μg/ml | 12.6 ± 1.4b | 4.5 ± 1.6b |
| Vpr 5 μg/ml | 3.5 ± 0.3c | 1.6 ± 0.2c |
Although untreated HFAs did not show any significant variations after in vitro cultivation for 24 or 48 h, exposure to rVpr induced a dose- and time-dependent decrease in the levels of reduced, overoxidized glutathione (GSH/GSSG). Ratios at the beginning of the experiment (0 h) are 54.2 ± 2.6.
p value < 0.05.
p value < 0.01.
Extracellular Vpr activates caspase-3 and -7
Previous studies reported activation of a caspase-induced cascade of events subsequent to Vpr exposure, leading to translocation of transcription factors to the nucleus, promoting DNA fragmentation and cell death in primary human astrocytes (Jones et al, 2007; Noorbakhsh et al, 2010). In similar manner, HFAs exposed to exogenous Vpr were assessed for caspase-3 and -7 activation through a luminescent assay (Figure 1). HFAs treated with the apoptogenic drug camptothecin (CPT) displayed almost a 2.5-fold increase in caspase-3 and -7 activity (Figure 1A), while addition of a caspase inhibitor, acetylated synthetic tetrapeptide, inhibitor of caspase (Ac-DEVD-CHO), abrogated the observed increase. Exposure of HFAs to rVpr promoted a 2-fold increase in caspase-3 and -7, a result consistent with previous results obtained from examining mRNA levels (Noorbakhsh et al, 2010). Conversely, addition of Ac-DEVD-CHO or an anti-Vpr antibody to Vpr-treated HFAs only partially diminished the observed increases (Figure 1A, columns 7 and 8). Western immunoblotting analyses confirmed expression of cleaved caspase in rVpr-treated HFAs (data not shown). These results demonstrated that, within astrocytes exposed to rVpr, a series of intracellular events was initiated, commencing with decreased availability of ATP and GSH and culminating with caspase-dependent apoptosis.
Figure 1. Extracellular Vpr applied to HFAs induces activation of caspase-3 and -7.

(A) HFAs were treated with different stimuli for 48 h and assayed for caspase activity using the Caspase-Glo 3/7 assay procedure. As a positive control, cells were treated for 48 h with CPT 2 μM, which inhibits DNA topoisomerase I in a caspase-dependent manner. Cells treated with rVpr (5 μg/ml) for 48 h similarly exhibited activation of caspase-3 and -7. This effect was only partially recovered after treatment with anti-Vpr antibody or a caspase inhibitor (Ac-DEVD-CHO). *p value <0.05 (Student paired t-test).
Secretion of cytokines in extracellular Vpr-treated astrocytes
HIV-1-induced neuropathogenesis is a concern among patients during the course of HIV-1 infection with an increasing number of individuals showing pathologies of neurodegeneration, such as excessive neuronal loss and abnormal astrocytic proliferation (Lipton, 1994). Several studies have identified molecules playing a role in the observed effects including a number of viral proteins or virus-induced cellular cytokines. Given this, studies were performed to determine whether exposure of primary HFAs to rVpr induced secretion of cellular cytokines using a cytokine array containing many of the cytokines most commonly altered during the course of HIV-1 disease.. Treatment of HFAs with rVpr induced increased secretion of IL-6, IL-8, MCP-1, and MIF and decreased secretion of serpin E1, a serine proteinase inhibitor (Figure 2A) also known as PAI-1. Semiquantitative densitometric analysis showed a significant increase in IL-6 (8-fold) as previously reported (Hoshino et al, 2010), as well as IL-8, MCP-1, and MIF (2-fold), and a significant decrease in PAI-1 (3-fold) (Figure 2B).
Figure 2. Exogenous Vpr-treated primary HFAs display a different pattern of cytokine secretion.
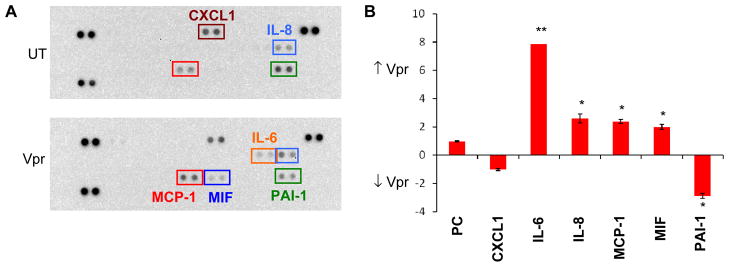
(A) Membranes spotted in duplicate with the most common cytokines were incubated with cell culture medium from either untreated (UT) or extracellular Vpr-treated HFAs (5 μg/ml). Cytokines differentially expressed in the two cell supernatants are shown in different colored squares (IL-6 = orange; MCP-1 = red; MIF = blue; IL-8 = light blue; PAI-1 = green, and CXCL1 = maroon). The doublet spots in the top left, top right, and bottom left corners are positive controls. (B) Densitometric analysis of the up- or downregulated cytokines present in the cell culture medium shows increased secretion of IL-6 (8-fold); increased secretion of IL-8, MCP-1, and MIF (2-fold); and decreased secretion of PAI-1 (3-fold). Levels of CXCL1 were unaltered. *p value <0.05; **p value <0.01 (Student paired t-test).
Vpr-induced decreases in the levels of ATP and GSH is not through ANT binding
We previously showed that exposure to extracellular Vpr affects both ATP and GSH concentrations (Ferrucci et al, 2012). Because ATP is involved in the synthesis of GSH from its precursor amino acid, we investigated whether the decreases in the two molecules were directly correlated. Previous reports showed binding between Vpr and the inner mitochondrial membrane protein ANT, responsible for the exchange of newly synthesized ATP molecules in the electron transport chain for cytoplasmic ADP (Pebay-Peyroula et al, 2003). A possible blockade of ADP/ATP exchange is detrimental intracellularly as shown by treatment with the ANT-binding bongkrekic acid (Halestrap et al, 1997). Indeed, a decline in the level of ATP could affect GSH concentrations, which is synthesized through two ATP-dependent reactions. For Vpr/ANT interaction to occur, two arginine residues at positions 73 and 80 of Vpr are of great importance (Jacotot et al, 2000; Sabbah et al, 2006). To test this hypothesis, a Vpr mutant protein (Vpr R73,80A) was constructed, wherein the ANT binding site was knocked out. HFAs were incubated in the absence or presence of rVpr or rVpr R73,80A, and the levels of both ATP and GSH were assayed (Figure 3). Both the rVpr and rVpr R73,80A mutant induced comparable decreases in the levels of ATP and GSH, without any statistical difference at 24 and 48 h after exposure (Figure 3A and B, respectively). These observations suggested that a Vpr-induced mechanism other than binding to the ANT protein causes the pseudosynchronous declines in ATP and GSH. Additionally, we evaluated whether the Vpr-induced declines in ATP and GSH were reversible or not: HFAs were initially exposed to rVpr or rVpr R73,80A mutant for 48 h and cultivated for an additional 24 h with fresh medium. As shown in Figure 3A and B, neither ATP nor GSH concentrations rebounded to sub-initial values upon removal of extracellular Vpr, which points to an irreversible effect. A kinetic analysis was then performed to determine whether the levels of ATP or GSH could be rescued after shorter exposure times. To address this point, HFAs were treated with rVpr for 4, 8, 16, or 24 h and subsequently cultivated for an additional 24 h with fresh Vpr-deficient medium. This experimental approach models removal of exogenous Vpr by antigen-presenting cells and allows analysis of the astrocytic ATP and GSH content at different exposure times. The concentration of ATP shows an initial steep drop for the first 4 h after exposure, which did not return to preexposure levels after removal of Vpr and further cultivation for 24 h with fresh medium (Figure 3C). Conversely, the concentration of GSH was steady for the first 4 h after exposure and commenced to decline in the 4- to 8-h window (Figure 3D). Addition of fresh medium for 24 h did not lead to recovery of either ATP or GSH for any exposure time examined (Figure 3C and D).
Figure 3. Exposure of primary HFAs to extracellular Vpr or Vpr R73,80A induces irreversible decreases in both ATP and GSH metabolites through an ANT-independent pathway.

(A and B) HFAs treated for 24 or 48 h with extracellular rVpr or rVpr R73,80A (with reduced binding affinity to ANT) were assayed for intracellular levels of ATP (A) and GSH (B). Upon removal of the protein in the extracellular medium and culture with fresh new medium for an additional 24 h, neither ATP nor GSH rebounded to initial values (doxorubicin [DOX] was used as a positive control). (C and D) A kinetic analysis denotes irreversible effects on the concentration of ATP and GSH induced by rVpr in HFAs. HFAs were incubated with rVpr (5 μg/ml) for 4, 8, 16, or 24 h, after which they were cultivated for an additional 24 h with fresh new medium. ATP levels dropped after exposure for 4 h (C) and to a greater extent after longer treatment periods, whereas the decrease in the level of GSH (D) was marginally delayed. Exposures for 24 h in the absence of Vpr did not lead to recovery of ATP or GSH levels.
Induction of OS by extracellular Vpr
We have previously shown that exposure to extracellular Vpr induced decreases in ATP and GSH levels and a shift in the intracellular GSH/GSSG ratio toward increased generation of ROS (Ferrucci et al, 2012), although the exact correlation remains elusive. Given this, the intracellular mechanisms involved in rVpr-induced OS were examined by identifying the genes within the antioxidant defense mechanism pathways that were differentially regulated. HFAs incubated in the absence or presence of extracellular rVpr were analyzed through a gene array containing the most relevant genes of the OS pathway. Six genes were significantly upregulated (APOE, CCL5, DGKK, GPX5, NOS2, and PXDNL), and 11 were significantly downregulated (BNIP3, CYGB, DUOX2, DUSP1, FOXM1, GAPDH, HPRT1, MT3, MTL5, PTGS2, and SCARA3) (Table II). Of immediate interest was the reduction in mRNA expression of a common housekeeping gene (GAPDH). All of the results reported herein were analyzed without considering GAPDH among the housekeeping genes in order not to skew the results. Four additional housekeeping genes (B2M, HPRT1, RPL13A, and ACTB) were used in the analysis as normalizing factors, because their expression was found unvaried between untreated and Vpr-exposed HFAs.
Table II.
Genes up- and downregulated in extracellular Vpr-exposed HFAs.
| Gene ID | Fold | Gene Name | |
|---|---|---|---|
| APOE | 7.9 | Apolipoprotein E | |
| CCL5 | 4.2 | RANTES | |
| ↑ | DGKK | 5.0 | Diacylglycerol kinase kappa |
| GPX5 | 5.1 | GSH peroxidase 5 | |
| NOS2 | 5.3 | Nitric oxide synthase 2 | |
| PXDNL | 16.3 | Peroxidasin homolog ligand | |
|
| |||
| BNIP3 | −10.7 | BCL2/adenovirus E1B 19 kDa interacting protein 3 | |
| CYGB | −17.6 | Cytoglobin | |
| DUOX2 | −16.3 | Dual oxidase 2 | |
| DUSP1 | −4.2 | Dual specificity phosphatase 1 | |
| ↓ | FOXM1 | −4.0 | Forkhead box M1 |
| MT3 | −120.1 | Metallothionein 3 | |
| MTL5 | −4.4 | Metallothionein-like 5 | |
| PTGS2 | −6.0 | Prostaglandin-endoperoxidase synthase 2 | |
| SCARA3 | −7.2 | Scavenge receptor class A, member 3 | |
| HPRT1 | −7.9 | Hypoxanthine phosphoribosyltransferase 1 | |
| GAPDH | −5.3 | Glyceraldehyde 3-phosphate dehydrogenase | |
Extracellular Vpr impairs the glycolytic pathway
Recent studies underscored the role of GAPDH in apoptosis and OS, possibly due to its propensity for nuclear translocation (Dastoor and Dreyer, 2001; Sukhanov et al, 2006; Thangima Zannat et al, 2011). Additionally, GAPDH activity was found to decline upon exposure to doxorubicin (a chemotherapeutic drug), directly correlating with a decline in ATP levels (Wolf and Baynes, 2006). Decline in GAPDH activity, a key regulator enzyme in glycolysis, has been shown to be detrimental for all intracellular ATP-dependent metabolic processes. Western immunoblotting analysis of HFAs either untreated or treated with rVpr or with rVpr R73,80A showed only a minimal decrease in GAPDH expression 24 h after exposure (Figure 4A). The decline was slightly more pronounced although not significant after 48 h of treatment, with about a 40% reduction in protein expression determined by semiquantitative densitometric analysis (Figure 4B). However, GAPDH activity was significantly decreased in HFAs treated with both rVpr or rVpr R73,80A (Figure 4C and D). As a positive control, HFAs treated with two different concentrations of doxorubicin displayed about 20% and 75% declines in GAPDH activity, respectively (Figure 4C), as previously reported (Cui et al, 2006).
Figure 4. Extracellular Vpr and Vpr R73,80A impair GAPDH activity.

(A) Immunoblot assay shows small downregulation of GAPDH in both rVpr- and rVpr R73,80A-treated HFAs (5 μg/ml each) at 24 and 48 h after exposure. (B) Densitometric analysis showed virtually no difference in GAPDH expression at 24 h in rVpr-, rVpr R73,80A-, and doxorubicin (DOX) (1 μM)-treated HFAs, whereas a slight decrease (~ ½ fold) was observed 48 h after exposure. (C) HFAs treated with doxorubicin at two different concentrations (0.5 and 1 μM) or exogenous rVpr (5 μg/ml) showed decreased activity of GAPDH 24 h after exposure as measured by fluorimetric assay. (D) Both rVpr and rVpr R73,80A (5 μg/ml each) proteins induced decreased GAPDH activity 48 h after treatment. *p value <0.05 (Student paired t-test).
Vpr-induced effects on astrocytes can impact neuron function
Astrocytes are key regulators in CNS homeostasis by tightly regulating brain homeostasis and supporting neurons architecturally and metabolically. One of the most important metabolites secreted by astrocytes for neuronal homeostasis is GSH, which aids in maintaining a balanced neuronal redox state. Secreted GSH is initially cleaved into cysteine-glycine and subsequently into cysteine, which is then taken up by neurons to synthesize GSH intracellularly (Garg et al, 2008). Increased astrocytic oxidation in the form of decreased GSH release is translated to the neuronal compartment, because less cysteine is available for uptake from the extracellular environment, unless supplementation with a cysteine-containing compound has been provided. To demonstrate that conditioned medium from astrocytes treated with rVpr or rVpr R73,80A perturbs neuronal oxidation status, the concentration levels of GSH were measured in an SK-N-SH neuroblastoma cell line subjected to different treatments. SK-N-SH neuroblastoma cells exposed to rVpr- or rVpr R73,80A-conditioned medium for 6 h displayed a 50% decrease in reduced GSH levels compared with untreated cells (Figure 5A). These values could be partially rescued on supplementation with N-acetyl-cysteine (NAC) (Figure 5A), supporting the hypothesis that conditioned medium containing Vpr indirectly affects neuronal synthesis of GSH through less availability of cysteine, at least in this cell line model. An anti-Vpr antibody (a kind gift of Dr. Jeffrey Kopp, NIH, Kidney Disease Section) was added to all conditioned media containing Vpr to demonstrate that the observed effects were not directly driven by the presence of residual exogenous Vpr in the supernatant. As a positive control, SK-N-SH neuroblastoma cells were treated for 6 h with 100 μM buthionine sulfoximine (BSO), which irreversibly inhibits the first enzyme in GSH synthesis and may not be rescued by cotreatment with NAC (Figure 5A). Additionally, to gain a better understanding of the downstream consequences caused by rVpr exposure, SK-N-SH neuroblastoma cell survival was assessed through an in situ TUNEL assay (terminal deoxynucleotidyl transferase dUTP nick end labeling). Untreated neuroblastoma cells showed minimal cell death after 6 h (Figure 5B), while the apoptogenic drug CPT (10 μM) induced significant cell death (Figure 5C). Similarly, cells exposed to conditioned medium treated with rVpr or rVpr R73,80A displayed increased apoptosis (Figure 5D and E), as shown by the increased number of apoptotic nuclei (overlapping green and blue staining).
Figure 5.

SK-N-SH neuroblastoma cells exposed to Vpr- or Vpr R73,80A-treated HFA-conditioned medium display decreased level of GSH synthesis and increased apoptosis. (A) SK-N-SH neuroblastoma cells were exposed for 6 h to different media conditioned with HFAs and assayed for intracellular reduced GSH content. Conditioned medium from Vpr- or Vpr R73,80A-treated HFAs (for 48 h) induced about a 50% decrease in neuronal GSH synthesis. These effects were recovered upon supplementation of NAC in the conditioned medium, while the oxidant BSO irreversibly inhibited GSH synthesis. Untreated (UT) SK-N-SH neuroblastoma cells (SK-N-SH cells exposed to conditioned medium from astrocytes that were not exposed to rVpr) also showed an increase in GSH concentrations with NAC treatment (~ 50%). Similarly, SK-N-SH neuroblastoma cells were exposed for 6 h to different HFA-conditioned media and subjected to the in situ TUNEL assay. Although untreated cells (UT) showed virtually a complete lack of apoptotic nuclei (B), SK-N-SH neuroblastoma cells exposed to the apoptogenic compound CPT (10 μM) (C), conditioned medium from Vpr- (D) or Vpr R73,80A (E) -treated HFAs (for 48 h) displayed an increase in the presence of apoptotic nuclei (FITC channels, second column), positively overlapping with DAPI staining (nuclei, first column), as shown in the merged figure (third column). All magnifications were ×10. Scale bars = 10 μm, *p value <0.05 (Student paired t-test).
Discussion
HIV-1 infection of the CNS and its pathogenesis in this compartment continues to be an area of intense interest to the scientific community due to an increase in HAND that has been noted for the past several years (Cysique et al, 2009; McArthur, 2004; Nath et al, 2008). The CNS compartment is separated from the peripheral blood by the blood-brain barrier (BBB) which restricts access of the brain to pathogens, drugs, and cells of the immune system (Strazza et al, 2011). Within the CNS there are a number of cells that are infected by HIV-1 or dysregulated by the virus or components of the virus, including Vpr. Within the CNS, previous studies of extracellular Vpr have centered their attention on neurons, which are gradually lost throughout disease and especially during late stages resulting in a wide range of HAND. With respect to the effect of extracellular Vpr on neurons,, studies have shown neurons exposed to rVpr are: (1) severely impaired, with inhibition of axonal outgrowth and death (Kitayama et al, 2008; Sabbah and Roques, 2005) and (2) able to form porous channels at the plasma membrane (Piller et al, 1996), which caused aberrant inward sodium current in neurons (Piller et al, 1998; Potter et al, 2004) as well as rapid intracellular Ca2+ flux in astrocytes (Noorbakhsh et al, 2010). Abnormal concentrations of cytoplasmic Ca2+ could then lead to alteration in mitochondrial Ca2+ uptake, which, along with downregulation of GAPDH within the glycolytic pathway, could impair the efficiency of oxidative phosphorylation (Brookes et al, 2004), leading to a decline in the level of ATP. In fact, in studies using HFAs, a dose-dependent decrease was detected in the levels of ATP and GSH (Ferrucci et al, 2012), (Table 1), products of the aerobic respiration and antioxidant pathways, respectively.
To further investigate this association, herein genes differentially regulated upon exposure to extracellular Vpr were examined with six genes upregulated and 11 downregulated (Table II). Among the upregulated genes,, GPX5 is well known for reducing lipid peroxide and hydrogen peroxide to water (Chabory et al, 2010), although its role outside the epididymis of the mammalian male reproductive tract has not yet been explored. DGKK was upregulated about 5-fold compared with untreated astrocytes (Table II): upon activation of the G-protein–coupled receptor, DGKs phosphorylate 1,2,diacylglycerol into phosphatidic acid and activate protein kinase C, thus leading to phosphorylation of several target proteins, such as kinases of the MAPK pathway. If confirmed at the protein level, upregulation of DGK could explain the observed increased phosphorylation of SAPK/JNK and p38 MAP kinases previously reported (Noorbakhsh et al, 2010). CCL5 upregulation at the mRNA level (about 4-fold) was not detected in the cytokine array (Figure 2A), possibly underlying a posttranslational sequestering effect or dampening of protein production. Vpr treatment also increased transcriptional expression of ApoE, essential in the catabolism of triglyceride-rich lipoproteins, recently associated with increased risk of Alzheimer’s disease (Ma et al, 1994) and accelerated progression toward AIDS-defining illnesses (Cutler et al, 2004).
Most of the downregulated genes code for ROS-scavenging proteins, such as metallothionein 3 (MT3), cytoglobin (CYGB), DUOX2, and SCARA3 (Table II), all of which confer cellular ability to detoxify ROS. Exogenous Vpr could possibly induce abundant ROS production by downregulating all these enzymes, thus promoting an oxidative intracellular status. Of particular interest was the downregulation (about 5-fold at the mRNA level) of GAPDH, a common housekeeping gene. Our initially proposed mode of action by extracellular Vpr through binding to the ANT protein and blocking the passage of ATP for ADP between the mitochondrial matrix and the cytoplasm was challenged using a Vpr mutant protein with lesser ANT binding affinity (Figure 3A and B). Instead theses results suggested a pathway through impairment of one of the enzymes in the glycolytic pathway (GAPDH), which may compromise the conversion of glucose to pyruvate. GAPDH activity was severely impaired upon exposure to both parental and mutant Vpr (Figure 4C and D) and only mildly underregulated at the mRNA and protein levels (Table II and Figure 4A), which leads to reduced ATP synthesis owing to a lesser availability of substrate for the Krebs cycle and oxidative phosphorylation. These observations point to either direct binding of Vpr to GAPDH or an indirect mechanism through which Vpr affects GAPDH activity by removal of its substrate glyceraldehyde-3-phosphate (G3P). An additional mechanism could involve suppressing the reduction of NAD+ to NADH, which occurs simultaneously with the conversion of G3P to D-glycerate-1,3-bisphosphate. These results suggest that the intracellular cascade of events does not involve interaction between Vpr and ANT but rather a GAPDH-dependent mechanism, where impairment of the glycolytic pathway leads to ROS accumulation and decline in GSH levels (Figure 6, panel 1).
Figure 6. Model of HIV-1 extracellular Vpr action on astrocytes.

As exogenous Vpr gains access to the intracellular environment, it induces an initial decrease in ATP concentration through impairment of GAPDH. Prolonged ATP deprivation promotes increased ROS accumulation, which may no longer be detoxified by intracellular GSH, thus leading to a highly oxidized status (1). ROS buildup is potentially harmful because it induces transcription of oxidative-sensitive genes along with NF-κB-dependent genes, including transcription driven from the HIV-1 LTR, which could enhance viral replication and spread from persistently infected astrocytes. ROS also induce caspase-dependent apoptosis (2). Additionally, exogenous Vpr induces secretion of the proinflammatory cytokines IL-6 and IL-8 and the chemoattractants MCP-1 and MIF, which in turn could facilitate migration of monocytes from the periphery. Decreased levels of PAI-1 in the CNS could also reduce the integrity of the BBB, allowing passage of otherwise excluded molecules. Increased chemotaxis of HIV-1-infected monocytes may also reseed viral infection in the CNS (3). Finally, GSH synthesized within astrocytes may also be secreted and, on cleavage by gamma-glutamyl transpeptidase (γ-GT) into Cys-Gly, may be taken up by neurons as Cys, thus igniting GSH production within neurons. A reduction of astrocytic GSH is then reverberated on the neuronal compartment, leading to decreased synthesis of GSH and neuronal demise (4).
Increased ROS accumulation could be either a direct consequence of prolonged ATP deprivation (Liu et al, 2005) or the result of a third partner molecule causing the observed effects. A likely candidate may be an increase in intracellular calcium concentrations. Not only has Vpr been shown to induce Ca2+ entry (Acharjee et al, 2010; Noorbakhsh et al, 2010; Rom et al, 2009), possibly through cell membrane permeabilization (Macreadie et al, 1996; Piller et al, 1996; Piller et al, 1998), which can trigger the opening of mitochondrial permeability transition pores (Gincel et al, 2001), but also Ca2+ ions can freely diffuse through the mitochondria owing to their molecular size. Although under physiological conditions, Ca2+ is beneficial for cell survival as it stimulates the Krebs cycle and electron flow through the electron transport chain, thus increasing ATP levels, excessive concentrations of Ca2+ impair oxidative phosphorylation and increase ROS buildup (Brookes et al, 2004), thus perturbing mitochondrial integrity. Indeed, mitochondrial transition pores are closed and ANT is in its matrix conformational state, whereby the voltage-dependent anion channel (VDAC) does not allow passage of solutes with a molecular weight greater than 1500 Da (Halestrap, 2009). Because there is no evidence that extracellular Vpr directly promotes opening of VDAC, which masks ANT from the cytoplasmic compartment, it is reasonable to hypothesize the existence of an intermediate molecule to promote pore opening. Overproduction of ROS then leads to a decrease in reduced GSH. Decreases in the intracellular levels of ATP and GSH explain mitochondrial membrane hyperpolarization (decreased Δψ) with induction of pore opening, leading to programmed cell death through a caspase cascade (Figs. 1A and B). Studies are under way to determine whether elimination of ROS (through supplementation of NAC or ATP-derived supplements) could restore GAPDH activity and functionality to counteract Vpr-induced downstream effects. Additionally, sustained ROS accumulation could trigger intracellular signaling and transcription of nuclear factor-κB (NF-κB)-dependent genes (Figure 6, panel 2). The dual action of ROS on both caspase-dependent apoptosis and transcription of NF-κB-dependent genes, including the HIV-1 long terminal repeat (LTR) promoter, is likely synchronized for the virus to take full advantage of the host cell machinery before its demise. Augmented viral production would then reseed infection in the CNS from persistently HIV-1-infected astrocytes.
The observed decrease in the levels of GSH could be either an immediate consequence of a decrease in ATP levels (although not through blockage of the ANT) or a consequence of an increased buildup of ROS within the intracellular environment. The interplay between ATP and GSH metabolites is particularly important, as different compounds might independently reduce the levels of ATP or GSH or both: for example, treatment with iodoacetate reduces ATP but not GSH (Verity et al, 1991). The role of GAPDH in OS has recently been evaluated and could be due to its nuclear translocation (Dastoor and Dreyer, 2001), thus disabling its specific role in glycolysis and inducing apoptosis. A lower expression of GAPDH and/or a decreased activity due to an abundant translocation to the nucleus could explain the observed loss in ATP production. An additional study wherein bovine pulmonary artery endothelial cells were exposed to doxorubicin identified decreased activity of GAPDH, which correlated positively with ATP concentration (Cui et al, 2006). GAPDH could promote initiation of apoptosis through nuclear translocation (Sen et al, 2008), further depleting cytoplasmic storage levels of ATP in a vicious cycle. The results herein support a role for GAPDH as an intracellular sensor of oxidation.
We subsequently evaluated any alteration in the cytokine profile of Vpr-exposed astrocytes, because HIV-1 has been shown to induce secretion of proinflammatory and chemotactic cytokines. We demonstrated increased secretion of IL-6 (Figure 2), as previously described in neuronal cells exposed to Vpr-treated, astrocytic-conditioned medium (Jones et al, 2007) or Vpr-treated monocyte-derived macrophages (Hoshino et al, 2010). IL-8 was also found to be increased as reported in a previous study in both T cells and macrophages infected with Vpr-containing HIV-1 virions (Muthumani et al, 2000). Two additional studies demonstrated increased IL-8 secretion in primary HFAs, either infected with a VSV-G (envelope glycoprotein G of vesicular stomatitis virus) pseudotyped virus (Li et al, 2007) or by coinfection with hepatitis C virus (Vivithanaporn et al, 2010). Two other chemotactic cytokines were found to be elevated in the extracellular medium of Vpr-treated HFAs: MCP-1 and MIF. Previous studies showed MCP-1 was found increased in astrocytes cocultured with HIV-1-infected macrophages (Muratori et al, 2010), thus possibly contributing to increased recruitment of T lymphocytes and HIV-1-infected macrophages migrating through the BBB, which could reseed infection within the CNS. Interestingly, astrocytes treated with exogenous HIV-1 Tat secreted abundant MCP-1, which was also found in excess in the brains and CSF of patients with HIV-associated dementia (Maingat et al, 2011). Given that Vpr may be acting through a similar mechanism as Tat.. MIF is upregulated in several different inflammatory and infectious diseases: peripheral blood mononuclear cells from HIV-1-infected patients have been found to release excessive amounts of MIF, which in turn activates viral transcription and replication in HIV-1-infected CD4+ T cells (Regis et al, 2010). Additionally, activation of the toll-like receptor 4 has been shown to induce the secretion of MIF by mature dendritic cells (Popa et al, 2006), macrophages (Roger et al, 2003), and astrocytes (Krasowska-Zoladek et al, 2007). Recombinant Vpr-treated macrophages have been shown to induce IL-6 production through toll-like receptor 4/MyD88 signaling (Hoshino et al, 2010). If substantiated in vitro, these results not only would confirm a similar mechanism of activation between monocyte-derived macrophages and astrocytes by exogenous Vpr but also would explain the cause of increased secretions of MIF and IL-6. The observed alteration in cytokine secretion could be either a direct consequence of Vpr exposure or an indirect effect of an autocrine/paracrine process by action of the secreted cytokines or a synergistic effect of Vpr with any of the aforementioned cytokines. Finally, decreased presence of PAI-1 in Vpr-exposed astrocytes could potentially cause deleterious effects in reducing the tightness of the BBB. PAI-1 is a member of the serine protease inhibitor 1 (serpin-1) family, the primary inhibitor of tissue-plasminogen activator, widely expressed both in the periphery and within the CNS, where astrocytes are the major source (Hino et al, 2001). A balanced production of tissue-plasminogen activator and PAI-1 is vital in a highly regulated and selective environment such as the CNS, through tight junctions within endothelial cells at the lining of the BBB (Tran et al, 1998). All of this put together may point to extracellular Vpr contributing to increased BBB permeability as well as increased recruitment of cells, especially monocyte-macrophage cells into the CNS through dysregulation of the astrocyte compartment (Figure 6, panel 3).
Kinetic analysis of rVpr-treated HFAs indicated that exogenous Vpr inhibited astrocytic ability to synthesize both ATP and GSH within 24 h of exposure (Figure 3). Short exposures to Vpr (4 h) were enough to compromise ATP pool levels (Figure 3C), although GSH content seemed unvaried (Figure 3D). However, the inability of HFAs to recover ATP values after a short 4-h exposure does not assure that Vpr-exposed astrocytes are similar to unexposed astrocytes. Fundamental functions such as removal of extracellular glutamate and neuronal support might be irreversibly compromised. Additionally, astrocytes preexposed to extracellular Vpr might be sensitized to future exposure because their ATP content has already declined and further treatment might lead to a larger and faster depletion. This difference in response to ATP and GSH at early time points after Vpr exposure could be explained by a delay in intracellular accumulation of ROS, whereas ATP decline is more rapid and caused by a direct impairment of GAPDH activity.
Lastly, augmented oxidation of GSH was shown to be detrimental for neuronal survival (Figure 6, panel 4): indeed, GSH is secreted into the extracellular medium and catabolized into Cys-Gly, subsequently taken up by neurons as cysteine, which triggers GSH synthesis (Garg et al, 2008). Increased shift from reduced (GSH) to oxidized (GSSG) glutathione within the astrocytes leads to diminished GSH secretion and uptake by the neuronal compartment (here modeled with the SK-N-SH neuroblastoma cell line), thus affecting their antioxidant pool (Figure 5A) and inevitably leading to apoptosis (Figure 5B–E; Figure 6, panel 4).
Given these and other studies, the importance of the role of astrocytes on support of the BBB and the neuronal population is once again highlighted. In the HAART era where patients are living longer with more subdued chronic disease including HAND, understanding the interplay of astrocyte dysfunction in conjunction with other cells of the CNS, especially neurons, is increasingly important. In fact, these results provide yet another piece of evidence to suggest that HIV-1 may be inducing neuronal pathogenesis leading to HAND.
Acknowledgments
We would like to thank Drs. Nathaniel Landau (NIH AIDS Research Program) and Jeffrey Kopp (NIH, Kidney Disease Section) for providing the Vpr plasmid and the anti-Vpr antibody, respectively. We also thank Dr. Bassel Sawaya (Temple University, Philadelphia, PA) for providing the GST-Vpr plasmid. This work was supported in part by funds from the Public Health Service, National Institutes of Health, through grants from the National Institute of Neurological Disorders and Stroke [NS32092 to B.W.] and the National Institute of Drug Abuse [DA19807 to B.W.]. Dr. Michael Nonnemacher was also supported by faculty development funds provided by the Department of Microbiology and Immunology and the Institute for Molecular Medicine and Infectious Disease.
List of abbreviations
- OS
oxidative stress
- ROS
reactive oxygen species
- GSH
glutathione
- GSSG
oxidized glutathione
- Vpr
viral protein R
- NAC
N-acetyl-l-cysteine
- ANT
adenine nucleotide translocator
- DOX
doxorubicin
- MCP-1
monocyte chemotactic protein-1
- MIF
migration inhibitor factor
- PAI-1
plasminogen activator inhibitor-1
- BSO
buthionine sulfoximine
- GST
glutathione S-transferase
- HA
hemagglutinin
- CPT
camptothecin
- qRT-PCR
quantitative real-time PCR
- HFA
human fetal astrocytes
- GFAP
glial fibrillary acidic protein
- SCARA3
scavenger receptor class A member 3
- IDV
integrated density value
- rVpr
recombinant Vpr
- DGK
diacylglycerol kinase
- BBB
blood-brain barrier
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ Contributions
AF was the principal experimentalist who was involved in the conception of this study, collected and analyzed data, and prepared the first draft of the manuscript. MRN and BW participated in the design and coordination of this study and critically reviewed the manuscript. All authors read and approved the final manuscript.
References
- Acharjee S, Noorbakhsh F, Stemkowski PL, Olechowski C, Cohen EA, Ballanyi K, Kerr B, Pardo C, Smith PA, Power C. HIV-1 viral protein R causes peripheral nervous system injury associated with in vivo neuropathic pain. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24:4343–53. doi: 10.1096/fj.10-162313. [DOI] [PubMed] [Google Scholar]
- Agrawal L, Louboutin JP, Marusich E, Reyes BA, Van Bockstaele EJ, Strayer DS. Dopaminergic neurotoxicity of HIV-1 gp120: Reactive oxygen species as signaling intermediates. Brain Res. 2009 doi: 10.1016/j.brainres.2009.09.113. [DOI] [PubMed] [Google Scholar]
- Baruchel S, Wainberg MA. The role of oxidative stress in disease progression in individuals infected by the human immunodeficiency virus. J Leukoc Biol. 1992;52:111–4. doi: 10.1002/jlb.52.1.111. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American journal of physiology Cell physiology. 2004;287:C817–33. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Chabory E, Damon C, Lenoir A, Henry-Berger J, Vernet P, Cadet R, Saez F, Drevet JR. Mammalian glutathione peroxidases control acquisition and maintenance of spermatozoa integrity. Journal of animal science. 2010;88:1321–31. doi: 10.2527/jas.2009-2583. [DOI] [PubMed] [Google Scholar]
- Cheng X, Mukhtar M, Acheampong EA, Srinivasan A, Rafi M, Pomerantz RJ, Parveen Z. HIV-1 Vpr potently induces programmed cell death in the CNS in vivo. DNA Cell Biol. 2007;26:116–31. doi: 10.1089/dna.2006.0541. [DOI] [PubMed] [Google Scholar]
- Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–44. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- Cui J, Tungaturthi PK, Ayyavoo V, Ghafouri M, Ariga H, Khalili K, Srinivasan A, Amini S, Sawaya BE. The role of Vpr in the regulation of HIV-1 gene expression. Cell Cycle. 2006;5:2626–38. doi: 10.4161/cc.5.22.3442. [DOI] [PubMed] [Google Scholar]
- Cutler RG, Haughey NJ, Tammara A, McArthur JC, Nath A, Reid R, Vargas DL, Pardo CA, Mattson MP. Dysregulation of sphingolipid and sterol metabolism by ApoE4 in HIV dementia. Neurology. 2004;63:626–30. doi: 10.1212/01.wnl.0000134662.19883.06. [DOI] [PubMed] [Google Scholar]
- Cysique LA, Vaida F, Letendre S, Gibson S, Cherner M, Woods SP, McCutchan JA, Heaton RK, Ellis RJ. Dynamics of cognitive change in impaired HIV-positive patients initiating antiretroviral therapy. Neurology. 2009;73:342–8. doi: 10.1212/WNL.0b013e3181ab2b3b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dastoor Z, Dreyer JL. Potential role of nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase in apoptosis and oxidative stress. Journal of cell science. 2001;114:1643–53. doi: 10.1242/jcs.114.9.1643. [DOI] [PubMed] [Google Scholar]
- Deshmane SL, Mukerjee R, Fan S, Del Valle L, Michiels C, Sweet T, Rom I, Khalili K, Rappaport J, Amini S, Sawaya BE. Activation of the oxidative stress pathway by HIV-1 Vpr leads to induction of hypoxia-inducible factor 1alpha expression. J Biol Chem. 2009;284:11364–73. doi: 10.1074/jbc.M809266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrucci A, Nonnemacher MR, Cohen EA, Wigdahl B. Extracellular human immunodeficiency virus type 1 viral protein R causes reductions in astrocytic ATP and glutathione levels compromising the antioxidant reservoir. Virus research. 2012;167:358–69. doi: 10.1016/j.virusres.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg SK, Banerjee R, Kipnis J. Neuroprotective immunity: T cell-derived glutamate endows astrocytes with a neuroprotective phenotype. J Immunol. 2008;180:3866–73. doi: 10.4049/jimmunol.180.6.3866. [DOI] [PubMed] [Google Scholar]
- Gincel D, Zaid H, Shoshan-Barmatz V. Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem J. 2001;358:147–55. doi: 10.1042/0264-6021:3580147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner VJ, Shvadchak V, Fritz J, Arntz Y, Didier P, Frisch B, Boudier C, Mely Y, de Rocquigny H. Characterization of the mechanisms of HIV-1 Vpr(52-96) internalization in cells. Biochimie. 2011;93:1647–58. doi: 10.1016/j.biochi.2011.05.033. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. What is the mitochondrial permeability transition pore? Journal of molecular and cellular cardiology. 2009;46:821–31. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. The Journal of biological chemistry. 1997;272:3346–54. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol. 1995;69:6705–11. doi: 10.1128/jvi.69.11.6705-6711.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzenberg LA, De Rosa SC, Dubs JG, Roederer M, Anderson MT, Ela SW, Deresinski SC. Glutathione deficiency is associated with impaired survival in HIV disease. Proc Natl Acad Sci U S A. 1997;94:1967–72. doi: 10.1073/pnas.94.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino H, Akiyama H, Iseki E, Kato M, Kondo H, Ikeda K, Kosaka K. Immunohistochemical localization of plasminogen activator inhibitor-1 in rat and human brain tissues. Neuroscience letters. 2001;297:105–8. doi: 10.1016/s0304-3940(00)01679-7. [DOI] [PubMed] [Google Scholar]
- Hoshino S, Konishi M, Mori M, Shimura M, Nishitani C, Kuroki Y, Koyanagi Y, Kano S, Itabe H, Ishizaka Y. HIV-1 Vpr induces TLR4/MyD88-mediated IL-6 production and reactivates viral production from latency. J Leukoc Biol. 2010;87:1133–43. doi: 10.1189/jlb.0809547. [DOI] [PubMed] [Google Scholar]
- Hoshino S, Sun B, Konishi M, Shimura M, Segawa T, Hagiwara Y, Koyanagi Y, Iwamoto A, Mimaya J, Terunuma H, Kano S, Ishizaka Y. Vpr in plasma of HIV type 1-positive patients is correlated with the HIV type 1 RNA titers. AIDS Res Hum Retroviruses. 2007;23:391–7. doi: 10.1089/aid.2006.0124. [DOI] [PubMed] [Google Scholar]
- Jacotot E, Ferri KF, El Hamel C, Brenner C, Druillennec S, Hoebeke J, Rustin P, Metivier D, Lenoir C, Geuskens M, Vieira HL, Loeffler M, Belzacq AS, Briand JP, Zamzami N, Edelman L, Xie ZH, Reed JC, Roques BP, Kroemer G. Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein rR and Bcl-2. J Exp Med. 2001;193:509–19. doi: 10.1084/jem.193.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacotot E, Ravagnan L, Loeffler M, Ferri KF, Vieira HL, Zamzami N, Costantini P, Druillennec S, Hoebeke J, Briand JP, Irinopoulou T, Daugas E, Susin SA, Cointe D, Xie ZH, Reed JC, Roques BP, Kroemer G. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J Exp Med. 2000;191:33–46. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GJ, Barsby NL, Cohen EA, Holden J, Harris K, Dickie P, Jhamandas J, Power C. HIV-1 Vpr causes neuronal apoptosis and in vivo neurodegeneration. J Neurosci. 2007;27:3703–11. doi: 10.1523/JNEUROSCI.5522-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitayama H, Miura Y, Ando Y, Hoshino S, Ishizaka Y, Koyanagi Y. Human immunodeficiency virus type 1 Vpr inhibits axonal outgrowth through induction of mitochondrial dysfunction. J Virol. 2008;82:2528–42. doi: 10.1128/JVI.02094-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klegeris A, Walker DG, McGeer PL. Regulation of glutamate in cultures of human monocytic THP-1 and astrocytoma U-373 MG cells. J Neuroimmunol. 1997;78:152–61. doi: 10.1016/s0165-5728(97)00094-5. [DOI] [PubMed] [Google Scholar]
- Krasowska-Zoladek A, Banaszewska M, Kraszpulski M, Konat GW. Kinetics of inflammatory response of astrocytes induced by TLR 3 and TLR4 ligation. Journal of neuroscience research. 2007;85:205–12. doi: 10.1002/jnr.21088. [DOI] [PubMed] [Google Scholar]
- Lassiter C, Fan X, Joshi PC, Jacob BA, Sutliff RL, Jones DP, Koval M, Guidot DM. HIV-1 transgene expression in rats causes oxidant stress and alveolar epithelial barrier dysfunction. AIDS Res Ther. 2009;6:1. doi: 10.1186/1742-6405-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DN, Refaeli Y, MacGregor RR, Weiner DB. Serum Vpr regulates productive infection and latency of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1994;91:10873–7. doi: 10.1073/pnas.91.23.10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DN, Refaeli Y, Weiner DB. Extracellular Vpr protein increases cellular permissiveness to human immunodeficiency virus replication and reactivates virus from latency. J Virol. 1995;69:1243–52. doi: 10.1128/jvi.69.2.1243-1252.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Bentsman G, Potash MJ, Volsky DJ. Human immunodeficiency virus type 1 efficiently binds to human fetal astrocytes and induces neuroinflammatory responses independent of infection. BMC Neurosci. 2007;8:31. doi: 10.1186/1471-2202-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA. AIDS-related dementia and calcium homeostasis. Annals of the New York Academy of Sciences. 1994;747:205–24. doi: 10.1111/j.1749-6632.1994.tb44411.x. [DOI] [PubMed] [Google Scholar]
- Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K. Human immunodeficiency virus type 1 (HIV-1) tat induces nitric-oxide synthase in human astroglia. J Biol Chem. 2002;277:39312–9. doi: 10.1074/jbc.M205107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu W, Song XD, Zuo J. Effect of GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Molecular and cellular biochemistry. 2005;268:45–51. doi: 10.1007/s11010-005-2996-1. [DOI] [PubMed] [Google Scholar]
- Ma J, Yee A, Brewer HB, Jr, Das S, Potter H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature. 1994;372:92–4. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- Macreadie IG, Arunagiri CK, Hewish DR, White JF, Azad AA. Extracellular addition of a domain of HIV-1 Vpr containing the amino acid sequence motif H(S/F)RIG causes cell membrane permeabilization and death. Mol Microbiol. 1996;19:1185–92. doi: 10.1111/j.1365-2958.1996.tb02464.x. [DOI] [PubMed] [Google Scholar]
- Maingat F, Halloran B, Acharjee S, van Marle G, Church D, Gill MJ, Uwiera RR, Cohen EA, Meddings J, Madsen K, Power C. Inflammation and epithelial cell injury in AIDS enteropathy: involvement of endoplasmic reticulum stress. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2011;25:2211–20. doi: 10.1096/fj.10-175992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur JC. HIV dementia: an evolving disease. J Neuroimmunol. 2004;157:3–10. doi: 10.1016/j.jneuroim.2004.08.042. [DOI] [PubMed] [Google Scholar]
- Muratori C, Mangino G, Affabris E, Federico M. Astrocytes contacting HIV-1-infected macrophages increase the release of CCL2 in response to the HIV-1-dependent enhancement of membrane-associated TNFalpha in macrophages. Glia. 2010;58:1893–904. doi: 10.1002/glia.21059. [DOI] [PubMed] [Google Scholar]
- Muthumani K, Kudchodkar S, Papasavvas E, Montaner LJ, Weiner DB, Ayyavoo V. HIV-1 Vpr regulates expression of beta chemokines in human primary lymphocytes and macrophages. Journal of leukocyte biology. 2000;68:366–72. [PubMed] [Google Scholar]
- Nath A, Schiess N, Venkatesan A, Rumbaugh J, Sacktor N, McArthur J. Evolution of HIV dementia with HIV infection. Int Rev Psychiatry. 2008;20:25–31. doi: 10.1080/09540260701861930. [DOI] [PubMed] [Google Scholar]
- Noorbakhsh F, Ramachandran R, Barsby N, Ellestad KK, LeBlanc A, Dickie P, Baker G, Hollenberg MD, Cohen EA, Power C. MicroRNA profiling reveals new aspects of HIV neurodegeneration: caspase-6 regulates astrocyte survival. FASEB J. 2010;24:1799–812. doi: 10.1096/fj.09-147819. [DOI] [PubMed] [Google Scholar]
- Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJ, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–9. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl A, Banki K. Genetic and metabolic control of the mitochondrial transmembrane potential and reactive oxygen intermediate production in HIV disease. Antioxid Redox Signal. 2000;2:551–73. doi: 10.1089/15230860050192323. [DOI] [PubMed] [Google Scholar]
- Piller SC, Ewart GD, Premkumar A, Cox GB, Gage PW. Vpr protein of human immunodeficiency virus type 1 forms cation-selective channels in planar lipid bilayers. Proc Natl Acad Sci U S A. 1996;93:111–5. doi: 10.1073/pnas.93.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piller SC, Jans P, Gage PW, Jans DA. Extracellular HIV-1 virus protein R causes a large inward current and cell death in cultured hippocampal neurons: implications for AIDS pathology. Proc Natl Acad Sci U S A. 1998;95:4595–600. doi: 10.1073/pnas.95.8.4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollicita M, Muscoli C, Sgura A, Biasin A, Granato T, Masuelli L, Mollace V, Tanzarella C, Del Duca C, Rodino P, Perno CF, Aquaro S. Apoptosis and telomeres shortening related to HIV-1 induced oxidative stress in an astrocytoma cell line. BMC Neurosci. 2009;10:51. doi: 10.1186/1471-2202-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa C, van Lieshout AW, Roelofs MF, Geurts-Moespot A, van Riel PL, Calandra T, Sweep FC, Radstake TR. MIF production by dendritic cells is differentially regulated by Toll-like receptors and increased during rheumatoid arthritis. Cytokine. 2006;36:51–6. doi: 10.1016/j.cyto.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Potter SJ, Lemey P, Achaz G, Chew CB, Vandamme AM, Dwyer DE, Saksena NK. HIV-1 compartmentalization in diverse leukocyte populations during antiretroviral therapy. Journal of leukocyte biology. 2004;76:562–70. doi: 10.1189/jlb.0404234. [DOI] [PubMed] [Google Scholar]
- Regis EG, Barreto-de-Souza V, Morgado MG, Bozza MT, Leng L, Bucala R, Bou-Habib DC. Elevated levels of macrophage migration inhibitory factor (MIF) in the plasma of HIV-1-infected patients and in HIV-1-infected cell cultures: a relevant role on viral replication. Virology. 2010;399:31–8. doi: 10.1016/j.virol.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger T, Froidevaux C, Martin C, Calandra T. Macrophage migration inhibitory factor (MIF) regulates host responses to endotoxin through modulation of Toll-like receptor 4 (TLR4) Journal of endotoxin research. 2003;9:119–23. doi: 10.1179/096805103125001513. [DOI] [PubMed] [Google Scholar]
- Rom I, Deshmane SL, Mukerjee R, Khalili K, Amini S, Sawaya BE. HIV-1 Vpr deregulates calcium secretion in neural cells. Brain Res. 2009;1275:81–6. doi: 10.1016/j.brainres.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah EN, Druillennec S, Morellet N, Bouaziz S, Kroemer G, Roques BP. Interaction between the HIV-1 protein Vpr and the adenine nucleotide translocator. Chem Biol Drug Des. 2006;67:145–54. doi: 10.1111/j.1747-0285.2006.00340.x. [DOI] [PubMed] [Google Scholar]
- Sabbah EN, Roques BP. Critical implication of the (70–96) domain of human immunodeficiency virus type 1 Vpr protein in apoptosis of primary rat cortical and striatal neurons. J Neurovirol. 2005;11:489–502. doi: 10.1080/13550280500384941. [DOI] [PubMed] [Google Scholar]
- Sen N, Hara MR, Kornberg MD, Cascio MB, Bae BI, Shahani N, Thomas B, Dawson TM, Dawson VL, Snyder SH, Sawa A. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nature cell biology. 2008;10:866–73. doi: 10.1038/ncb1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman MP, Schubert U, Williams SA, de Noronha CM, Kreisberg JF, Henklein P, Greene WC. HIV-1 Vpr displays natural protein-transducing properties: implications for viral pathogenesis. Virology. 2002;302:95–105. doi: 10.1006/viro.2002.1576. [DOI] [PubMed] [Google Scholar]
- Shinya E, Owaki A, Shimizu M, Takeuchi J, Kawashima T, Hidaka C, Satomi M, Watari E, Sugita M, Takahashi H. Endogenously expressed HIV-1 nef down-regulates antigen-presenting molecules, not only class I MHC but also CD1a, in immature dendritic cells. Virology. 2004;326:79–89. doi: 10.1016/j.virol.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Sorg O, Horn TF, Yu N, Gruol DL, Bloom FE. Inhibition of astrocyte glutamate uptake by reactive oxygen species: role of antioxidant enzymes. Mol Med. 1997;3:431–40. [PMC free article] [PubMed] [Google Scholar]
- Strazza M, Pirrone V, Wigdahl B, Nonnemacher MR. Breaking down the barrier: the effects of HIV-1 on the blood-brain barrier. Brain research. 2011;1399:96–115. doi: 10.1016/j.brainres.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhanov S, Higashi Y, Shai SY, Itabe H, Ono K, Parthasarathy S, Delafontaine P. Novel effect of oxidized low-density lipoprotein: cellular ATP depletion via downregulation of glyceraldehyde-3-phosphate dehydrogenase. Circulation research. 2006;99:191–200. doi: 10.1161/01.RES.0000232319.02303.8c. [DOI] [PubMed] [Google Scholar]
- Thangima Zannat M, Bhattacharjee RB, Bag J. In the absence of cellular poly (A) binding protein, the glycolytic enzyme GAPDH translocated to the cell nucleus and activated the GAPDH mediated apoptotic pathway by enhancing acetylation and serine 46 phosphorylation of p53. Biochemical and biophysical research communications. 2011;409:171–6. doi: 10.1016/j.bbrc.2011.04.094. [DOI] [PubMed] [Google Scholar]
- Tran ND, Schreiber SS, Fisher M. Astrocyte regulation of endothelial tissue plasminogen activator in a blood-brain barrier model. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1998;18:1316–24. doi: 10.1097/00004647-199812000-00006. [DOI] [PubMed] [Google Scholar]
- Treitinger A, Spada C, Ferreira LA, Neto MS, Reis M, Verdi JC, de Miranda AF, de Oliveira OV, Van der Sander Silveira M, Abdalla DS. Hepatitis B and hepatitis C prevalence among blood donors and HIV-1 infected patients in Florianopolis--Brazil. Braz J Infect Dis. 2000;4:192–6. [PubMed] [Google Scholar]
- Trotti D, Rossi D, Gjesdal O, Levy LM, Racagni G, Danbolt NC, Volterra A. Peroxynitrite inhibits glutamate transporter subtypes. J Biol Chem. 1996;271:5976–9. doi: 10.1074/jbc.271.11.5976. [DOI] [PubMed] [Google Scholar]
- Verity MA, Torres M, Sarafian T. Paradoxical potentiation by low extracellular Ca2+ of acute chemical anoxic neuronal injury in cerebellar granule cell culture. Molecular and chemical neuropathology/sponsored by the International Society for Neurochemistry and the World Federation of Neurology and research groups on neurochemistry and cerebrospinal fluid. 1991;15:217–33. doi: 10.1007/BF03161061. [DOI] [PubMed] [Google Scholar]
- Vivithanaporn P, Maingat F, Lin LT, Na H, Richardson CD, Agrawal B, Cohen EA, Jhamandas JH, Power C. Hepatitis C virus core protein induces neuroimmune activation and potentiates Human Immunodeficiency Virus-1 neurotoxicity. PLoS One. 2010;5:e12856. doi: 10.1371/journal.pone.0012856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf MB, Baynes JW. The anti-cancer drug, doxorubicin, causes oxidant stress-induced endothelial dysfunction. Biochimica et biophysica acta. 2006;1760:267–71. doi: 10.1016/j.bbagen.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Zhang HS, Li HY, Zhou Y, Wu MR, Zhou HS. Nrf2 is involved in inhibiting Tat-induced HIV-1 long terminal repeat transactivation. Free Radic Biol Med. 2009;47:261–8. doi: 10.1016/j.freeradbiomed.2009.04.028. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Yau C, Gray JW, Chew K, Dairkee SH, Moore DH, Eppenberger U, Eppenberger-Castori S, Benz CC. Enhanced NF kappa B and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer. 2007;7:59. doi: 10.1186/1471-2407-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]