

Abstract
The ability to control pre-mRNA splicing with small molecules could facilitate the development of therapeutics or cell-based circuits that control gene function. Myotonic dystrophy type 1 (DM1) is caused by the dysregulation of alternative pre-mRNA splicing due to sequestration of muscleblind-like 1 protein (MBNL1) by expanded, non-coding r(CUG) repeats (r(CUG)exp). Here we report two small molecules that induce or ameliorate alternative splicing dysregulation. The thiophene-containing small molecule (1) inhibits the interaction of MBNL1 with its natural pre-mRNA substrates. Compound (2), a substituted naphthyridine, binds r(CUG)exp and displaces MBNL1. Structural models show that 1 binds MBNL1 in the Zn-finger domain and that 2 interacts with UU loops in r(CUG)exp. This study provides a structural framework for small molecules that target MBNL1 by mimicking r(CUG)exp and shows that targeting MBNL1 causes dysregulation of alternative splicing, suggesting that MBNL1 is thus not a suitable therapeutic target for the treatment of DM1.
INTRODUCTION
Pre-mRNA splicing defects cause a wide variety of diseases including β-thalassemia, inherited breast cancer, fragile x-associated tremor ataxia syndrome (FXTAS), and myotonic dystrophy types 1 and 2 (DM1 and DM2, respectively) 1–6. In the former two cases, pre-mRNA splicing defects are caused by single nucleotide polymorphisms within intronic regions of their mRNAs, yielding cryptic alternative splice sites. In the latter three cases, pre-mRNA splicing defects are caused by sequestration of proteins that regulate alternative pre-mRNA splicing 5, 7.
DM1 is caused by sequestration of MBNL1 by expanded r(CUG) repeats (r(CUG)exp) present in the 3’ untranslated region (UTR) of the dystrophia myotonica protein kinase (DMPK) mRNA 8. r(CUG)exp folds into an extended hairpin that displays multiple copies of a 5’CUG/3’GUC motif, which are high affinity binding sites for MBNL1. Studies in patient-derived cell lines and in mouse models have shown that pre-mRNA splicing events are dysregulated in DM1, including alternative splicing of the insulin receptor (IR) 9, the cardiac troponin T (cTNT) 10, and the muscle-specific chloride ion channel (Clcn1) 11, 12. These defects explain symptoms suffered by DM1 patients such as insulin insensitivity (IR splicing), myotonia (Clcn1 splicing), and cardiac defects (cTNT splicing). Moreover, translational defects of the DMPK mRNA and the presence of nuclear foci are observed in DM patients 13, 14. Both defects are due to the binding of r(CUG)exp to various proteins, leading to poor nucleocytoplasmic transport 13, 15.
To identify small molecules that disrupt the r(CUG)exp-MBNL1 complex, we previously developed an assay amendable for quantitative high throughput screening (qHTS) 16. The small molecules that comprise the Molecular Libraries Small Molecule Repository (MLSMR) (>300,000 members) were screened, and the results have been published online at PubChem 17 and in reference 16.
The high throughput screen identifies compounds that inhibit the r(CUG)-MBNL1 interaction by binding the RNA or the protein. Thus, a small molecule binds r(CUG)exp and displaces MBNL1 could theoretically improve DM1-associated pre-mRNA splicing defects (Fig. 1A). However, small molecules that bind MBNL1 (and disrupt the r(CUG)exp-MBNL1 interaction) may inactivate the protein and induce DM1-like pre-mRNA splicing defects (Fig. 1A). Importantly, the screen could yield chemotypes that reverse DM1-associated pre-mRNA splicing defects as a first step toward development of therapeutics for this devastating disease.
Fig. 1.
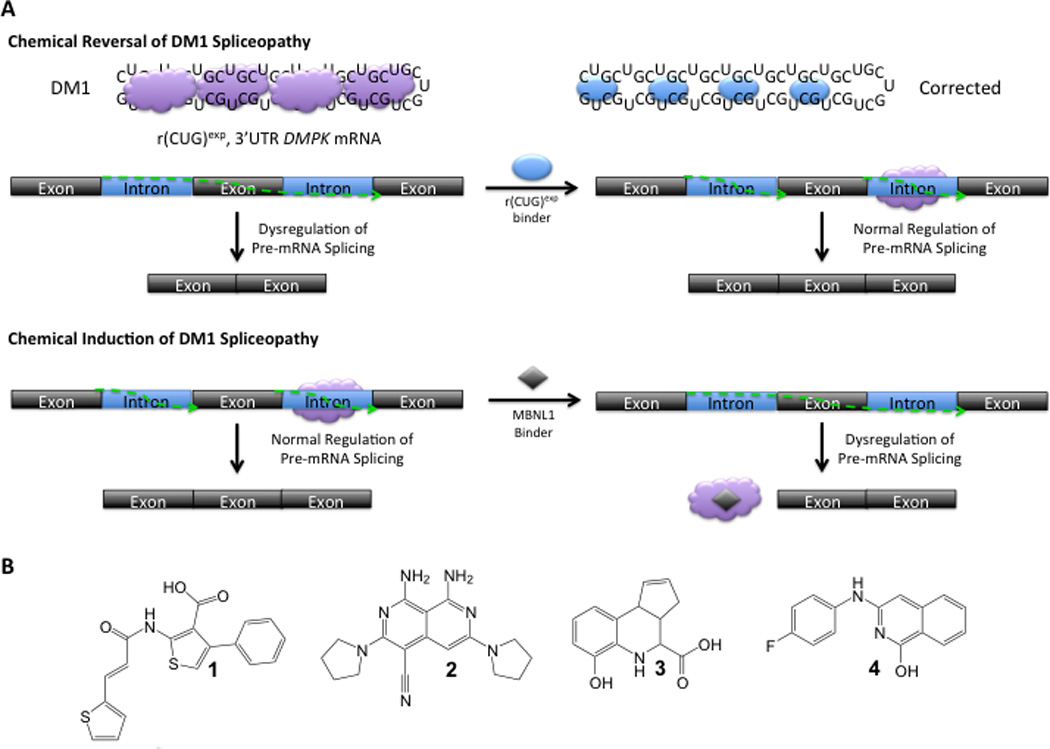
Identification of small molecules that inhibit the formation of the r(CUG)12-MBNL1 complex (A top) The biochemical outcome of a small molecule that targets r(CUG)exp would be the improvement of DM1-associated pre-mRNA splicing defects. (A bottom) The biochemical outcome of a small molecule that targets MBNL1 would be the induction of DM1-associated pre-mRNA splicing defects. (B) The small molecules that inhibit formation of the r(CUG)12-MBNL1 complex as identified by screening of the NIH’s MLPCN library.
Herein, we describe the results from our investigation of two hits from our qHTS: one that reverses DM1 pre-mRNA spliceopathy and another that induces DM1-like pre-mRNA spliceopathy. These compounds can serve as useful tools to study the effect of DM1 splicing on disease and to understand how to control pre-mRNA splicing or translation with cell-permeable small molecules.
RESULTS
High throughput screening for lead compounds against DM1
Previously, our team reported a screen for inhibitors of the toxic RNA-protein interaction that causes myotonic dystrophy type 1 16. The screen was designed to identify ligands that inhibit formation of an r(CUG)-MBNL1 complex that causes DM1. The assay utilizes biotinylated r(CUG)12 and MBNL1-His6. After formation of the RNA-protein complex, a ligand of interest is added followed by the addition of fluorescently labeled streptavidin (recognizes the biotinylated RNA) and a fluorescently labeled anti-His6 antibody (recognizes MBNL1). When the r(CUG)12-MBNL1 complex forms, the two fluorophores are within close enough proximity to form a fluorescence resonance energy transfer (FRET) pair, and time resolved (TR)-FRET can be measured. If, however, a small molecule inhibits r(CUG)12-MBNL1 complex formation, no TR-FRET would be observed. Four compounds identified from the NCGC screen were further investigated: (E)-4-phenyl-2-(3-(thiophen-2-yl)acrylamido)thiophene-3-carboxylic acid (1), 1,8-diamino-3,6-di(pyrrolidin-1-yl)-2,7-naphthyridine-4-carbonitrile (2), 6-hydroxy-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-4-carboxylic acid (3), and 3-((4-fluorophenyl)amino)isoquinolin-1-ol (4) (Fig. 1B). These compounds inhibit formation of the r(CUG)12-MBNL1 complex with IC50’s that range from 2 to 242 µM when the ligand and RNA are incubated first followed by addition of MBNL1. Compounds 2 and 3 are the most and least potent, respectively (Table 1).
Table 1.
Potencies and selectivities of compounds 1 – 4a
| Potencies & Selectivities of Small Molecules | ||||
|---|---|---|---|---|
| Compound | IC50b (µM) |
Complex | Compound 1; Selectivity for R(CUG)exp-MBNL1c |
Compound 2; Selectivity for r(CUG)exp-MBNL1d |
| 1 | 52 ± 12 | r(CUG)12-MBNL1 | 54 ± 2; - | 2 ± 0.4; - |
| 2 | 2 ± 0.4 | r(CAG)12-MBNL1 | 66 ± 13; 1.2 | 37 ± 5; 20 |
| 3 | 242 ± 1 | r(CGG)12-MBNL1 | 54 ± 4; 1.0 | 45 ± 5; 24 |
| 4 | 12 ± 1 | r(CCUG)12- MBNL1 |
75 ± 6; 1.4 | 29 ± 3; 15 |
Secondary structures of the RNAs are provided in Supplementary Figure S1.
The RNA and small molecule were pre-incubated followed by addition of MBNL1.
For studies with compound 1, the ligand was pre-incubated with MBNL1 followed by addition of RNA.
For studies with compound 2, the ligand was pre-incubated with the RNA followed by addition of MBNL1.
Improvement of DM1-associated translational defects
As an initial test for bioactivity, a previously described cellular model system that mimics the DMPK translational defects observed in DM1 was used (Fig. 2A) 18. In this model, firefly luciferase mRNA containing 800 r(CUG) repeats (r(CUG)800) in the 3’ UTR is stably expressed in C2C12 cells. Formation of the r(CUG)800-MBNL1 complex retains the firefly luciferase mRNA in the nucleus, disallowing nucleocytoplasmic transport. If a small molecule enters the nucleus and displaces MBNL1 from r(CUG)800, then the luciferase mRNA may be translocated to the cytoplasm and translated, resulting in increased luciferase activity. An increase in luciferase activity should be observed if compounds bind to r(CUG)exp or MBNL1, as both inhibit the formation of the r(CUG)800-MBNL1 complex.
Fig. 2.
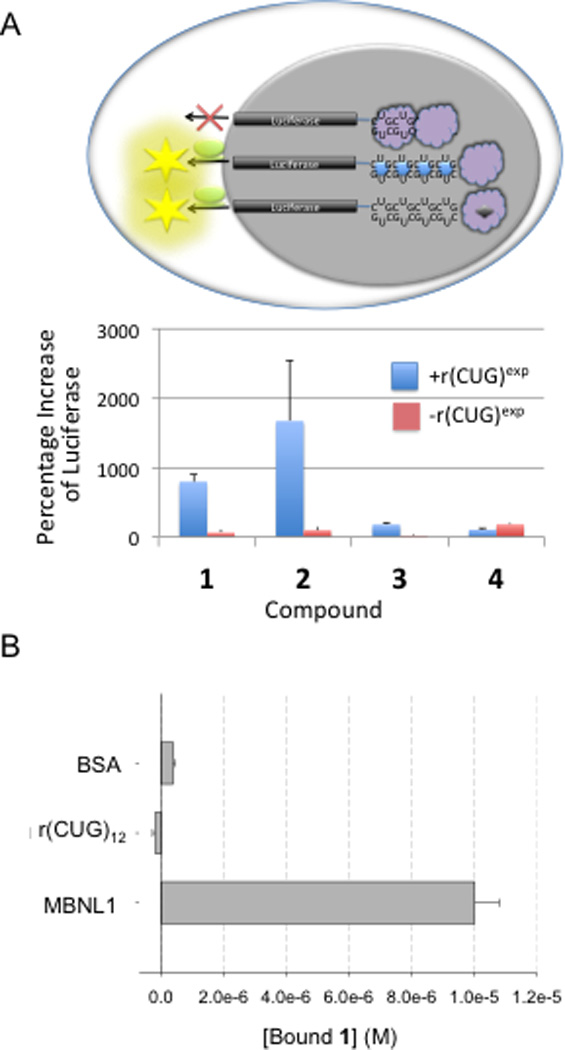
Compounds 1 – 4 improve translational defects caused by r(CUG)exp. Improvement of translational could occur if a compound binds to r(CUG)exp (compound 2) or MBNL1 (compound 1), as both modes of action disrupt the r(CUG)exp-MBNL1 complex and could allow for more efficient nucleocytoplasmic export. (A, top) A schematic of the luciferase reporter system that models the DM1 translational defect. The presence of r(CUG)exp in the 3’ UTR of firefly luciferase reduces nucleocytoplasmic transport and thereby suppresses luciferase expression. However, if a small molecule binds r(CUG)exp and displaces proteins, nucleocytoplasmic transport is improved and luciferase activity increases. Likewise, if a small molecule binds proteins and displaces them from r(CUG)exp, nucleocytoplasmic transport is also improved and luciferase activity increases. (A, bottom) Effects on luciferase activity when cells are dosed with 20 µM compound of interest. “+r(CUG)exp” indicates that the cell line expresses luciferase with r(CUG)exp in the 3′ UTR, “-r(CUG)exp” indicates that the cell line expresses luciferase without r(CUG)exp. Results are expressed as the percentage increase of luciferase activity relative to untreated cells, where a value of “0” denotes no change in activity. Experiments were completed in triplicate. Values shown in the plot are the averages of those experiments, and the errors reported are the standard deviations. (B), competition dialysis data for binding of 1 to BSA, MBNL1, and r(CUG)12. The data clearly show that the preferred target is MBNL1 as there is no measureable binding to r(CUG)12 or to BSA. Experiments were completed in duplicate. The values reported are the averages of those experiments, and the errors reported are the standard deviations.
All four compounds shown in Fig. 1B were tested in this system at 20 µM (Fig. 2A). Compounds 1 and 2 stimulated luciferase production most effectively, with increases in luciferase activity of ~700% and ~1600%, respectively. (Please note that no change in luciferase activity is equal to 0%.) Compounds 3 and 4 also increase luciferase activity but to a lesser extent (ca. 125%). Control experiments were completed using an analogous cellular model system that stably expresses firefly luciferase mRNA lacking r(CUG)exp. A slight increase in luciferase activity was observed for compounds 1, 2, and 3, although the increase is small compared to the increase observed in the presence of r(CUG)exp. In the case of compound 4, however, we observed similar increases in luciferase activity in cell lines with and without r(CUG)exp, indicating a non-specific effect. Therefore, 4 was no longer pursued as a lead compound. Due to the enhanced biological activity of 1 and 2, these two compounds were further investigated.
Affinity and Selectivity of 1 and 2 for r(CUG)exp and MBNL1
The binding of 1 and 2 to a mimic r(CUG)exp, r(CUG)12, and MBNL1 were determined by competition dialysis 19 and fluorescence anisotropy, respectively. Compound 2 non-specifically adheres to dialysis membranes, and thus is incompatible with competition dialysis.
Competition dialysis shows that 1 binds specifically to MBNL1 over both r(CUG)12 and a protein control, bovine serum albumin (BSA) (Fig. 2B). Almost no detectable partitioning of 1 was observed into dialysis tubes that contained BSA or r(CUG)12. In contrast, a large amount of partitioning was observed in tubes that contained MBNL1, affording a Kd of 470±20 nM. These data suggest that 1 inhibits r(CUG)exp-MBNL1 complex formation by binding to MBNL1 (Fig. 1A).
Fluorescence anisotropy measurements with 2 show that it binds specifically to r(CUG)12 with a Kd of 125 nM and a stoichiometry of ~6 small molecules per RNA (Table 2). No change in anisotropy was observed when up to two times the concentration of MBNL1 (650 nM) and ~35 times the concentration of BSA (12.5 µM) were added to 2 (325 nM), indicating weak protein binding. The affinity of 2 for another repeating RNA, r(CAG)12 (Supplementary Fig. S1), and a fully paired RNA (AU, Supplementary Fig. S1) were also determined. Compound 2 binds r(CAG)12 with a dissociation constant of 1400 µM and a 1:1 stoichiometry while no binding was observed to the AU RNA (Kd >> 25000 nM) (Table 2).
Table 2.
Affinity of 2 for various biomolecules
| Biomolecule | Kd (nM); stoichiometry | Biomolecule | Kd (nM); stoichiometry |
|---|---|---|---|
| MBNL1 | >> 2,000 | r(CAG)12 | 1400 ± 0.2; 1.2 ± 0.1 |
| BSA | >> 2,000 | 1×1 UU | 530 ± 0.1; 1.0 ± 0.1 |
| r(CUG)12 | 125 ± 0.052; 6 ± 0.4 | AU | >>25,000 |
The binding of 2 to an RNA containing a single 1×1 nucleotide UU internal loop (1×1 UU, Supplementary Fig. S1) was also measured. The compound binds 1×1 UU with a 1:1 stoichiometry and a Kd of 500 nM. There is an approximately 4-fold difference in affinity of 2 for r(CUG)12 and 1×1 UU. These data suggest that the ligand binds with positive cooperativity to longer RNAs. Such properties are favorable for selectively recognizing r(CUG)exp in vivo since many other genomic RNA targets contain single 1×1 nucleotide UU internal loop, notably the A-site in human ribosomes 20.
Selectivity of 1 and 2 for inhibiting RNA-MBNL1 complexes
Since 1 binds to MBNL1, its potency for disrupting RNA-MBNL1 interactions should be independent of the RNA’s sequence. In contrast, 2 (binds specifically to r(CUG)exp) should more potently inhibit r(CUG)12-MBNL1 complex formation as compared to complexes formed with other RNAs that bind MBNL1 (r(CAG)12, r(CCUG)12, and r(CGG)12, for example) 21, 22. (r(CAG)exp-MBNL1 and r(CCUG)exp-MBNL1 complexes cause pre-mRNA splicing defects in Huntington Disease patient-derived cell lines and in myotonic dystrophy type 2 cellular model, respectively 2, 6, 15, 23.) Therefore, we studied the potencies of compounds 1 and 2 for preventing RNA-MBNL1 complex formation (Table 1). In these experiments, 1 was pre-incubated with MBNL1, followed by addition of RNA while 2 was pre-incubated with the RNA of interest followed by addition of MBNL1.
As expected, our results indicate that compound 1 is a general inhibitor of RNA-MBNL1 complexes with an average IC50 of 62±10 µM (Table 1). In contrast, the IC50’s for 2 vary from 2 µM (r(CUG)12) to 45 µM (r(CGG)12). Compound 2 is at least 10-fold more potent for inhibiting the r(CUG)12-MBNL1 interaction than for inhibiting r(CAG)12-, r(CCUG)12-, and r(CGG)12-MBNL1 interactions. These data are in good agreement with the results of the binding measurements summarized in Table 2 and Fig. 2B and further support the hypothesis that the potencies of 1 and 2 are derived from binding MBNL1 and r(CUG)exp, respectively.
The contrasting modes of in vitro inhibition and binding data support the results observed in the luciferase assay. The luciferase model system cannot discriminate between compounds that disrupt r(CUG)exp-protein complexes by binding r(CUG)exp from those that disrupt the complexes by binding to proteins. If a small molecule binds to either protein or RNA, then the complex will be disrupted, allowing the mRNA to be translocated to the cytoplasm and translated. Our previously described competition dialysis, anisotropy, and potency data suggest that compounds 1 and 2 could have very different effects on pre-mRNA splicing. Compound 1, which binds MBNL1, could prevent MBNL1 from binding to its natural pre-mRNA substrates, inducing DM1-like splicing dysregulation in non-DM1 cells (i.e., like that observed when MBNL1 is sequestered by r(CUG)exp) (Fig. 1A). Compound 2, which binds r(CUG)exp, may displace MBNL1, freeing it to regulate pre-mRNA splicing as though r(CUG)exp had been removed (Fig. 1A).
Compound 1 induces splicing defects
MBNL1 regulates the alternative splicing of many pre-mRNAs 7, 9–12, 24, 25. Numerous splicing defects are observed in DM1 patients including the insulin receptor (IR) and the cardiac troponin T (cTNT) pre-mRNAs 9, 10, 24, 25. It has been shown that co-transfection of HeLa cells with a DM1 mini-gene that expresses 960 interrupted r(CUG) repeats causes dysregulation of mini-genes regulated by MBNL1; in particular splicing defects in the IR and cTNT pre-mRNAs have been observed by RT-PCR 10, 21, 26, 27. The percentage of exon 11 included in the IR mini-gene transcripts decreases from ≈97% in the absence of r(CUG)exp to ≈75% in the presence of r(CUG)exp (Fig. 3). Regarding the alternative splicing of the cTNT pre-mRNA, the percentage of exon 5 inclusion increases from ≈75% in the absence of r(CUG)exp to ≈90% in the presence of r(CUG)960 (Fig. 3).
Fig. 3.
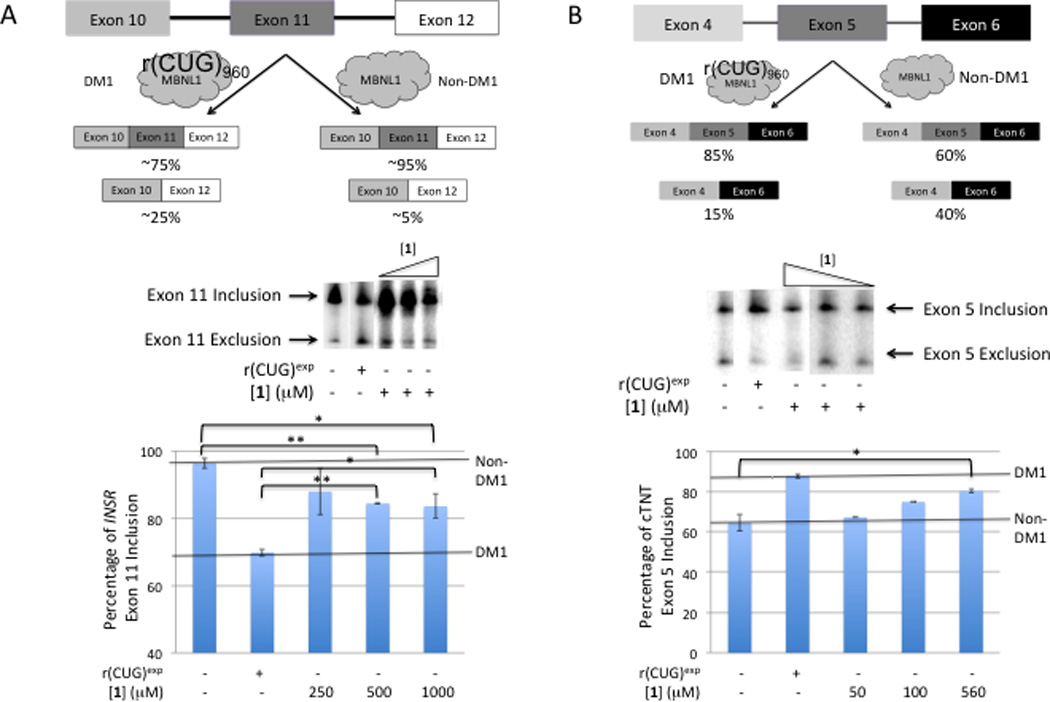
Compound 1 induces DM1-like splicing defects in HeLa cells. Briefly, HeLa cells were co-transfected with the mini-gene of interest and a plasmid that expresses r(CUG)exp or an empty vector. Varying concentrations of 1 were added in growth medium post-transfection, and total RNA was harvested 16–20 h later. The percentage of each splicing isoform was determined by RT-PCR using a radioactively labeled forward PCR primer. (A, top) Schematic of the pre-mRNA splicing isoforms observed for the IR mini-gene. (A, middle) representative gel image of IR alternative splicing in the presence of varying concentrations of 1. (A, bottom) Quantification of exon inclusion for IR alternative splicing in the presence or absence of r(CUG)exp and 1 (n ≥ 3). Error bars are the standard deviations in the measurements. (B, top) Schematic of the pre-mRNA splicing isoforms observed for the cTNT mini-gene. (A, middle) representative gel image of cTNT alternative splicing in the presence of varying concentrations of 1. (A, bottom) Quantification of exon inclusion for cTNT alternative splicing in the presence or absence of r(CUG)exp and 1 (n ≥ 3). Error bars are the standard deviations in the measurements. “*” indicates p ≤ 0.05 and “**” indicates p ≤ 0.01 as determined by a two-tailed student t-test.
Treatment of HeLa cells that do not express r(CUG)exp with compound 1 causes a shift in the alternative splicing patterns of the IR and cTNT mini-genes towards a DM1-like phenotype (Fig. 3). Dysregulation of IR pre-mRNA splicing is observed when cells are treated with 500 and 1000 µM of 1. Likewise, alternative splicing of the cTNT mini-gene is shifted towards DM-like phenotype when cells are dosed with compound 1, and a dose response is observed from 50 to 560 µM. Statistically significant changes in cTNT alternative splicing are only observed at 560 µM when compared to cells that do not express r(CUG)exp.
In order to determine if compound 1 generally affects the regulation of alternative splicing, additional experiments were completed with three mini-genes, the alternative splicing of which are not controlled by MBNL1 (Supplementary Fig. S2) 5, 27–29. These include pleckstrin homology domain containing, family H member 2 (PLEKHH2) 27, survival of motor neuron 2 (SMN2) 29, and B-cell lymphoma x (Bcl-x) 28. The alternative splicing of SMN2 and Bcl-x is regulated by the pre-mRNA splicing regulatory protein Sam68 5. Sam68 is sequestered by r(CGG)exp in Fragile X-associated tremor ataxia syndrome (FXTAS), causing dysregulation of SMN2 and Bcl-x alternative splicing 5. HeLa cells were co-transfected with the splicing reporter of interest and a DM1 mini-gene or a mini-gene that expresses only five r(CUG) repeats. Exposure of transfected cells to 500 µM of 1 had no effect on alternative splicing of PLEKHH2 or Bcl-x (Supplementary Fig. S2). A modest change in SMN2 alternative splicing was observed when cells were treated with 1 but this effect was independent of r(CUG)exp (Supplementary Fig. S2). Taken together, 1 primarily affects pre-mRNA splicing of genes controlled by MBNL1 but not those controlled by other proteins.
Next, we tested the effect of 1 on normal and DM1-affected human fibroblasts. Normal human fibroblasts were treated with 1 for 48 h, and its effect on the splicing of several MBNL1-dependent exons were determined including MBNL1, muscleblind-like 2 (MBNL2), nuclear receptor co-repressor (Ncor), nuclear factor I/X (CCAAT-binding transcription factor) (Nfix), and calcium/calmodulin-dependent protein kinase II gamma (Camk2g). Splicing abnormalities (towards the DM-like phenotype) were observed when cells were treated with 250 or 500 µM of 1 for all five MBNL1-dependent exons (Fig. 4A & B). The strongest effects were observed for MBNL1 and MBNL2 exons, in which splicing dysregulation is the same as in DM1-affected fibroblasts. The DM1-like splicing shift in NCOR2, NFIX and CAMK2G was also evident upon 500 µM 1, although more subtle than in the case of MBNLs (Figs. 4A & B).
Fig. 4.
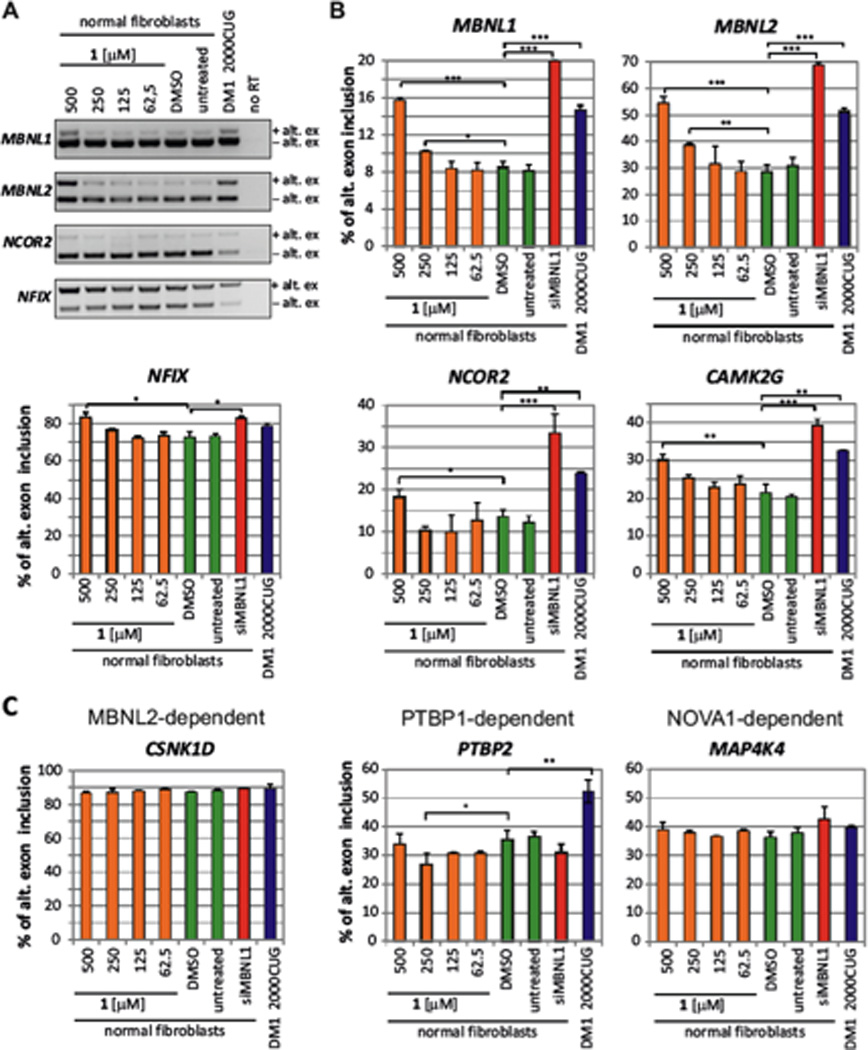
Compound 1 induces an MBNL1-dependent DM1-like splicing shift in normal human fibroblasts. (A) Representative RT-PCR assays showing splicing changes in MBNL1-dependent exons in normal fibroblasts treated with increasing amounts of 1 (n=2). The DM1-like splicing shift is depicted as alternative exon inclusion (+alt. ex), while the normal splicing isoform is depicted as alternative exon exclusion (−alt. ex). As a control, untreated and DMSO-treated (DMSO) normal fibroblasts (n=5) and DM1 fibroblasts expressing 2000 CUG repeats (DM1 2000CUG) (n=4) were used. No RT lane refers to RT-PCR control without reverse transcriptase. (B–C) Quantification of alternative splicing shift towards the DM1-like phenotype (% of alternative exon inclusion) in normal fibroblasts treated with increasing concentrations of 1 (orange bars; n=2), untreated and DMSO treated normal fibroblasts (green bars; n=5), siMBNL1 treated normal fibroblasts (red bars; n=2) and DM1-affected human fibroblasts expressing 2000 r(CUG) repeats (blue bars; n=2). Splicing of MBNL1-dependent exons is shown in (B) while splicing of MBNL1-independent exons is shown in (C). Each sample was subjected to RT-PCR twice. The errors reported are the standard deviations derived from analysis of all samples. “*” indicates p ≤ 0.05; “**” indicates p ≤ 0.01; and “***” indicates p ≤ 0.001 as determined by a two-tailed student t-test.
Notably, efficient siRNA knock-down of MBNL1 (by approximately 70% relative to untreated controls) in normal fibroblasts triggered only slightly more pronounced DM1-like splicing shift in MBNL1-dependent exons than treatment with 1 or than DM1-affected fibroblasts (Fig. 4B, Supplementary Fig. S3A). This suggests that 500 µM of 1 is not a saturating concentration required to induce a maximum possible effect on the splicing activity of MBNL1. To explore whether compound 1-induced DM-like splicing alterations can be further exacerbated, we next knocked-down MBNL1 in normal human fibroblasts using siRNA and then, 24h post siMBNL1 transfection, treated the cells with 1. Combined treatment with siMBNL1 (which reduced the level of MBNL1 mRNA by approximately 70% relative to untreated controls) and compound 1 allowed us to test whether reducing the pool of available MBNL1 exerts any effects on the splicing dysregulation induced by 1, and also assess whether 1 induces DM1-like splicing shift via MBNL1-dependent mechanism. Importantly, compound 1-induced dysregulation of preµmRNA alternative splicing of MBNL1-regulated exons (MBNL1, MBNL2, NCOR2, and NFIX) was significantly exacerbated by MBNL1 knock-down, strongly suggesting that the effects of 1 are indeed MBNL1-dependent (Fig. 5). For all analyzed MBNL1-regulated exons, splicing changes induced by highest concentration of 1 (i.e. 500 µM) combined with an siRNA against MBNL1 were much more pronounced than those induced by 1 alone (Figs. 4 & 5). We hypothesize that knocking-down MBNL1 with siRNA makes it possible to “saturate” the remaining pool of MBNL1 with compound 1, and thus augments the effect of 1 on the alternative splicing dysregulation of MBNL1-regulated exons.
Fig. 5.
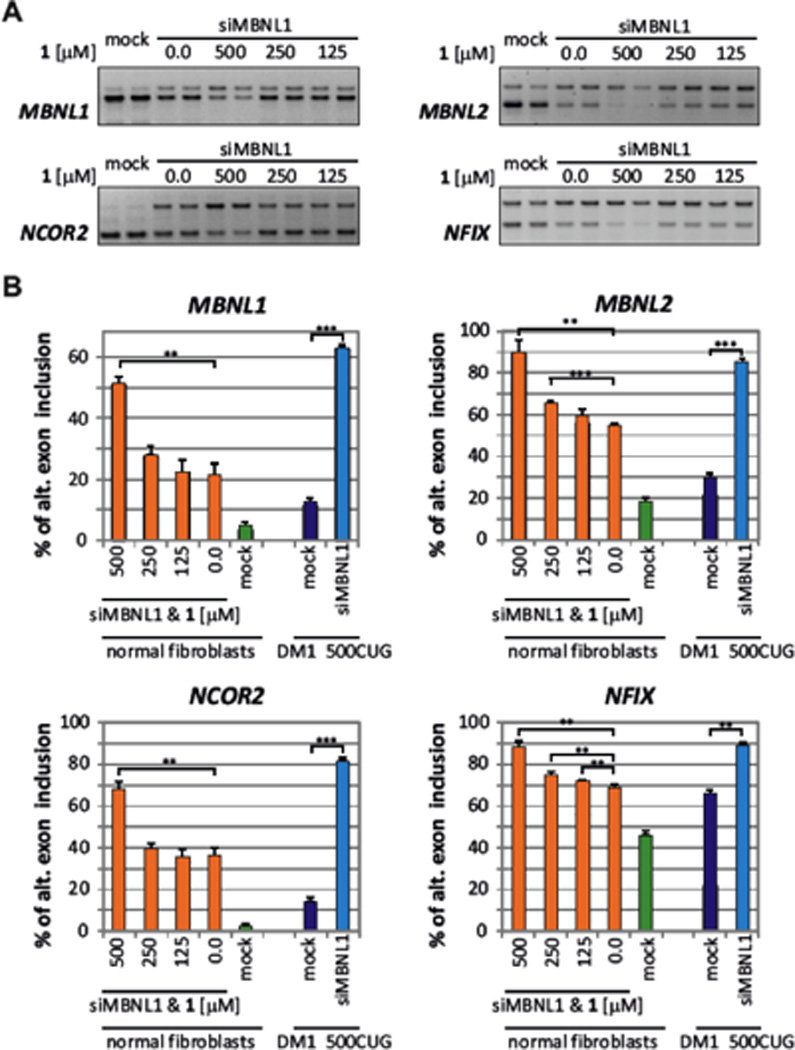
MBNL1 knock-down exacerbates DM1-like splicing shift induced by compound 1 in normal human fibroblasts. (A) Representative RT-PCR assays showing splicing changes in MBNL1-dependent exons of four genes in normal fibroblasts treated with siRNA against MBNL1 (siMBNL1) and increasing amounts of 1 (n=2). Mock controls are lipofectamine treated samples. (B) Quantification of alternative splicing shift towards the DM1-like phenotype (% of alternative exon inclusion) in four MBNL1-regulated exons (MBNL1, MBNL2, NCOR2, NFIX) upon combined treatment of normal human fibroblasts with an siRNA against MBNL1 and increasing concentrations of 1 (orange bars; n=2). For comparison, splicing shifts in siMBNL1-treated DM1-affected 500CUG fibroblasts are shown (blue bars; n=2). Mock controls represent splicing changes in lipofectamine treated cells (green bars - normal fibroblasts; violet bars - 500CUG fibroblast; n=2). “*” indicates p ≤ 0.05; “**” indicates p ≤ 0.01; and “***” indicates p ≤ 0.001 as determined by a two-tailed student t-test.
In agreement with our hypothesis, the combined effect of siMBNL1 and 500 µM 1 in normal fibroblast resulted in a roughly similar level of splicing shifts as those induced by MBNL1 knock-down in DM1µaffected fibroblasts expressing 500 r(CUG) repeats (500CUG fibroblasts) (Fig. 5). Likewise, 500CUG fibroblasts showed augmented preµmRNA alternative splicing dysregulation of MBNL1, MBNL2, NCOR2, and NFIX exons when treated with high concentration of 1 alone (i.e. 400 µM but not 200 µM) (Supplementary Figs. S4–S7). The extent of MBNL1 and MBNL2 splicing dysregulation in these cells was similar to DM1µaffected cells that express 1000 or 2000 r(CUG) repeats, and reached the level observed in normal fibroblasts treated with siMBNL1 (Fig. 4B & Supplementary Fig. S3). Taken together, these data strongly support our hypothesis that the DM1-like splicing shift exerted by 1 is mediated via MBNL1-dependent mechanism.
To investigate the specificity of 1 towards MBNL1 activity, we analyzed the effects of the compound on the alternative splicing of seven exons regulated independently of MBNL1, rather controlled by the splicing regulators MBNL2, PTBP1 (polypyrimidine tract-binding protein 1) or NOVA1 (neuro-oncological ventral antigen 1). Importantly, 1 did not affect the alternative splicing of previously established MBNL2-dependent exons of casein kinase 1 delta (CSNK1D), spectrin alpha chain, non-erythrocytic 1 (SPTAN1) and coactivator-associated arginine methyltransferase 1 (CARM1) even though MBNL1 and MBNL2 show high similarity of amino acid sequence (Fig. 4C and Supplementary Fig. S8) 30, 31. Furthermore, the splicing of NOVA1-dependent exons of mitogenµactivated protein kinase kinase kinase kinase 4 (MAP4K4) and amyloid beta (A4) precursorµlike protein 2 (APLP2) as well as PTBP1-regulated exons of polypyrimidine tract binding protein 2 (PTBP2) and fibroblast growth factor receptor 1 (FGFR1) were also unaffected in normal human fibroblasts treated with 1 (Fig. 4C and Supplementary Fig. S8).
To confirm that the exons under investigation are regulated independently of MBNL1, we analyzed their alternative splicing upon siRNA-mediated MBNL1 knock-down in normal and DM1-affected human fibroblasts. Indeed, none of these exons showed significantly altered splicing pattern upon siRNA knock-down of MBNL1 (Supplementary Fig. S3). Intriguingly, however, DM1-affected fibroblasts expressing 2000 r(CUG) repeats displayed marked splicing shift in PTBP2 when compared to compound 1-treated normal human fibroblasts, even though siRNA knock-down experiments have shown that this particular splicing event is apparently MBNL1-independent (Fig. 4C and Supplementary Figs. S3 & S8). Our interpretation of these results is that DM1 affected cells, in addition to MBNL1 sensitivity, might have additional inherent differences in their ability to regulate MBNL1-independent splicing events, for example via CUGBP1 or other splicing regulators. This hypothesis is bolstered by a recent report by Jog et al. in which they show that number and severity of alternative pre-mRNA splicing defects is reflective of the amount of free MBNL1 32. That is, modest changes in repeat number or the extent of r(CUG)exp hairpin structure formation (and thus the amount of active MBNL1) causes phenotypic variations 32. Taken together, our data strongly suggest that 1 specifically affects the function of MBNL1 protein in vivo.
Compound 2 improves splicing defects
In contrast, compound 2 improves DM1-associated pre-mRNA splicing defects in HeLa cells transfected with the DM mini-gene (Fig. 6). Addition of as little as 125 µM of 2 to cells that express r(CUG)exp restores IR pre-mRNA splicing back to levels those observed in the absence of r(CUG)exp. Similarly, improvement in cTNT alternative splicing is also observed. When cells are treated with 300 µM of 2, pre-mRNA splicing levels are statistically indistinguishable from those observed in the absence of r(CUG)exp. Alternative splicing levels are statistically different from cells that express r(CUG)exp.
Fig. 6.
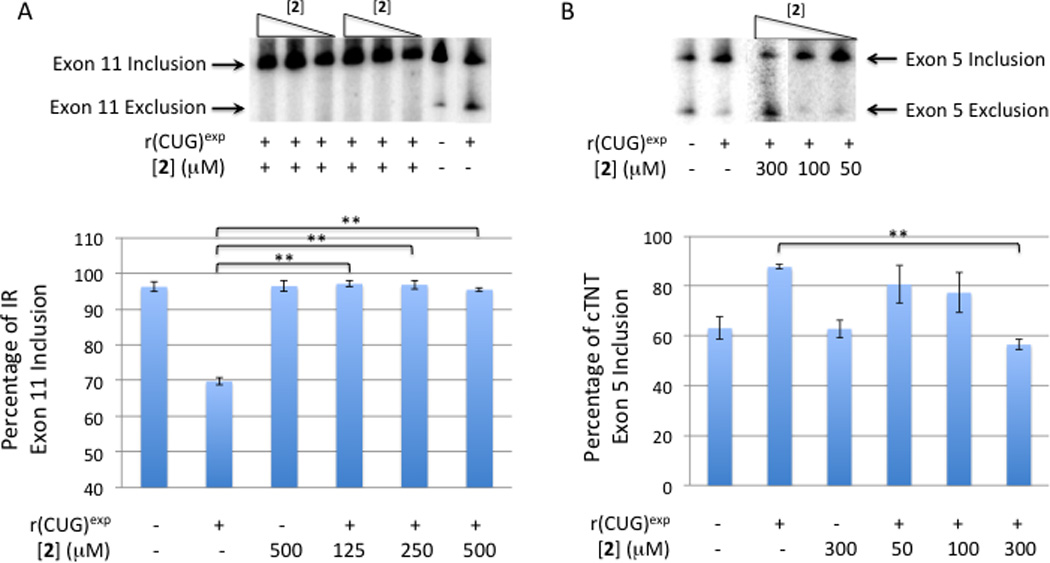
Compound 2 improves DM1-like splicing defects induced in HeLa cells. (A) Compound 2 improves dysregulation of IR alternative splicing in a DM1 model cellular system. (A, top) Representative gel image of IR alternative splicing in the presence of varying concentrations of 2. (A, bottom) Quantification of exon inclusion for IR alternative splicing in the presence or absence of r(CUG)exp and 2 (n ≥ 3). Error bars are the standard deviations in the measurements. (B) Compound 2 improves dysregulation of cTNT alternative splicing in a DM1 model cellular system. (B, top) Representative gel image of cTNT alternative splicing in the presence of varying concentrations of 2. (B, bottom) Quantification of exon inclusion for cTNT alternative splicing in the presence or absence of r(CUG)exp and 2 (n ≥ 3). Error bars are the standard deviations in the measurements. The percentage of each isoform was determined by RT-PCR using a radioactively labeled forward PCR primer. “**” indicates p ≤ 0.01 as determined by a two-tailed student t-test.
Fluorescence in situ hybridization (FISH) was employed to determine if 2 decreases the number or intensity of nuclear foci. Although foci are still present, they are much more dispersed when cells are treated with 2, and it appears that there is less MBNL1 within the foci as determined by immunostaining (MB1a antibody; Supplementary Fig. S9) 33.
To test the specificity of splicing modulation by compound 2, we examined its effect on exons not regulated by MBNL1. HeLa cells were co-transfected with a DM1 mini-gene and splicing reporter constructs for PLEKHH2, SMN2, and Bcl-x, the alternative splicing of which have been shown previously not to be regulated by MBNL1 5, 27. Treatment with 500 µM 2 had no effect on the alternative splicing of PLEKHH2, SMN2, or Bcl-x mini-genes (Supplementary Fig. S2), regardless of whether cells expressed r(CUG)exp. The alternative splicing of endogenous mRNAs not regulated by MBNL1, CAMKK2 (calcium/calmodulin-dependent protein kinase kinase 2) and TTC8 (tetratricopeptide repeat domain 8) 27, were also not affected (Supplementary Fig. S-2). Thus, the effect of compound 2 appears to be specific to its ability to bind r(CUG)exp and displace MBNL1.
Next, we determined if 2 also improves defects associated with MBNL2 sequestration by r(CUG)exp as they contribute to toxicity in the central nervous systems/brains of DM1 patients 30. For this purpose, the HEK cell line was used as it expresses MBNL proteins at very low levels 34. HEK cells were co-transfected with the DM1 and cTNT mini-genes and increasing amounts of a plasmid encoding MBNL1 or MBNL2. As expected, increased expression levels of MBNLs suppress cTNT exon 5 inclusion 10, 24, confirming that cTNT alternative splicing is regulated by both proteins in our model system (Supplementary Fig. S10). Dysregulation of cTNT alternative splicing is improved when HEK cells that express r(CUG)exp and MBNL1 or MBNL2 are treated with 300 µM of 2, although the effect is somewhat less for splicing regulated by MBNL2 (Supplementary Fig. S11). (Dysregulation of splicing is not improved when cells are treated with 75 µM or 150 µM of 2.) Taken together, 2 inhibits r(CUG)exp-MBNL1 and r(CUG)exp-MBNL2 complexes.
Molecular modeling and docking studies
A set of docking studies support our experimental evidence that 1 binds to MBNL1 and 2 binds to r(CUG) repeats. Figure 7A shows the results of the molecular modeling of 1 and MBNL1. The model of the complex shows extensive interactions between MBNL1 and 1 that occurs in the MBNL1 RNA binding pocket. The ethenyl group and the phenyl and thiofuran rings of 1 are stabilized by extensive cation-π interactions with Arg186, Arg195 and Arg201 in MBNL1. The cation-π interaction between 1 and Arg195 is reminiscent of the interaction between MBNL1 and RNA 35. In addition, compound 1’s carboxyl group forms a bifurcated hydrogen bond with backbone amino groups of Arg186 and Glu187 as well as a hydrogen bond with side chain of Arg186. Noticeably, the carbonyl oxygen of 1 is within the coordination distance of a bound Zn ion. This suggests that the oxygen provides the fifth coordination site, completing the trigonal bipyramidal geometry of the Zn ion.
Fig. 7.
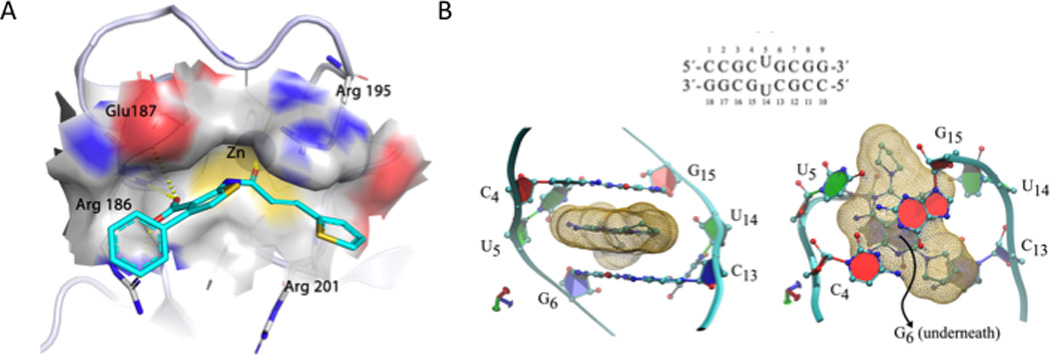
Docking studies support the binding of 1 to MBNL1 and 2 to r(CUG) repeats. (A) Extensive interactions are observed between MBNL1 and 1 at the RNA binding pocket. The ethenyl group and the phenyl and thiofuran rings of 1 are stabilized by extensive cation-π interactions with arginine residues. The cation-π interaction between 1 and Arg195 is reminiscent of the MBNL1-RNA interaction 35. The ligand binding pocket is shown as a transparent surface. Hydrogen bonds are shown as yellow dash lines. (B) Lowest free energy conformation of the 2-r(CUG) complex (binding mode p in Supplementary Table S4 and Supplementary Fig. S13). (B, top) RNA sequence used in MD simulations. Side (B, bottom left) and top (B, bottom right) views of the 2-r(CUG) complex. The yellow wireframe represents the molecular surface of compound 2. The RNA backbone is represented in light blue. For simplicity, hydrogen atoms are not shown. Note that in this binding mode, the interactions in the UU pair (green) are fully lost, and compound 2 stacks between the flanking GC base pairs (represented in red and blue).
We previously reported the structure of r(CCGCUGCGG)2, which was determined by NMR spectroscopy 36. These studies showed that the UU mismatch stacks within the helix, maintaining an overall A-form geometry. The UU mismatch is dynamic, however, and can interconvert between structures that contain 0, 1, or 2 hydrogen bonds 36. This dynamic nature could provide a means for small molecule binding. Docking studies of 2 and a r(CUG) repeat were completed using the structure of r(CCGCUGCGG)2. (Because of the dynamic nature of the RNA, a new method to simulate the binding of 2 to r(CUG) repeats was developed (Supplementary Methods). The lowest free energy conformation of the 2-r(CUG) complex is shown in Figure 7B. The complex is stabilized by stacking of 2 on the 5’ and 3’ closing GC pairs (shown in red and blue, respectively; Fig. 7B) and formation of three hydrogen bonds with the uridine bases. Both of 2’s amino groups hydrogen bond with one uridine base: one hydrogen bonds with the uridine’s N1 while the other hydrogen bonds with a non-bridging oxygen. Compound 2’s nitrile N hydrogen bonds with another U’s H2’.
Non-canonical 1×1 UU base pairs have weaker hydrogen bonds compared to Watson-Crick base pairs, which makes them more sensitive to the environment such as in the presence of 2. The pathway for the binding mode shown in Figure 7B is via the minor groove side as no major groove side binding was seen in the molecular dynamics simulation trajectories (Supplementary Figs. S13 & S14). Initial contact between 2 and the UU mismatch occurs at the minor groove side where 2’s amino groups interact with the free carboxyl group of the uridine bases. While compound 2 tries to stack within the helix, the weak hydrogen bonds of the UU mismatch break, resulting in one of the uridines unstacking from the helix. Later, the second uridine unstacks from helix and yields the final binding mode where 2 is fully stacked between the flanking GC base pairs (Fig. 7B). The simulation indicates that the complex is dynamic (Supplementary Figs. S13 & S14) and that stacking is the predominant interaction driving complex formation.
DISCUSSION
Only very few compounds have been previously reported to target the protein in an RNA-protein complex. A notable example includes the development of T5626448, a small molecule that inhibits the formation of the Toll-like receptor 3 (TLR3)-dsRNA complex 37. This interaction activates TLR3 and causes induction of pro-inflammatory cytokines. Interestingly, T5626448 shares several similar chemotypes with 1; both have carboxylate groups and thiophene-like side chains. Collectively, these data suggest that these chemotypes may be biased for targeting Zn-finger-containing proteins that form RNA-protein complexes.
Previous studies have shown that a variety of small molecules bind the expanded triplet repeat in the r(CUG)exp-MBNL1 complex and improve pre-mRNA splicing and other defects in cellular models of DM1. For example, both pentamidine and a bis-benzimidazole improve pre-mRNA splicing defects in DM1 cellular model systems and a transgenic mouse model 27, 38. These compounds have modest potencies with pentamidine requiring dosage of 25 µM and the bis-benzimidazole requiring dosage of 100 µM in cellular models. Modularly assembled small molecules that target the repeating 5’CUG/3’GUC motifs in r(CUG)exp require lower concentrations (as low as 5 µM) to improve splicing and translational defects in cellular models 18, 39. Thus, even though 2 is a modestly potent modulator of r(CUG)exp toxicity, its bioactivity could be enhanced via modular assembly. It is likely that sites in 2 can be identified to enable such an approach, perhaps via reduction of the cyano group to an amine handle.
Compound 2 is more selective than previously reported small molecules that bind to expanded repeats. It binds with ~11-fold selectivity for an RNA that displays 12 5'CUG/3'GUC motifs when compared to an RNA that displays 12 5'CAG/3'GAC and >200-fold selectivity over a fully paired RNA (Table 2). A bis-benzimidazole that also improves DM1-associated defects shows at best 3-fold selectivity for RNAs with a single 5'CUG/3'GUC 38. A 4',6-diamidino-2-phenylindole (DAPI) derivative binds to r(CAG)exp and improves associated pre-mRNA splicing defects 40. DAPI has modest specificity for the 5'CAG/3'GAC motif displayed by r(CAG)exp, ranging from approximately 2–5 fold selectivity over RNAs that display other 1×1 nucleotide mismatches and >10-fold selectivity over fully paired RNAs 40.
Compounds 1 and 2 have utility as probes and tools. The ability to reverse or induce a disease-like state chemically can enable studies to determine the repertoire of pre-mRNAs whose splicing function is dysregulated in DM1 or other diseases by using RNA-seq. Studies could also be completed in animal models of DM1, such as in Drosophila, to determine if 1 can induce DM1-like pre-mRNA splicing defects. The compound can then be withdrawn to measure recovery back to wild type. Lastly, these compounds could have a use in the development of artificial gene circuits. Traditionally, such circuits have been controlled through aptamer-small molecule interactions in which the aptamer has been selected or excised from a riboswitch 41–44.
METHODS
Compounds
Compounds 1 and 4 were procured from Enamine (catalog numbers T0306–5218 and T0504–3845, respectively). Compound 2 was purchased from eMolecules (Specs) (catalog number AN-584/43420506), and compound 3 was purchased from Vitas-M Laboratory, Ltd (catalog number STK676326). All compounds were >95% pure.
Competition Dialysis
Competition dialysis was completed as described previously 45. Briefly, 5’-biotinylated r(CUG)12, MBNL1 protein and BSA were diluted separately to a final concentration of 2.5 µM in 1X Phosphate Buffer EDTA Saline (1X PBES, 8 mM Na2HPO4 (pH = 7.0), 185 mM NaCl, 1 mM EDTA). The RNA was folded by heating at 60 °C for 5 min and slow cooling to room temperature. Compound 1 was then added at a final concentration of 2.5 µM to the folded RNA, MBNL1 or BSA solution. A 150 µL aliquot of each solution was added to a dialysis unit (Pierce Slide-A-Lyzer MINI dialyzer unit (7000 kDa MWCO; Pierce Biotechnology, Inc.). Dialysis units were placed in 200 mL of dialysate, which consisted of 2.5 µM of 1 in 1X PBES. The samples were allowed to equilibrate with dialysate by stirring at 70 rpm for 48 h at room temperature (20 – 22°C). At the end of the equilibration period, sodium dodecyl sulfate (SDS) was added to the sample solution to the final concentration of 1% (v/v). Addition of SDS was necessary to ensure complete dissociation of 1 from the RNA or protein, as binding can affect its spectroscopic properties. The total concentration of 1 (Ct) within the dialysis unit was determined spectrophotometrically using an appropriate absorbance wavelength and extinction coefficient. A dilution factor was applied to account for the addition of SDS.
The free concentration of 1 (Cf) was determined from an aliquot of the dialysate solution, which did not vary appreciable from the initial 2.5 µM. The bound ligand concentration (Cb) was then determined using (1):
| (1) |
where Cb, Ct and Cf are concentration of 1 in bound, total and free states, respectively. The dissociation constant was determined using (2) 45:
| (2) |
where Kapp is the apparent association constant (equivalent to 1/Kd) and [MBNL1] is the concentration of MBNL1.
Fluorescence Binding Assays
Dissociation constants of 2 to various 1×1 internal loop RNAs were measured by an in-solution, fluorescence-based assay. RNA was folded in 1X PBES at 60 °C for 5 min and allowed to slowly cool to room temperature. 2 was then added to a final concentration of 1250 nM. This solution was then serially diluted (2X) in to 1X PBES solution containing 1250 nM of 2. The solutions were incubated at room temperature for 10 min and then transferred to a 96-well plate. Anisotropy signal was recorded using a Spectra Max M5 plate reader (excitation 300 nm; emission: 470 nm; cutoff: 420 nm). The change in anisotropy signal as a function of RNA concentration was fitted to a standard ligand binding for one site saturation for r(CAG)12, 1×1 UU and AU RNAs (3):
| (3) |
where ∆r is the change in anisotropy signal, Bmax represents the maximum number on binding sites, [RNA] is the RNA concentration and Kd is the dissociation constant. The dissociation constant for binding to r(CUG)12 was determined via a Hill plot.
RNA-MBNL1 Displacement Assays
The RNA-MBNL1 binding studies were completed as previously described 38, 40. Briefly, 5’-biotinylated r(CUG)12 (folded by heating at 60 °C for 5 min then slowly cooling to room temperature) or MBNL1-His6 was incubated with compound for 30 min at room temperature. Next, r(CUG)12 was added to MBNL1-containing samples or vice versa, and the samples were incubated for 15 min at room temperature. Streptavidin-XL665 (binds to the biotinylated RNA oligonucleotide) and Tb-Anti-His6 (binds to MBNL1) were then added. If the compound does not disrupt the r(CUG)12-MBNL1 interaction, then XL665 and Tb can form a FRET pair and TR-FRET can be measured. Conversely, TR-FRET is not observed if the compound inhibits formation of the r(CUG)12-MBNL1 complex. IC50’s were determined by fitting the corresponding curve to SigmaPlot’s four parameter logistic curve fit.
Assessment of DM1-associated Translational Defects
The model systems used to detect the DM1-associated translational have been previously described 18. Briefly, a C2C12 cell line that stably expresses the firefly luciferase gene containing a (CTG)800 expansion in the 3’ UTR was employed. r(CUG)800 causes nuclear retention of the luciferase mRNA and thus decreased expression of luciferase. Compounds that disrupt the r(CUG)800-MBNL1 interaction therefore have the potential to allow for nucleocytoplasmic export, which is correlated to the luciferase activity in cell extracts Luciferase activity was measured using a Luciferase Assay Kit (Promega) as previously described 18.
Assessment of pre-mRNA Splicing Defects in cellular models
DM1-associated pre-mRNA splicing defects using a HeLa model system were completed as previously described 18, 27, 38 using a DM1 mini-gene that express 960 interrupted r(CUG) repeats 10. Briefly, HeLa cells were co-transfected with the mini-gene of interest and a plasmid that expresses r(CUG)exp or an empty vector. Varying concentrations of compound were added in growth medium post-transfection, and total RNA was harvested 16–20 h later. The percentage of each splicing isoform was determined by RT-PCR. Statistical significance was determined using a two-tailed t-test.
HEK 293T cells were grown as monolayers in 96-well plates to approximately 95% confluency in growth medium (1X DMEM, 10% FBS, and 1X GlutaMax (Invitrogen)). In order to show that the cTNT mini-gene was responsive to MBNL1 and MBNL2 in the HEK model system, cells were co-transfected with plasmids expressing the DM1 mini-gene (100 ng), the cTNT mini-gene (100 ng), and MBNL1 or MBNL2 (varying amounts; 0–60 ng) using Lipofectamine 2000 reagent per the manufacture’s protocol. The effects of compound 2 on MBNL1- and MBNL2-dependent alternative splicing were then determined using 100 ng DM1 mini-gene plasmid, 100 ng cTNT mini-gene plasmid, and 5 ng of MBNL plasmid. Approximately 5 h post transfection, the transfection cocktail was replaced with grown medium containing various concentrations of 2 (0, 75, 150 and 300 µM). After 16–24 h, the cells were lysed and the total RNA was collected using a GelElute Mammalian Total RNA Miniprep Kit (Sigma-Aldrich). RT-PCR analysis was completed as previously described 18, 27, 38.
DM1 patient-derived cell lines and compound 1 treatment
The following cell lines were obtained from the Coriell Cell repositories: GM07492 - fibroblasts from unaffected individual (normal fibroblasts); GM03987, GM04033 and GM03989 - fibroblasts from myotonic dystrophy type 1 probands expressing mutant DMPK transcript containing ~500, ~1000 and ~2000 CUG repeat tracts, respectively (referred to as 500CUG, 1000CUG and 2000CUG, respectively). Cells were grown in a high-glucose EMEM (Lonza) supplemented with 10% fetal bovine serum, 1x antibiotic-antimycotic (Gibco) and 1X non-essential amino acids solution (Sigma), in a humidified incubator containing 5% CO2, at 37°C. Appropriate amounts of compound 1 were mixed with medium and added directly onto 70–80% confluent cells. All splicing changes were analyzed 48 h post treatment.
siRNA knock-down of MBNL1
siRNA oligos targeting human MBNL1 were obtained from Future Synthesis and the sequences were as follows: MBNL1_AS 5’[P]-UCUCUACAUACUUCCAGUGdTdT, MBNL1_S 5’[P]-CACUGGAAGUAUGUAGAGAdTdT 32. At 80% confluence, fibroblasts were transfected with 25 nM annealed siRNA directed against MBNL1 transcript (referred to as siMBNL1) using Lipofectamine2000 (Invitrogen). All splicing changes were analyzed 48 h post siMBNL1 treatment. For combined siMBNL1 and compound 1 treatment, normal human fibroblasts were successively treated with 25 nM siMBNL1 and 125, 250 or 500 µM compound 1. Cells were first transfected with 25 nM siMBNL1 at 70–80% confluence. Then, appropriate amounts of compound 1 were added 24 h post siMBNL1 treatment. The following controls were used for combined siMBNL1 and 1 treatment: lipofectamine treated cells (mock), siMBNL1 only and compound 1 only treated cells. All splicing changes were analyzed 48 h post compound 1 treatment (72h post siMBNL1 treatment).
RNA isolation and RT-PCR
RNA was isolated 48 h post 1 treatment or siRNA treatment using TRI reagent (Sigma-Aldrich) per the manufacturer’s recommended protocol. cDNA was synthesized using SuperscriptTM III Reverse Transcriptase kit (Invitrogen) according to the manufacturer’s protocol, and diluted with 50 µL water prior to PCR. Standard RT-PCR reactions were performed using 2 µL diluted cDNA and GoTaq Flexi DNA Polymerase kit (Promega). PCR primer sequences are listed in Supplementary Table S1. PCR products were separated on a 2% agarose gel stained with 0.5 µg/mL ethidium bromide, photographed using G-Box (SynGene) and quantified with the use of GeneTools Software (SynGene). Quantified splicing changes were plotted on a bar graph as a percentage of respective exon spliced in (% of alternative exon inclusion), which in the case of MBNL1-dependent exons represents splicing isoform characteristic of DM1.
Experimental setup and statistical analyses
The effect of 1 on normal and DM1 (CUG500) human fibroblasts was analyzed in duplicate biological repeats (n=2 for each given concentration of 1). As controls, we used n=5 each for untreated and DMSO-treated normal human fibroblasts, and n=2 for the untreated CUG500 fibroblasts. For additional DM1 controls, we used n=4 each of the untreated CUG1000 and CUG2000 human fibroblasts. The effects of siMBNL1 knock-down as well as combined siMBNL1 and compound 1 treatment were assessed in duplicate biological repeats. All RT-PCR reactions were performed and analyzed at least twice to verify results. The fractions of alternatively spliced isoforms (exon inclusions representing the DM1-like splicing isoform) were calculated by dividing the intensity of a PCR product band corresponding to the splicing variant representing exon inclusion (upper band) by the total intensity of both splicing isoforms (upper and lower band). Error bars represent standard deviation (±SD) obtained from a single experiment with at least two independent biological repeats. Statistical significance was determined by a two-tailed Student t-test using Microsoft Excel (* for P≤0.05, ** for P≤0.01 and *** for P≤0.001).
Docking of 1 and MBNL1
Compound 1 was prepared for docking simulations using LigPrep v2.5 (Schrodinger, LLC) 46, affording a single ligand pose. PDB entry 3D2S (MBNL1) was prepared for molecular modeling by using Maestro’s protein preparation wizard (v9.3; Schrodinger, LLC). Bound RNA and water molecules were removed, the bond order was assigned, and hydrogen atoms were added. The rotamer states of Asn and Gln residues, and the charge states of His residues were also optimized. The docking grid was generated around the Zn coordinate with a box size of 15 × 15 × 15 Å3. No constraints were used for grid generation. Compound 1 was then docked into MBNL1 35 using Glide SP v5.6 (Schrodinger, LLC). Energy minimization was completed using Prime v3.1 under the Maestro interface (Schrodinger, LLC). Figure 7A was generated using PyMol (Schrodinger, LLC).
Docking of 2 and r(CUG) repeats
Please see the Supplementary Methods for details related to molecular modeling of 2 and r(CUG) repeats.
Supplementary Material
ACKNOWLEDGMENT
We thank Don Mackenzie from the Marigold Foundation for his support of this research project in particular and DM1 research in general and Amit Kumar for preliminary studies. We also thank Prof. Glenn E. Morris (Wolfson Centre for Inherited Neuromuscular Disease) for generously providing the MB1a antibody (anti-MBNL1). Funding for this work was provided by the Scripps Research Institute, the Muscular Dystrophy Association (158552 to MDD), the National Institutes of Health (3R01GM079235-02S1 and 1R01GM079235-01A2 to MDD; AR049077 and U54NS48843 to CAT), the Molecular Libraries Initiative of the NIH Roadmap for Medical Research, the Marigold Foundation postdoctoral fellowship. IY and GCS were supported by the PS-OC Center of the NIH/NCI (grant 1U54CA143869-01). ES-K and KS were supported by the Foundation for Polish Science - TEAM programme co-financed by the European Union within the European Regional Development Fund.
Footnotes
Author Contributions. JLC-D and MDD wrote the manuscript, which was also edited by co-authors. JLC-D, ES-K, TT, IY, HP, CZC, JH, NS, JJM, SP, and WZ completed experiments. All authors contributed to project planning and data analysis.
Conflicts of Interest. The authors declare no conflicts of interest including no competing financial interests.
REFERENCES
- 1.Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 2.Savkur RS. Insulin receptor splicing alteration in myotonic dystrophy type 2. Am. J. Hum. Genet. 2004;74:1309–1313. doi: 10.1086/421528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 4.Fugier C, et al. Misregulated alternative splicing of BIN1 is associated with Ttubule alterations and muscle weakness in myotonic dystrophy. Nat. Med. 2011;17:720–725. doi: 10.1038/nm.2374. [DOI] [PubMed] [Google Scholar]
- 5.Sellier C, et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010;29:1248–1261. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liquori CL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9 . Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 7.Du H, et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 2010;17:187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orengo JP, Ward AJ, Cooper TA. Alternative splicing dysregulation secondary to skeletal muscle regeneration. Ann. Neurol. 2011;69:681–690. doi: 10.1002/ana.22278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 10.Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 11.Charlet BN, et al. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 12.Mankodi A, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 13.Mastroyiannopoulos NP, Feldman ML, Uney JB, Mahadevan MS, Phylactou LA. Woodchuck post-transcriptional element induces nuclear export of myotonic dystrophy 3' untranslated region transcripts. EMBO Rep. 2005;6:458–463. doi: 10.1038/sj.embor.7400390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amack JD, Paguio AP, Mahadevan MS. Cis and trans effects of the myotonic dystrophy (DM) mutation in a cell culture model. Hum. Mol. Genet. 1999;8:1975–1984. doi: 10.1093/hmg/8.11.1975. [DOI] [PubMed] [Google Scholar]
- 15.Cardani R, Mancinelli E, Rotondo G, Sansone V, Meola G. Muscleblind-like protein 1 nuclear sequestration is a molecular pathology marker of DM1 and DM2. Eur. J. Histochem. 2006;50:177–182. [PubMed] [Google Scholar]
- 16.Chen CZ, et al. Two high-throughput screening assays for aberrant RNA-protein interactions in myotonic dystrophy type 1. Anal. Bioanal. Chem. 2012;402:1889–1898. doi: 10.1007/s00216-011-5604-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=2675&loc=ea_ras.
- 18.Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem. Biol. 2012;7:856–862. doi: 10.1021/cb200408a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaires JB, Ragazzon PA, Garbett NC. A competition dialysis assay for the study of structure-selective ligand binding to nucleic acids. Curr. Protoc. Nucleic Acid Chem. 2003 doi: 10.1002/0471142700.nc0803s11. Chapter 8, Unit 8.3. [DOI] [PubMed] [Google Scholar]
- 20.Kumar A, et al. Myotonic dystrophy type 1 RNA crystal structures reveal heterogeneous 1 × 1 nucleotide UU internal loop conformations. Biochemistry. 2011;50:9928–9935. doi: 10.1021/bi2013068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Warf MB, Berglund JA. MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA. 2007;13:2238–2251. doi: 10.1261/rna.610607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee MM, Pushechnikov A, Disney MD. Rational and modular design of potent ligands targeting the RNA that causes myotonic dystrophy 2. ACS Chem. Biol. 2009;4:345–355. doi: 10.1021/cb900025w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mykowska A, Sobczak K, Wojciechowska M, Kozlowski P, Krzyzosiak WJ. CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res. 2011;39:8938–8951. doi: 10.1093/nar/gkr608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho TH, et al. Muscleblind proteins regulate alternative splicing. EMBO J. 2004;23:3103–3112. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dansithong W, Paul S, Comai L, Reddy S. MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1. J. Biol. Chem. 2005;280:5773–5780. doi: 10.1074/jbc.M410781200. [DOI] [PubMed] [Google Scholar]
- 26.Orengo JP, Bundman D, Cooper TA. A bichromatic fluorescent reporter for cell-based screens of alternative splicing. Nucleic Acids Res. 2006;34:e148. doi: 10.1093/nar/gkl967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warf MB, Nakamori M, Matthys CM, Thornton CA, Berglund JA. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. U. S. A. 2009;106:18551–18556. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x . J. Cell. Biol. 2007;176:929–939. doi: 10.1083/jcb.200701005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pedrotti S, et al. The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J. 2010;29:1235–1247. doi: 10.1038/emboj.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Charizanis K, et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron. 2012;75:437–450. doi: 10.1016/j.neuron.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang ET, et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–724. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jog SP, et al. RNA splicing is responsive to MBNL1 dose. PLoS One. 2012;7:e48825. doi: 10.1371/journal.pone.0048825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holt I, et al. Defective mRNA in myotonic dystrophy accumulates at the periphery of nuclear splicing speckles. Genes Cells. 2007;12:1035–1048. doi: 10.1111/j.1365-2443.2007.01112.x. [DOI] [PubMed] [Google Scholar]
- 34.Xiao X, et al. Splice site strength-dependent activity and genetic buffering by poly-G runs. Nat. Struct. Mol. Biol. 2009;16:1094–1100. doi: 10.1038/nsmb.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teplova M, Patel DJ. Structural insights into RNA recognition by the alternative-splicing regulator muscleblind-like MBNL1. Nat. Struct. Mol Biol. 2008;15:1343–1351. doi: 10.1038/nsmb.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parkesh R, Fountain M, Disney MD. NMR spectroscopy and molecular dynamics simulation of r(CCGCUGCGG)2 reveal a dynamic UU internal loop found in myotonic dystrophy type 1. Biochemistry. 2011;50:599–601. doi: 10.1021/bi101896j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng K, Wang X, Yin H. Small-molecule inhibitors of the TLR3/dsRNA complex. J. Am. Chem. Soc. 2011;133:3764–3767. doi: 10.1021/ja111312h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parkesh R, et al. Design of a bioactive small molecule that targets the myotonic dystrophy type 1 RNA via an RNA motif-ligand database & chemical similarity searching. J. Am. Chem. Soc. 2012;134:4731–4742. doi: 10.1021/ja210088v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Childs-Disney JL, Parkesh R, Nakamori M, Thornton CA, Disney MD. Rational design of bioactive, modularly assembled aminoglycosides targeting the RNA that causes myotonic dystrophy type 1. ACS Chem. Biol. 2012;7:1984–1993. doi: 10.1021/cb3001606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar A, et al. Chemical correction of pre-mRNA splicing defects associated with sequestration of muscleblind-like 1 protein by expanded r(CAG)-containing transcripts. ACS Chem. Biol. 2012;7:496–505. doi: 10.1021/cb200413a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sinha J, Reyes SJ, Gallivan JP. Reprogramming bacteria to seek and destroy an herbicide. Nat. Chem. Biol. 2010;6:464–470. doi: 10.1038/nchembio.369. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Topp S, Gallivan JP. Emerging applications of riboswitches in chemical biology. ACS Chem. Biol. 2010;5:139–148. doi: 10.1021/cb900278x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Werstuck G, Green MR. Controlling gene expression in living cells through small molecule-RNA interactions. Science. 1998;282:296–298. doi: 10.1126/science.282.5387.296. [DOI] [PubMed] [Google Scholar]
- 44.Culler SJ, Hoff KG, Smolke CD. Reprogramming cellular behavior with RNA controllers responsive to endogenous proteins. Science. 2010;330:1251–1255. doi: 10.1126/science.1192128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaires JB. A Competition Dialysis Assay for the Study of Structure-Selective Ligand Binding to Nucleic Acids. John Wiley & Sons, Inc. 2001 doi: 10.1002/0471142700.nc0803s11. [DOI] [PubMed] [Google Scholar]
- 46.Podvinec M, et al. Novel inhibitors of dengue virus methyltransferase: discovery by in vitro-driven virtual screening on a desktop computer grid. J. Med. Chem. 2010;53:1483–1495. doi: 10.1021/jm900776m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.