

Abstract
Rett syndrome (RTT) results from loss-of-function mutations in the gene encoding the methyl-CpG-binding protein 2 (MeCP2) and is characterized by abnormal motor, respiratory and autonomic control, cognitive impairment, autistic-like behaviors and increased risk of seizures. RTT patients and Mecp2-null mice exhibit reduced expression of brain-derived neurotrophic factor (BDNF), which has been linked in mice to increased respiratory frequency, a hallmark of RTT. The present study was undertaken to test the hypotheses that BDNF deficits in Mecp2 mutants are associated with reduced activation of the BDNF receptor, TrkB, and that pharmacologic activation of TrkB would improve respiratory function. We characterized BDNF protein expression, TrkB activation and respiration in heterozygous female Mecp2 mutant mice (Het), a model that recapitulates the somatic mosaicism for mutant MECP2 found in typical RTT patients, and evaluated the ability of a small molecule TrkB agonist, LM22A-4, to ameliorate biochemical and functional abnormalities in these animals. We found that Het mice exhibit (1) reduced BDNF expression and TrkB activation in the medulla and pons and (2) breathing dysfunction, characterized by increased frequency due to periods of tachypnea, and increased apneas, as in RTT patients. Treatment of Het mice with LM22A-4 for 4 weeks rescued wild-type levels of TrkB phosphorylation in the medulla and pons and restored wild-type breathing frequency. These data provide new insight into the role of BDNF signaling deficits in the pathophysiology of RTT and highlight TrkB as a possible therapeutic target in this disease.
Introduction
Rett syndrome (RTT; OMIM 312750) is a complex autism spectrum disorder caused by loss-of-function mutations in the gene encoding methyl-CpG-binding protein 2 (MeCP2) (Amir et al., 1999; Shahbazian et al., 2002). In addition to cognitive and behavioral impairments, RTT patients exhibit severe motor, respiratory, and autonomic dysfunction (Chahrour and Zoghbi, 2007) and ∼25% die of apparent cardiorespiratory failure (Kerr et al., 1997). Development of RTT therapies has been complicated by the fact that loss of MeCP2 function is associated with dysregulation of large numbers of genes and downstream effectors (Chahrour et al., 2008) and the relationship between specific signaling pathways and individual RTT endophenotypes is poorly understood.
Brain-derived neurotrophic factor (BDNF) has emerged as a candidate for treatment of RTT based on findings that (1) BDNF expression is abnormally low in RTT mouse models (Chang et al., 2006; Wang et al., 2006; Ogier et al., 2007; Fyffe et al., 2008), and in RTT patients (Deng et al., 2007) and (2) increasing BDNF levels in RTT mice by genetic means or with agents that nonspecifically promote endogenous BDNF production improves physiologic function and/or survival (Chang et al., 2006; Ogier et al., 2007). Mature BDNF acts via two receptors, the p75 neurotrophin receptor (p75NTR) and the TrkB tyrosine kinase receptor (Chao and Hempstead, 1995). BDNF itself is limited as a therapeutic tool due to an unfavorable pharmacokinetic profile and because, through actions at more than one receptor, may have unwanted pleiotropic effects (Massa et al., 2010; Chao et al., 2006). Similarly, agents that increase BDNF, including ampakines (Lynch et al., 2008), inhibitors of BDNF gene repression (Rigamonti et al., 2007), mixed-lineage kinase inhibitors (Apostol et al., 2008), and antidepressants (Duman and Monteggia, 2006; Zanella et al., 2008), or that transactivate TrkB (Lee and Chao, 2001), are not specific for BDNF or TrkB, respectively. An alternative approach is to directly activate TrkB; potential strategies include TrkB activating antibodies (Qian et al., 2006) and small molecules that function as direct TrkB ligands (Xie and Longo, 2000; Jang et al., 2010; Massa et al., 2010). In studies in which enhancement of BDNF expression in RTT mice has been shown to improve function (Chang et al., 2006; Ogier et al., 2007), it is unknown which BDNF receptor target is required. The present study was undertaken to evaluate the ability of a small molecule, non-peptide BDNF loop 2 domain mimetic, LM22A-4, which functions as a direct and specific partial agonist of TrkB, but not p75 (Massa et al., 2010), to increase TrkB activation and improve breathing in a mouse model of RTT. LM22A-4 was developed by Longo, Massa and colleagues by in silico screening for mimetics of BDNF loop domains that selectively activate TrkB in vitro and in vivo and promote recovery of motor function in a rodent model of brain trauma (Massa et al., 2010). Using heterozygous female Mecp2 mutant mice (Chen et al., 2003), we show that daily treatment with LM22A-4 is well tolerated and rescues wild-type levels of TrkB phosphorylation and wild-type breathing frequency.
Materials and Methods
Animals.
Mecp2tm1.1Jae mice, developed by Dr. R. Jaenisch (Whitehead Institute, Massachusetts Institute of Technology, Cambridge, MA), were purchased from the Mutant Mouse Regional Resource Center (University of California Davis, Davis, CA) and maintained on a mixed background (129Sv, C57BL/6, BALB/c) by crossing Mecp2tm1.1Jae heterozygous females (Mecp2−/+, Het) with Mecp2tm1.1Jae wild-type males (Mecp2+/y). Experimental procedures were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University.
BDNF protein measurements.
BDNF concentrations were quantified by ELISA using the BDNF Emax Immunoassay System (Promega) according to the manufacturer's protocol. The sensitivity of this BDNF ELISA assay is ∼1–3 pg BDNF/ml. Brain tissues and nodose ganglia were rapidly dissected and quick-frozen on dry-ice. Brain tissue samples were homogenized in 200 μl of RIPA buffer (containing, in mm: 50 Tris-HCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 NaCl, 1 EDTA, pH 7.4) containing a mixture of protease inhibitors (Roche), and centrifuged at 16,000 × g for 15 min at 4°C. Nodose ganglia were homogenized in 100 μl of the same buffer and centrifuged at 14,000 × g for 30 s at room temperature. The supernatant from each sample was collected and stored at −80°C until further use. For brain tissue samples, an aliquot of the supernatant was used to determine total protein content using the Bradford technique (Bradford et al., 1976) and 300 μg of total protein were used for BDNF ELISA. For BDNF ELISA, the entire supernatant from each individual nodose ganglion was loaded.
TrkB, AKT, and ERK phosphorylation assays.
The ratios of (1) phospho-TrkBY817/TrkB (full-length), phospho-ERK/ERK and phospho-AKT/AKT, (2) phospho-TrkBY817/actin, phospho-ERK/actin and phospho-AKT/actin, (3) TrkB (full-length)/actin, ERK/actin and AKT/actin and (4) TrkB (truncated)/actin were measured by Western blot using the ECL Chemiluminescence System (GE Healthcare) as described previously (Yang et al., 2003). Site-specific rabbit monoclonal anti-phospho-TrkBY817 antibody was obtained from Epitomics and rabbit polyclonal TrkB antibody was obtained from Millipore. Rabbit polyclonal ERK and AKT, mouse monoclonal phospho-ERK and phospho-AKT antibodies were obtained from Cell Signaling Technology). Tissues were homogenized in a lysis buffer (containing, in mm: 20 Tris, pH 8.0, 137 NaCl, 1% Igepal CA-630, 10% glycerol, 1 PMSF, 10 μg/ml aprotinin, 1 μg/ml leupeptin, 500 μm orthovanadate). Lysates were centrifuged at 14,000 × g for 10 min, then the supernatant was collected and protein concentration was determined using the BCA Protein Assay Reagent (Pierce).
Spectrometric analysis of LM22A-4 levels in the brain.
The brain concentration of LM22A-4 (custom synthesized by Ricerca Biosciences, LLC) was evaluated in Het mice 1 h after a 50 mg/kg intraperitoneal dose using reverse-phase liquid chromatography with triple-quadrupole tandem mass spectrometric (LC-MS/MS) detection. Atenolol, a drug that does not cross the blood–brain barrier, was administered orally as a control to correct for contamination by blood present in the brain vascular space. Brainstem and forebrain samples were homogenized with a Virsonic 100 ultrasonic homogenizer and prepared for analysis using acetonitrile precipitation by combining one volume of sample with three volumes of acetonitrile. Samples were centrifuged and the resulting supernatant was sampled for analysis using a CTC Leap PAL autosampler (Leap Technologies) and two PerkinElmer series 200 micro pumps. Chromatography was performed at ambient temperature using a 50 × 2.0 mm inner diameter, 4 μm Synergi Polar-RP analytical column (Phenomenex). The aqueous mobile phase (A) was 4 mm ammonium formate, pH 3.5 and the organic mobile phase (B) was 10:90 (v/v) 4 mm ammonium formate, pH 3.5/acetonitrile. The analyte was eluted with a gradient which changed linearly from 0 to 100% B in 3 min at a flow rate of 300 μl/min. The total run time was 4.5 min and the injection volume was 10 μl. The analyte was detected on a Sciex API 3000 triple-quadrupole mass spectrometer equipped with a TurboIonSpray interface in the positive electrospray ionization mode (Applied Biosystems/MDS). The multiple reaction monitoring transitions and instrument settings were optimized for LM22A-4. Equipment operation, data acquisition, and data integration were performed using Analyst version 1.4.2. software (Applied Biosystems). The drug injections, tissue extraction and LC-MS/MS analysis were performed by Absorption Systems.
Respiratory function.
Respiratory patterns were analyzed at 8, 10, and 12 weeks after birth in unrestrained mice using whole-body plethysmography as described previously (Ogier et al., 2007). Measurements were taken from quiet breathing periods of at least 5 min total duration, and apneas were defined as pauses in breathing greater than two times the average breath duration calculated for each animal.
LM22A-4 treatment.
Wt and Het littermates were divided into 4 groups: Wt vehicle-treated (100 μl of 0.9% NaCl, i.p., b.i.d.), Wt drug-treated (50 mg/kg LM22A-4 in 0.9% NaCl, i.p., b.i.d.), Het vehicle-treated (100 μl of 0.9% NaCl, i.p., b.i.d.), and Het drug-treated (50 mg/kg LM22A-4 in 0.9% NaCl, i.p., b.i.d.). Each mouse was treated from 8 to 13 weeks of age. Whole-body plethysmography was performed during week 12 and animals were subsequently killed and tissues harvested for TrkB Western blots and BDNF ELISA during week 13.
Statistical analyses.
Comparison of respiratory parameters between Wt and Het animals in the initial phenotyping experiments were performed using unpaired Student's t test. Comparisons among Wt vehicle-treated, Wt drug-treated, Het vehicle-treated, and Het drug-treated groups in the LM22A-4 trials, including plethysmography and Western blots were performed using one-way ANOVA with post hoc Least Significant Difference test (LSD) for intergroup comparisons. Results were considered significant if the p-value was <0.05. Data are presented as the mean ± SEM.
Results
Development of respiratory dysfunction in Het mice
The development of respiratory dysfunction in heterozygous Mecp2tm1.1Jae mice has not previously been described. Significant differences in breathing between Wt and Het mice first appeared between 8 and 10 weeks after birth (Fig. 1). At 10 weeks, Het mice exhibited an abnormally high breathing frequency associated with marked decreases in expiratory time (Te) and total breath duration (Ttot) and a small but significant decrease in inspiratory time (Ti; Fig. 1A1–A4). Significant differences in respiratory frequency, Te and Ttot (but not Ti) persisted at 12 weeks. The number of apneas in the Het population increased between 8 and 12 weeks of age (Fig. 1B1). This was due to a progressive increase in the proportion of Hets exhibiting significantly more apneas than Wt (20% at 8 weeks vs 50% at 12 weeks; Fig. 1B2).
Figure 1.

Development of respiratory dysfunction in 8- to 12-week-old Mecp2−/+ (Het) mice. A1–A4, Breathing frequency (A1), Ttot (A2), Ti (A3), and Te (A4) are not significantly different between Wt (open bars) and Het (gray bars) mice at 8 weeks of age (Wt, n = 7; Het, n = 5). Significant increases in frequency, associated with decreased Ti, Te, and Ttot, are observed in 10-week-old Hets compared with Wt controls (Wt, n = 22; Het, n = 14); these differences persist at 12 weeks (Wt, n = 16; Het, n = 14) with the exception of Ti. Results are expressed as the mean in percentage Wt ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, unpaired t test. B1, Box-and-whisker plots showing the number of apneas in Wt (open bars) and Het (gray bars) mice at 8, 10, and 12 weeks of age. B2, The proportion of Hets exhibiting significantly more apneas than Wt increased from 20% at 8 weeks to 50% at 12 weeks of age.
BDNF expression deficits in the brainstem of Het mice
Mecp2tm1.1Jae-null mice exhibit deficits in BDNF expression in structures critical for respiratory control, including the cranial sensory nodose ganglion (NG) and brainstem (Katz et al., 2009). To determine whether the development of respiratory dysfunction in Het mice is associated with changes in BDNF expression, we compared BDNF protein levels in these regions in Wt and Het mice at 8, 10, and 12 weeks of age (Fig. 2). BDNF levels in the Het NG were significantly below Wt at all 3 ages (Fig. 2A). Accordingly, we found reduced immunostaining for BDNF in the medullary nucleus tractus solitarius (nTS), the primary target of afferent projections from NG sensory neurons to the brainstem (Fig. 2D; 12 weeks of age). Despite this selective deficit within the Het nTS, differences in BDNF level between the Wt and Het medulla as a whole were only detectable by ELISA at 8 weeks of age (Fig. 2B). In the Het pons, the level of BDNF was normal at 8 weeks and fell below Wt values at 10 and 12 weeks (Fig. 2C).
Figure 2.
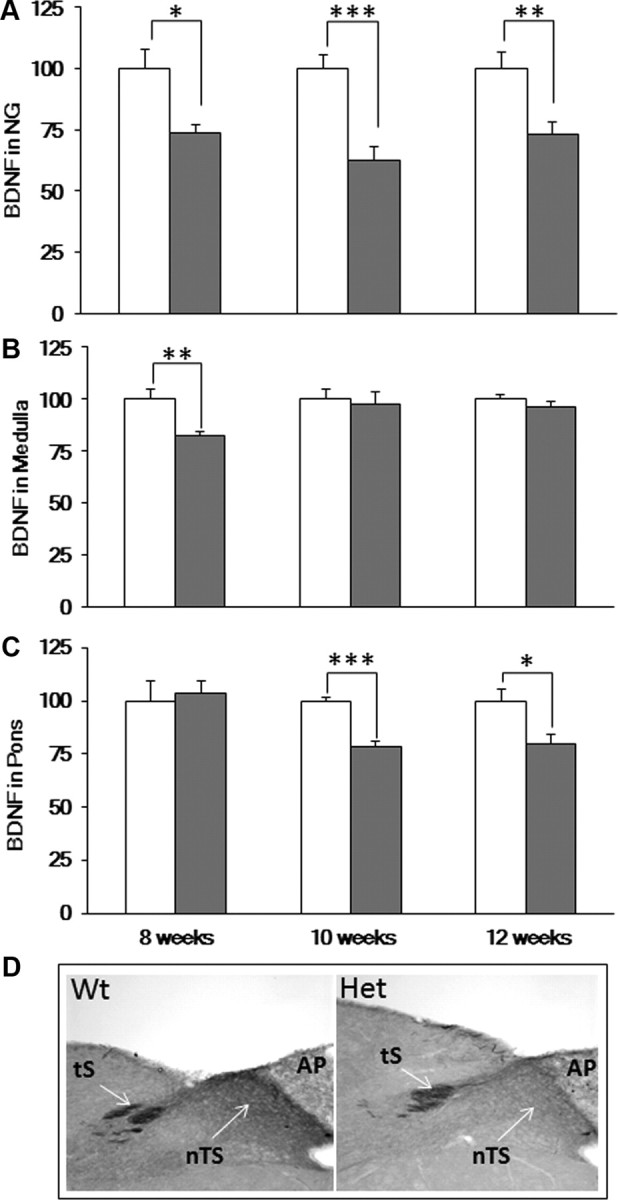
BDNF protein levels in 8-, 10-, and 12-week-old Mecp2 Het mice. A–C, BDNF levels, measured by ELISA, in the NG (A), medulla (B), and pons (C) of Wt (open bars) and Het mice (gray bars). Results are expressed as the mean in percentage Wt ± SEM (n = 5–19 NG per group). *p < 0.05, **p < 0.01, ***p < 0.001, unpaired t test. D, BDNF immunostaining, representative of 3 Wt and 3 Het littermate pairs, demonstrates reduced BDNF levels in the nTS subregion of the dorsomedial medulla in 12-week-old Het animals compared with Wt. AP, area postrema; tS, tractus solitarius.
Improved respiratory function following LM22A-4 treatment
In light of our finding that Het mice exhibit BDNF deficits in the nTS and pons, regions in which BDNF/TrkB signaling is important for respiratory control (Ogier and Katz, 2008), we next sought to examine whether or not systemic administration of LM22A-4, a small molecule BDNF loop domain mimetic that acts as a selective TrkB agonist (Massa et al., 2010), could improve the Het breathing phenotype. To determine the brain penetrance of LM22A-4 following systemic administration, brainstem and forebrain samples were analyzed by LC-MS/MS 1 h after a single intraperitoneal injection of 50 mg/kg LM22A-4 as described under Materials and Methods. These experiments demonstrated brain tissue concentrations (corrected for blood contamination) of 2.9 and 4.1 nm LM22A-4, respectively, which is well within the range at which LM22A-4 exhibits biological activity in assays of TrkB function (Massa et al., 2010). To evaluate the effects of systemic LM22A-4 administration on respiratory function, we used whole-body plethysmography to compare resting ventilation in 12-week-old Wt vehicle-treated, Wt drug-treated, Het vehicle-treated and Het drug-treated animals following 4 weeks of twice daily injections of LM22A-4 (50 mg/kg, i.p.) (Fig. 3A,B). In contrast to vehicle-treated Het controls, LM22A-4-treated Hets exhibited values of breathing frequency, Te and Ttot that were not significantly different from those of Wt (Fig. 3A,B; pooled results of 4 independent experiments). The percentage of apneic animals was unaffected by drug treatment (data not shown), and there were no significant differences in respiratory function between vehicle- and drug-treated Wt mice. Moreover, drug treatment had no effect on body weight in either Wt or Het mice throughout the treatment period (Fig. 3C).
Figure 3.
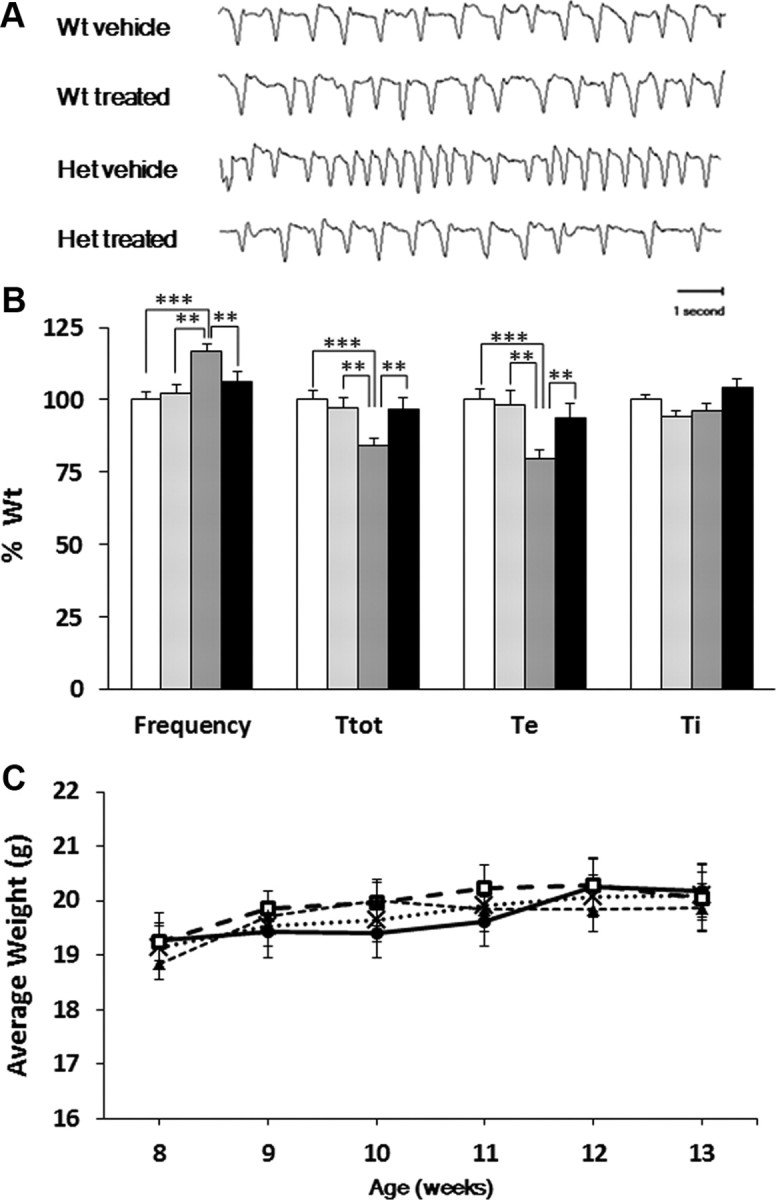
Treatment of Mecp2 Het mice with LM22A-4 restores normal respiratory frequency by increasing Te and Ttot. Animals were treated from 8 to 12 weeks of age as described in Materials and Methods. A, Representative plethysmographic traces showing the breathing pattern of Wt vehicle-treated, Wt LM22A-4-treated, Het vehicle-treated, and Het LM22A-4-treated mice. B, Comparison of the breathing frequency, Ttot, Ti, and Te among all four treatment groups. Results are expressed as the mean in percentage Wt ± SEM (Wt vehicle-treated, n = 29, open bars; Wt LM22A-4 treated, n = 13, light gray bars; Het vehicle-treated, n = 22, dark gray bars; Het LM22A-4-treated, n = 22, black bars). *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA I with post hoc LSD test. C, Body weight is unaffected by 5 weeks of treatment with LM22A-4 (50 mg/kg, i.p., b.i.d.) Results are shown as mean ± SEM (Wt vehicle-treated, n = 26, black cross marker; Wt LM22A-4 treated, n = 11, dark circle markers; Het vehicle-treated, n = 24, open square markers; Het LM22A-4 treated, n = 29, dark triangle markers).
LM22A-4 treatment reverses TrkB phosphorylation deficits in the brainstem of Het mice
We next sought to determine whether or not improved respiratory function in LM22A-4-treated Hets is associated with increased TrkB signaling in the brainstem. To address this issue, we used Western blots to compare the ratio of phosphorylated TrkB to total full-length TrkB (p-TrkBY817/TrkB) in 13-week-old Wt vehicle-treated, Wt drug-treated, Het vehicle-treated, and Het drug-treated animals following 5 weeks of systemic treatment with LM22A-4 (50 mg/kg, i.p., b.i.d.) (Fig. 4). To control for possible changes in total TrkB levels between Wt and Het samples, or with LM22A-4 treatment, we also compared the ratios of p-TrkBY817, total full-length TrkB and total truncated TrkB to actin (p-TrkBY817/actin, TrkB/actin, TrkB-T/actin, respectively).
Figure 4.
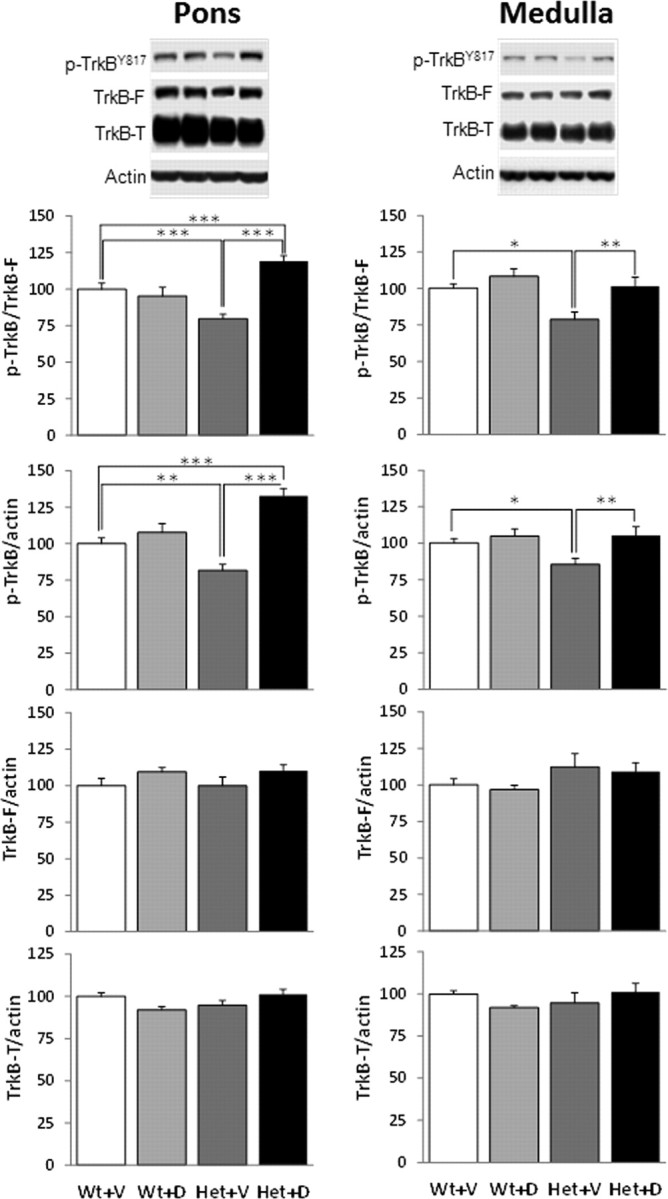
TrkB phosphorylation deficits in Mecp2 Het mice are reversed by chronic treatment with LM22A-4. Top, Representative Western blots showing phosphorylated TrkBY817 (p-TrkB), full-length TrkB (TrkB-F), truncated TrkB (TrkB-T), and actin in medulla and pons samples from (left to right) Wt vehicle-treated, Wt drug-treated, Het vehicle-treated, and Het drug-treated mice. Graphs, Summary results showing the ratios of p-TrkB/TrkB-F, p-TrkB-F/actin, TrkB-F/actin and TrkB-T/actin, respectively, in medulla and pons samples from all four treatment groups [open bars, Wt vehicle-treated (Wt+V); light gray bars, Wt drug-treated (Wt+D); dark gray bars, Het vehicle-treated (Het+V); black bars, Het drug-treated (Het+D)]. Results are expressed as the mean ± SEM (medulla; Wt+V, n = 9; Wt+D, n = 3; Het+V, n = 8; Het+D, n = 13; pons; Wt+V, n = 13; Wt+D, n = 7; Het+V, n = 11; Het+D, n = 15). *p < 0.05, ** p < 0.01, ***p < 0.001, ANOVA I with post hoc LSD test.
Pons and medulla samples from vehicle-treated (control) Hets exhibited significant decreases in p-TrkBY817/TrkB and p-TrkBY817/actin compared with vehicle-treated (control) Wt animals with no change in the levels of total full-length TrkB or truncated TrkB. These deficits in TrkB phosphorylation in Het mice were completely reversed by treatment with LM22A-4 (Fig. 4). In fact, drug-treated Hets showed significantly higher levels of p-TrkB/TrkB and p-TrkB/actin than wild-type controls in the pons. We also examined whether or not genotype and drug effects on TrkB phosphorylation were reflected in changes in phosphorylation of the serine/threonine protein kinase AKT (p-AKT) and extracellular signal-regulated kinase ERK/MAPK (p-ERK), key downstream mediators of the biological effects of TrkB activation (Lee et al., 2002). These experiments demonstrated significant decreases in p-AKT/AKT and p-AKT/actin in the pons of Het mice compared with Wt controls that were reversed by LM22A-4 treatment, with no change in total AKT levels (Fig. 5). In contrast, there was no significant effect of genotype or drug treatment on the level of p-ERK/ERK in pons samples from Wt and Het mice. In the medulla, although p-AKT/AKT and p-AKT/actin tended to be lower in Het mice compared with Wt, these trends were not statistically significant. p-ERK/ERK in the medulla was significantly lower in Het animals compared with Wt controls in both vehicle- and drug-treated animals (Fig. 6).
Figure 5.

AKT phosphorylation deficits in the pons of Mecp2 Het mice are reversed by chronic treatment with LM22A-4. Top, Representative Western blots showing p-AKT, total AKT (AKT), p-ERK, total ERK (ERK), and actin in pons samples from (left to right in each blot) Wt+V, Wt+D, Het+V, and Het+D mice. Graphs, Summary results showing the ratios of p-AKT/AKT, p-ERK/ERK, p-AKT/actin, p-ERK/actin, AKT/actin and ERK/actin in pons samples from all four treatment groups (open bars, Wt+V; light gray bars, Wt+D; dark gray bars, Het+V; black bars, Het+D). Results are expressed as the mean ± SEM (Wt+V, n = 13; Wt+D, n = 7; Het+V, n = 11; Het+D, n = 14). *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA I with post hoc LSD test.
Figure 6.

AKT and ERK phosphorylation in the medulla. Top, Representative Western blots showing p-AKT, total AKT (AKT), p-ERK, total ERK (ERK) and actin in medulla samples from (left to right in each blot) Wt+V, Wt+D, Het+V, and Het+D mice. Graphs, Summary results showing the ratios of p-AKT/AKT, p-ERK/ERK, p-AKT/actin, p-ERK/actin, AKT/actin and ERK/actin in medulla samples from all four treatment groups (open bars, Wt+V; light gray bars, Wt+D; dark gray bars, Het+V; black bars, Het+D). Results are expressed as the mean ± SEM (Wt+V, n = 9; Wt+D, n = 3; Het+V, n = 8; Het+D, n = 13). *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA I with post hoc LSD test.
Discussion
Increasing evidence indicates that symptoms of autism spectrum disorders in mouse models can be significantly improved by therapeutic interventions designed either to reverse genetic defects or restore normal signaling downstream of defective genes (Silva and Ehninger, 2009). In the case of RTT, identification of potential therapeutic targets has been complicated by the fact that loss-of-function mutations in Mecp2 result in dysregulation of large numbers of genes and downstream effectors (Chahrour et al., 2008), and the relationship between specific signaling pathways and individual RTT endophenotypes is poorly understood. The present finding that pharmacologic activation of TrkB in Mecp2 Het mice eliminates respiratory tachypnea supports the role of BDNF/TrkB signaling deficits in respiratory dysfunction in RTT and provides proof-of-concept for the therapeutic potential of TrkB agonists to improve this, and possibly other aspects of the disease.
Although we cannot yet ascribe reversal of respiratory tachypnea in treated Hets to specific populations of neurons, BDNF is known to regulate synaptic function in medullary and pontine cell groups important for respiratory control, including nTS (Balkowiec et al., 2000; Kline et al., 2010), the Kolliker-Fuse nucleus (Kron et al., 2007, 2008) and the pre-Bötzinger complex (Thoby-Brisson et al., 2003). We found that the medulla and pons of Het mice exhibit significant deficits in p-TrkB/TrkB, consistent with reduced expression of BDNF in these tissues, which are reversed by chronic treatment with LM22A-4. The fact that functional recovery in Het mice is associated with restoration of Wt levels of TrkB phosphorylation in the medulla and pons is consistent with previous findings that (1) exogenous BDNF can rescue wild-type synaptic function in brainstem slices from Mecp2-null mice in vitro (Kline et al., 2010) and (2) enhanced BDNF expression in ampakine-treated Mecp2-null mice is associated with normalization of respiratory frequency (Ogier et al., 2007). Moreover, the fact that the onset of significant respiratory dysfunction in Het mice between 8 and 10 weeks of age is temporally correlated with the appearance of reduced BDNF levels in the pons raises the possibility that pontine deficits in TrkB signaling, alone or in combination with BDNF deficits in the NG and nTS, are particularly important for expression of the Het respiratory phenotype. Of course, BDNF deficits elsewhere in the brain may also contribute to the Het phenotype, and LM22A-4 may act at multiple sites throughout the neuraxis to improve neurologic function in these animals. This possibility is supported by recent findings from our laboratory that, in addition to rescue of respiratory tachypnea, LM22A-4 treatment also improves sensorimotor function (D. Schmid and D. Katz, unpublished observation).
Analysis of signaling pathways downstream of TrkB revealed some apparently tissue-specific effects of genotype and drug treatment. In the pons, for example, vehicle-treated Het mice exhibited significant deficits in p-AKT/AKT and p-AKT/actin (and, to a lesser degree, AKT/actin) that paralleled the reduction in TrkB activation, whereas levels of p-ERK/ERK and p-ERK/actin were not significantly different from Wt controls. In the medulla, on the other hand, there was a trend, though no significant effect of genotype on levels of p-AKT/AKT or p-AKT/actin, whereas p-ERK/ERK was significantly below Wt controls. LM22A-4 treatment completely rescued the deficits in p-AKT/AKT and p-AKT/actin in the pons without affecting levels of p-ERK/ERK (although there was an increase in p-ERK/actin in the pons of drug-treated Hets, this was proportional to an increase in overall ERK levels (ERK/actin). p-AKT levels in the pons and medulla of Wt mice were unaffected by LM22A-4 treatment, perhaps indicating that AKT activation by TrkB is already maximal in these animals. Drug treatment also had no effect on levels of ERK phosphorylation in the Het medulla, where levels of p-ERK/ERK are reduced compared with Wt controls. These findings contrast with those of Massa et al. (2010) who showed that activation of TrkB by LM22A-4 in fetal hippocampal neurons from normal mice is accompanied by increased phosphorylation of both AKT and ERK. This apparent discrepancy may indicate that reduced expression of MeCP2, as in Het mice, interferes with pathways regulating ERK activation downstream of TrkB. These data also raise the possibility that restoration of AKT signaling in the pons may be particularly important for the phenotypic effects of LM22A-4 treatment. In addition, it is possible that the activity of other TrkB signaling partners not examined here, such as PLCγ (Huang and Reichardt, 2003) are also affected by reduced TrkB signaling in Het mice and play a role in the response to LM22A-4.
Our data highlight important differences between the respiratory phenotype of Mecp2tm1.1Jae mice, used in the present study, and studies of Mecp2tm1.1Bird mice, which are often used to model breathing dysfunction in RTT (Katz et al., 2009). According to published studies, Mecp2tm1.1Bird mice, in contrast to Mecp2tm1.1Jae mice (present study and Ogier et al., 2007), and RTT patients (Weese-Mayer et al., 2006), exhibit reduced respiratory frequency (Bissonnette and Knopp, 2008) and progressive slowing of breathing until death (Viemari et al., 2005). It is currently unknown whether this discrepancy results from differences in genetic background [(C57BL/6 (Mecp2tm1.1Bird) vs 129Sv,C57BL/6,BALB/c (Mecp2tm1.1Jae)], gene targeting strategies, or both. However, Ward et al. (2011) recently showed that animals with the Mecp2tm1.1Bird allele on a 129S6B6F1 background exhibit increased respiratory frequency, arguing strongly for genetic background effects on the expression of respiratory tachypnea in this model. Apneas, on the other hand, are characteristic of both Mecp2tm1.1Bird and Mecp2tm1.1Jae mice (Katz et al., 2009; Abdala et al., 2010; Ward et al., 2011). Our finding that LM22A-4 treatment at 50 mg/kg, b.i.d. resolves respiratory tachypnea, without affecting apneas, suggests possible differences in the mechanism underlying these two components of respiratory dysfunction in RTT. However, recent studies in our laboratory indicate that acute administration of LM22A-4 at higher doses than used in the present study can significantly reduce apneas in Het mice, suggesting that deficits in TrkB signaling contribute to respiratory pauses as well (I. Adams and D. Katz, unpublished observation). Other groups have shown that apneas can be reduced or eliminated in Mecp2tm1.1Bird mice by various pharmacologic agents, including desipramine (Zanella et al., 2008), 8-hydroxy-dipropyl-aminotetralin, a serotonin 1a agonist (Abdala et al., 2010), or enhancers of GABA function (Abdala et al., 2010). Thus, it will now be important to determine whether or not different LM22A-4 dosing regimens, or combined strategies targeting BDNF/TrkB signaling and other transmitter systems together, can ameliorate both respiratory tachypnea and apnea in appropriate mouse models.
In summary, our data demonstrate the ability of a BDNF loop domain mimetic to enhance TrkB activation and restore wild-type respiratory frequency in Mecp2 Het mice. These findings provide validation of TrkB as a therapeutic target in mouse models of RTT and indicate that BDNF loop domain mimetics can be effective at overcoming functional deficits associated with reduced BDNF expression.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke, including funding from the American Recovery and Reinvestment Act (D.M.K.), the International Rett Syndrome Foundation (D.M.K.), the Jean Perkins Foundation (F.M.L.), Taube Philanthropies (F.M.L.), the Koret Foundation (F.M.L.), the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory R&D (S.M.M.). We thank James Howell, and Elizabeth Shick for outstanding technical assistance, Dr. David Baekey (University of Florida) for validating the selection of quiet breathing periods used for analysis of the plethysmographic results, and Drs. Sharyl Fyffe-Maricich and Steve Maricich for their thoughtful comments on an earlier version of the manuscript.
F.M.L. is a founder of PharmatrophiX, a company focused on development of neurotrophin receptor ligands.
References
- Abdala AP, Dutschmann M, Bissonnette JM, Paton JF. Correction of respiratory disorders in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2010;107:18208–18213. doi: 10.1073/pnas.1012104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Apostol BL, Simmons DA, Zuccato C, Illes K, Pallos J, Casale M, Conforti P, Ramos C, Roarke M, Kathuria S, Cattaneo E, Marsh JL, Thompson LM. CEP-1347 reduces mutant huntingtin-associated neurotoxicity and restores BDNF levels in R6/2 mice. Mol Cell Neurosci. 2008;39:8–20. doi: 10.1016/j.mcn.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Balkowiec A, Kunze DL, Katz DM. Brain-derived neurotrophic factor acutely inhibits AMPA-mediated currents in developing sensory relay neurons. J Neurosci. 2000;20:1904–1911. doi: 10.1523/JNEUROSCI.20-05-01904.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonnette JM, Knopp SJ. Effect of inspired oxygen on periodic breathing in methyl-CpG-binding protein 2 (Mecp2) deficient mice. J Appl Physiol. 2008;104:198–204. doi: 10.1152/japplphysiol.00843.2007. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Chao MV, Hempstead BL. p75 and Trk: a two-receptor system. Trends Neurosci. 1995;18:321–326. [PubMed] [Google Scholar]
- Chao MV, Rajagopal R, Lee FS. Neurotrophin signalling in health and disease. Clin Sci (Lond) 2006;110:167–173. doi: 10.1042/CS20050163. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Deng V, Matagne V, Banine F, Frerking M, Ohliger P, Budden S, Pevsner J, Dissen GA, Sherman LS, Ojeda SR. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum Mol Genet. 2007;16:640–650. doi: 10.1093/hmg/ddm007. [DOI] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Fyffe SL, Neul JL, Samaco RC, Chao HT, Ben-Shachar S, Moretti P, McGill BE, Goulding EH, Sullivan E, Tecott LH, Zoghbi HY. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior; aggression, and the response to stress. Neuron. 2008;59:947–958. doi: 10.1016/j.neuron.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A. 2010;107:2687–2692. doi: 10.1073/pnas.0913572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz DM, Dutschmann M, Ramirez JM, Hilaire G. Breathing disorders in Rett syndrome: progressive neurochemical dysfunction in the respiratory network after birth. Respir Physiol Neurobiol. 2009;168:101–108. doi: 10.1016/j.resp.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr AM, Armstrong DD, Prescott RJ, Doyle D, Kearney DL. Rett syndrome: analysis of deaths in the British survey. Eur Child Adolesc Psychiatry. 1997;6:71–74. [PubMed] [Google Scholar]
- Kline DD, Ogier M, Kunze DL, Katz DM. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J Neurosci. 2010;30:5303–5310. doi: 10.1523/JNEUROSCI.5503-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron M, Zhang W, Dutschmann M. Developmental changes in the BDNF-induced modulation of inhibitory synaptic transmission in the Kolliker-Fuse nucleus of rat. Eur J Neurosci. 2007;26:3449–3457. doi: 10.1111/j.1460-9568.2007.05960.x. [DOI] [PubMed] [Google Scholar]
- Kron M, Reuter J, Gerhardt E, Manzke T, Zhang W, Dutschmann M. Emergence of brain-derived neurotrophic factor-induced postsynaptic potentiation of NMDA currents during the postnatal maturation of the Kolliker-Fuse nucleus of rat. J Physiol. 2008;586:2331–2343. doi: 10.1113/jphysiol.2007.148916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FS, Chao MV. Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc Natl Acad Sci U S A. 2001;98:3555–3560. doi: 10.1073/pnas.061020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FS, Rajagopal R, Chao MV. Distinctive features of Trk neurotrophin receptor transactivation by G protein-coupled receptors. Cytokine Growth Factor Rev. 2002;13:11–17. doi: 10.1016/s1359-6101(01)00024-7. [DOI] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Chen LY, Gall CM. The substrates of memory: defects, treatments, and enhancement. Eur J Pharmacol. 2008;585:2–13. doi: 10.1016/j.ejphar.2007.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, Nehama D, Rajadas J, Longo FM. Small molecule BDNF mimetics that activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest. 2010;120:1774–1785. doi: 10.1172/JCI41356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett Syndrome. J Neurosci. 2007;27:10912–10917. doi: 10.1523/JNEUROSCI.1869-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogier M, Katz DM. Breathing dysfunction in Rett syndrome: understanding epigenetic regulation of the respiratory network. Respir Physiol Neurobiol. 2008;164:55–63. doi: 10.1016/j.resp.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian MD, Zhang J, Tan XY, Wood A, Gill D, Cho S. Novel agonist monoclonal antibodies activate TrkB receptors and demonstrate potent neurotrophic activities. J Neurosci. 2006;26:9394–9403. doi: 10.1523/JNEUROSCI.1118-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti D, Bolognini D, Mutti C, Zuccato C, Tartari M, Sola F, Valenza M, Kazantsev AG, Cattaneo E. Loss of huntingtin function complemented by small molecules acting as repressor element 1/neuron restrictive silencer element silencer modulators. J Biol Chem. 2007;282:24554–24562. doi: 10.1074/jbc.M609885200. [DOI] [PubMed] [Google Scholar]
- Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Ehninger D. Adult reversal of cognitive phenotypes in neurodevelopmental disorders. J Neurodev Disord. 2009;1:150–157. doi: 10.1007/s11689-009-9018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoby-Brisson M, Cauli B, Champagnat J, Fortin G, Katz DM. Expression of functional tyrosine kinase B receptors by rhythmically active respiratory neurons in the pre-Botzinger complex of neonatal mice. J Neurosci. 2003;23:7685–7689. doi: 10.1523/JNEUROSCI.23-20-07685.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viemari JC, Roux JC, Tryba AK, Saywell V, Burnet H, Peña F, Zanella S, Bévengut M, Barthelemy-Requin M, Herzing LB, Moncla A, Mancini J, Ramirez JM, Villard L, Hilaire G. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci. 2005;25:11521–11530. doi: 10.1523/JNEUROSCI.4373-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Chan SA, Ogier M, Hellard D, Wang Q, Smith C, Katz DM. Dysregulation of brain-derived neurotrophic factor expression and neurosecretory function in Mecp2 null mice. J Neurosci. 2006;26:10911–10915. doi: 10.1523/JNEUROSCI.1810-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CS, Arvide EM, Huang TW, Yoo J, Noebels JL, Neul JL. MeCP2 Is critical within HoxB1-derived tissues of mice for normal lifespan. J Neurosci. 2011;31:10359–10370. doi: 10.1523/JNEUROSCI.0057-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weese-Mayer DE, Lieske SP, Boothby CM, Kenny AS, Bennett HL, Silvestri JM, Ramirez JM. Autonomic nervous system dysregulation: breathing and heart rate perturbation during wakefulness in young girls with Rett syndrome. Pediatr Res. 2006;60:443–449. doi: 10.1203/01.pdr.0000238302.84552.d0. [DOI] [PubMed] [Google Scholar]
- Xie Y, Longo FM. Neurotrophin small-molecule mimetics. Prog Brain Res. 2000;128:333–347. doi: 10.1016/s0079-6123(00)28030-8. [DOI] [PubMed] [Google Scholar]
- Yang T, Bernabeu R, Xie Y, Zhang JS, Massa SM, Rempel HC, Longo FM. Leukocyte antigen-related protein tyrosine phosphatase receptor: a small ectodomain isoform functions as a hemophilic ligand and promotes neurite outgrowth. J Neurosci. 2003;23:3353–3363. doi: 10.1523/JNEUROSCI.23-08-03353.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanella S, Mebarek S, Lajard AM, Picard N, Dutschmann M, Hilaire G. Oral treatment with desipramine improves breathing and life span in Rett Syndrome mouse model. Respir Physiol Neurobiol. 2008;160:116–121. doi: 10.1016/j.resp.2007.08.009. [DOI] [PubMed] [Google Scholar]