

SUMMARY
Host response to viral RNA genomes and replication products represents an effective strategy to combat viral invasion. PKR is a Ser/Thr protein kinase that binds to dsRNA, autophosphorylates its kinase domain, and subsequently phosphorylates eukaryotic initiation factor 2α (eIF2α). This results in attenuation of protein translation, preventing synthesis of necessary viral proteins. In certain DNA viruses, PKR function can be evaded by transcription of highly structured virus-encoded dsRNA inhibitors that bind to and inactivate PKR. We probe here the mechanism of PKR inhibition by two viral inhibitor RNAs, EBERI (from Epstein-Barr) and VAI (from human adenovirus). Native gel shift mobility assays and isothermal titration calorimetry experiments confirmed that the RNA-binding domains of PKR are sufficient and necessary for the interaction with dsRNA inhibitors. Both EBERI and VAI are effective inhibitors of PKR activation by preventing trans-autophosphorylation between two PKR molecules. The RNA inhibitors prevent self-association of PKR molecules, providing a mechanistic basis for kinase inhibition. A variety of approaches indicated that dsRNA inhibitors remain associated with PKR under activating conditions, as opposed to activator dsRNA molecules that dissociate due to reduced affinity for the phosphorylated form of PKR. Finally, we show using a HeLa cell extract system that inhibitors of PKR result in translational recovery by the protein synthesis machinery. These data indicate that inhibitory dsRNAs bind preferentially to the latent, dephosphorylated form of PKR and prevent dimerization that is required for trans-autophosphorylation.
Keywords: RNA, PKR, kinase, inhibition, virus
INTRODUCTION
A hallmark of many viral infections is the presence of viral genomes and replication by products containing highly structured double-stranded RNA molecules (dsRNA). Recognition of these dsRNA molecules by cellular pattern-recognition receptors (PRRs) results in the induced interferon response, thereby modulating gene expression of enzymes that mediate innate immunity 1; 2; 3. The interferon-inducible enzyme PKR (Protein Kinase RNA-dependent) specifically serves as an intracellular sensor for viral genomic contamination to down-regulate translation initiation 4. Activated by the binding of dsRNA, PKR regulates protein synthesis by the phosphorylation of eukaryotic initiation factor 2α (eIF2α) at Ser51, thereby forming a sequestered inhibitory complex with its guanine nucleotide exchange factor eIF2B 5. As a result, the pool of active eIF2 ternary complex is reduced, causing a general decrease in both cellular and viral protein synthesis 6. Therefore, the cellular interactions between dsRNA and members of the interferon response pathway represent a fundamental anti-viral response.
Human PKR is a 551-residue enzyme comprising an N-terminal RNA binding and C-terminal kinase domain bridged by an 80-residue interdomain linker 7; 8. The N-terminal domain contains two highly basic, conserved 70-residue motifs capable of dsRNA recognition (dsRBDs); high-affinity binding requires each of the dsRBDs 9; 10; 11; 12. The C-terminal domain of PKR is a Ser-Thr Kinase domain responsible for both substrate recognition and phosphorylation activity of the enzyme 13; 14; 15. Defining characteristics of the kinase domain include the ATP coordination site required to mediate phosphoryl transfer, activation loop phosphorylation sites important for activity (T446 and T451), and substrate recognition sites 12; 13; 15. In solution, the interdomain linker is highly unstructured, resulting in a highly dynamic protein in which the domains behave as independent units 12; 16. Upon dsRNA binding, latent PKR undergoes autophosphorylation, which greatly increases the kinase activity of the enzyme 17; 18. PKR autophophorylation proceeds through a bimolecular reaction mechanism, in which the self-association of PKR is modulated by both dsRNA interactions and the phosphorylation state of the enzyme itself 12; 13; 19; 20; 21. Although the structural basis for self-association remains unclear, either conformational or electrostatic changes upon dsRNA binding are thought to unmask domains within both the interdomain linker and kinase domain to facilitate self-association 12; 13; 15. RNA-PKR complex stability is significantly reduced upon autophosphorylation of PKR 12; 22.
PKR is specifically activated by dsRNA molecules compared to other nucleic acid substrates. RNA-DNA hybrids, dsDNA, and single-stranded polynucleotides are not recognized by the enzyme 4. Single dRBDs recognize dsRNA through structure-specific, sequence independent contacts to the 2'-hydroxyl groups of the ribose sugar 23; 24; 25, and the two dsRBDs and linker likely make similar contacts. All known viral ligands of PKR are distorted dsRNA helices, possessing structural features beyond simple duplex, such as bulges and hairpin loops 26; 27; 28; 29; 30; 31. These structural features tune RNA ligand recognition by the dsRBDs, as significantly reduced affinity is observed when distortions from the dsRNA molecules are removed 10; 32. A minimum dsRNA duplex length (15−50 bp) is required for high-affinity interaction between the dsRBDs and dsRNAs 26; 32; 33; 34.
Many viruses counteract the PKR response to viral infection by encoding protein and dsRNA molecules that inhibit PKR function 3; 35; 36; 37; 38. Human adenovirus and Epstein-Barr virus encode RNA polymerase III-directed, highly structured RNAs (VA and EBER respectively) that are required for efficient translation of viral mRNAs. These inhibitory dsRNAs bind directly to PKR with similar affinity to dsRNA activators 10, but prevent subsequent activation and substrate phosphorylation, allowing protein synthesis to proceed 10; 37; 39. The EBER and VA RNAs form roughly similar 160-nt stem-loop secondary structures containing an apical stem-loop (AS), central stem-loop (CS), and distal stem (DS) (Fig.1). Thermodynamically, a single stem-loop domain from each inhibitory dsRNA mediates the interaction with PKR; the apical stem from VAI (VAI-AS), and the central stem from EBERI (EBERI-CS) 10, although low affinity interactions with other stem-loop domains have been reported 40. Both dsRBDs of PKR are required for high-affinity interaction with the inhibitor, and identical surfaces of the protein mediate the interaction with dsRNA whether activator or inhibitor is bound 10. Isolated dsRBD binding stem loops from inhibitors serve to activate PKR 10, indicating that dsRNA binding and inhibition are non-equivalent 41. Therefore, regions other than the dsRBD binding stem-loops must be responsible for mediating the inhibitory effect.
Figure 1.

Sequences and secondary structures of viral dsRNAs. Secondary structures of (A) adenovirus VAI inhibitor 51, (B) Epstein-Barr virus EBERI inhibitor 52, (C) the apical stem truncation of VAI (VAI-AS), and (D) HIV TAR dsRNA 53.
The mechanism of viral RNA inhibition of PKR remains poorly understood. Here we characterized viral inhibitor-PKR complexes by employing a variety of techniques, including native gel shift mobility assays, isothermal titration calorimetry (ITC), autophosphorylation assays, dynamic light scattering, and in vitro translation assays. These data support a model for the inactivation of PKR autophophorylation by dsRNA inhibitors in which inhibitory dsRNAs bind preferentially to the latent, dephosphorylated form of PKR and prevent dimerization that is required for efficient trans-autophosphorylation.
RESULTS
The dsRBDs of dephosphorylated PKR are sufficient and required for interaction with dsRNA inhibitors.
To extend our understanding of inhibition of PKR by viral dsRNA, two inhibitors of PKR were synthesized in vitro; adenovirus derived VAI (Fig. 1A), and EBERI from Epstein-Barr virus (Fig. 1B). Both inhibitors contain stem-loop motifs that have been implicated in mediating the interaction with PKR 10; 40; 42. To serve as positive controls for PKR activity, two ligands of PKR were also produced that lead to activation; the apical stem-loop of VAI (VAI-AS, Fig. 1C) and HIV trans-acting response element (HIV-TAR) (Fig. 1D) 10; 32. RNA-PKR complexes were characterized biophysically to determine the mechanistic basis for inhibition.
A series of PKR domains and truncations were used to delineate regions of PKR that interact with inhibitory dsRNAs using native gel shift mobility assays (Fig. 2A). Addition of full-length PKR or truncated protein containing only the dsRBDs of PKR (dsRBD1/2) to VAI results in formation of a higher-molecular weight species, corresponding to a 1:1 PKR:RNA complex in both cases (Fig. 2B). A functional ATP binding site in PKR is not a requirement for interaction, as mutation of the nucleotide coordination site (PKRK296R) does not affect complex formation. Constructs lacking the dsRBDs (PKR170−551, PKR252−551 and PKR170−252) do not discernibly interact with VAI, indicating that the interdomain linker and/or kinase domains do not mediate the high-affinity association with PKR. Addition of the domains of PKR in trans (dsRBD1/2 and PKR170−551) results only in formation of a complex with equivalent migration to a VAI-dsRBD1/2 complex; again only the dsRBDs mediate the interaction. In contrast, phosphorylated PKR (PKRP) does not form an observable RNA-protein complex with VAI. Identical results were obtained when EBERI was used instead of VAI (data not shown). In summary, these results suggest that the dsRBDs of PKR are required and sufficient for interaction with inhibitory RNAs, and that phosphorylation of PKR blocks the interaction with the inhibitors.
Figure 2.

dsRBDs of PKR are sufficient and required for interaction with inhibitory dsRNAs. (A) Domain organization of PKR. N-terminal dsRBDs, C-terminal kinase domain, and the interdomain linker are shown. Critical autophosphorylation sites (T446, T451) in the kinase domain are indicated. (B) Native gel mobility shift assay for PKR derivatives (600 nM) binding to VAI (200 nM). (C) Summary of dissociation constants (μM) at 30 °C for titration of dsRNA (10 μM, sample cell) with PKR derivatives added in trans (150 μM, syringe). Thermodynamic parameters are included in the supplemental materials.
Gel shift mobility assays were confirmed and extended by isothermal titration calorimetry (ITC), which determines the affinity and thermodynamics of complex formation. A single, high-affinity binding-site within dsRNA inhibitors (VAI or EBERI) or activators (VAI-AS) (Fig. 2C) is observed for both dsRBD1/2 and full-length PKR; the affinities of inhibitor and activator RNA-protein interactions are similar. Mutations at the ATP coordination site (PKRK296R) or activation loop phosphorylation sites (PKRT446A/T451A) do not affect RNA inhibitor-PKR affinity. As expected from the gel shift assay results, phosphorylated PKR has a significantly reduced affinity for dsRNA inhibitors or activators (>15-fold decrease). Deletion mutants of PKR lacking the dsRBDs have similarly reduced affinities relative to either the full-length protein or dsRBDs alone. Thus, dsRBDs mediate interaction of inhibitors with PKR.
Inhibitors prevent trans-autophosphorylation of latent PKR
Characteristic of an autocatalytic process, a sigmoidal buildup of product with a lag phase prior to maximal rates of autophosphorylation has been observed for the bimolecular kinetics of PKR autophosphorylation 10; 12; 32. Inhibitors could be effective against the latent form of PKR, the phosphorylated form, or both. Given that inhibitors do not interact significantly with phosphorylated PKR (Fig. 2), we expected that only the latent form of the enzyme would be inhibited. To test our hypothesis, a kinase activation assay was established based on the autophosphorylation of PKR in the presence of a dsRNA activator, HIV-TAR. A buffered reaction containing 32P-γATP, Mg2+, HIV-TAR, and full-length PKR was incubated for a total of 2 hours, with either EDTA or dsRNA ligands added at various points in the time course. After 2 hours, reaction components were separated by SDS-PAGE under denaturing conditions, and the resulting incorporation of radiolabeled phosphate into PKR was quantified, thereby providing a direct measurement of inhibition efficiency. EDTA chelates all available Mg2+ in the reaction mixture and therefore quenches the reaction; EDTA acts as the idealized inhibitor of PKR as the amount of phosphorylation detected is a direct result of the bimolecular activation kinetics of PKR (Fig. 3A, dashed line).
Figure 3.

Inhibitors prevent latent PKR from trans-phosphorylation. (A) Inhibition of PKR autophosphorylation in the presence of HIV-TAR activator. Either EDTA, VAI, or EBERI was added to a reaction mixture containing PKR, TAR, ATP and MgCl2 at various time points as indicated. The reaction was then allowed to proceed for the complete 2 hours. Each time point was performed in triplicate, resolved by SDS-PAGE, and quantitated for 32P incorporation by autoradiography. (B) Trans-autophosphorylation assays in which PKRP was used to phosphorylate PKR in the presence of increasing amounts of inhibitor (VAI or EBERI). Reaction mixtures were incubated in the presence of [γ-32P]ATP at 30°C for 15 minutes, and quenched by addition of EDTA. SDS-PAGE separation followed by autoradiography is shown.
Addition of PKR inhibitors (VAI or EBERI) is as effective as EDTA at inhibiting phosphorylation of PKR when added at earlier time points (< 15 minutes), but less effective as the pool of PKR becomes increasingly phosphorylated. This indicates that VAI and EBERI preferentially inhibit the latent form of PKR. As expected, addition of another dsRNA activator to the reaction mixture, VAI-AS, has no inhibitory effect (Fig. 3A). RNA inhibitors specifically block autophosphorylation by the phosphorylated form of PKR.
PKR activation proceeds through a bimolecular reaction mechanism in which one PKR phosphorylates another in trans 12; inhibitor-bound PKR may not be an efficient substrate for trans-autophosphorylation. Phosphorylated PKR (PKRP) is capable of trans-phosphorylating PKR in buffered reactions containing 32P-γATP and Mg2+ 12. These experiments remove the need for RNA activators completely, and therefore one can dissect the ability of PKR to serve as a substrate for trans-phosphorylation. After 15-minute incubation, minimal phosphorylation of PKRP itself is observed (Fig. 3B) indicating that 32P incorporation in other lanes is attributable to substrate phosphorylation only. PKRP is capable of trans-phosphorylating both latent PKR and activator-bound PKR; RNA binding to the dsRBDs does not itself preclude phosphorylation. Significantly, trans-autophosphorylation is strongly attenuated upon inclusion of increasing amounts of VAI or EBERI to the reaction mixture, indicating that the inhibitors protect PKR from phosphorylation. A 3 to 10-fold excess of inhibitory dsRNA relative to PKR is required to protect PKR fully from trans-autophosphorylation, consistent with the known dissociation constants established for these complexes 10. Thus, RNA inhibitors block bimolecular phosphorylation by PKR.
Inhibitors prevent self-association of PKR
Multiple lines of evidence support the importance of PKR self-association in kinase activation 12; 13; 15; 19; 43; 44. As RNA inhibitors block phosphorylation of latent PKR, they may directly block self-association of PKR monomers. To probe the formation of bimolecular complexes under non-activating conditions (i.e. no prior incubation at 30 °C), we employed dynamic light scattering (DLS) to determine the apparent molecular weight (Mr) of complexes containing either wild-type or catalytically inactive (PKRK296R) PKR at 5 μM concentration. Mr determinations for both VAI (∼55 kDa) and PKR (∼83 kDa) alone were close to expected values, indicating that each molecule behaves as a monomeric species at low μM concentrations (Fig. 4A). Addition of excess ATP and Mg2+ did not impact the hyrdrodynamic radius of PKR; no global distortion to the protein is observed. Interaction between VAI and PKR results in complex formation with an apparent Mr of 128 kDa, and again, addition of excess ATP and Mg2+ did not impact the DLS results. The catalytically inactive mutant, PKRK296R, which is unable to self-associate 13, behaves in an identical manner to wild-type PKR, indicating that a 1:1 VAI:PKR complex forms.
Figure 4.

Inhibitors of PKR prevent its self-association (A) Molecular weight of PKR-VAI complexes (5 μM) as determined by DLS without incubation at 30 °C. (B) Concentration-dependent dimerization of PKR was examined by determining the molecular weight at the specified concentration of PKR (solid black line, squares), PKR-VAI (solid black line, circles), PKR-EBERI (dashed grey line, crosses), PKR-VAI-AS (solid black line, diamonds), or PKR-TAR (dashed grey line, triangles). Each data point was repeated in triplicate and error bars reflect standard deviation associated with measurements. (C) Superdex 200 HiLoad 26/60 size exclusion chromatography elution profiles of complexes at 5 μM (bottom) and 80 μM (top).
To examine the association state of PKR complexes, we next determined the concentration dependence (2−80 μM) of apparent Mr for wild-type PKR, PKR-VAI, PKR-EBERI, PKR-VAI-AS and PKR-TAR complexes (Fig. 4B). No significant increase in Mr is observed at high concentration for PKR alone, whereas a significant increase in apparent Mr is observed upon increasing concentration of either PKR-VAI-AS or PKR-TAR complex. The increase in hydrodynamic radius likely reflects an equilibrium between monomer and dimer forms of PKR. These results are consistent with previously observed PKR-activator complexes 10. Significantly, no such increase in apparent Mr is observed for PKR-VAI or PKR-EBERI; the inhibited PKR behaves remains monomeric at all concentrations examined (Fig. 4B). The hydrodynamic behavior established by DLS was confirmed using size exclusion chromatography, where molecular weight determinations were in good accordance between the two methodologies (Fig. 4C). These data indicate that PKR bound to inhibitory dsRNA is not capable of bimolecular associations required for trans-autophosphorylation.
dsRNA inhibitors remain associated with PKR under activating conditions
Activator dsRNAs demonstrate reduced affinity for PKR upon PKR phosphorylation, and reagents that limit phosphorylation typically prevent RNA dissociation 12; 22. As inhibitor binding to PKR prevents phosphorylation of the kinase, we hypothesized that activating conditions should not modulate the affinity of RNA inhibitors for PKR. To observe complex stability directly, native gel shift mobility assays were performed on activator (VAI-AS) and inhibitor (VAI or EBERI) dsRNA in the presence of PKR under conditions suitable and unsuitable for autophosphosphorylation (Fig. 5A). Each dsRNA ligand is shifted to a higher molecular weight species when PKR is added, indicating successful complex formation. As expected, addition of ATP/Mg2+ to the reaction mixtures resulted in a marked decrease in affinity between PKR and VAI-AS, whereas no change in complex stability is observed for complexes containing either VAI or EBERI. Stability of PKR-TAR and PKR-VAI-AS complexes is restored using mutants of the ATP coordination site (PKRK296R) or critical phosphorylation sites in the kinase domain of PKR (PKRT446A/T451A) (Fig. 5B). As expected, neither of these mutations affected inhibitor-containing complexes (Fig. 5B). Therefore, the RNA inhibitors maintain their affinity for PKR under kinase activating conditions.
Figure 5.
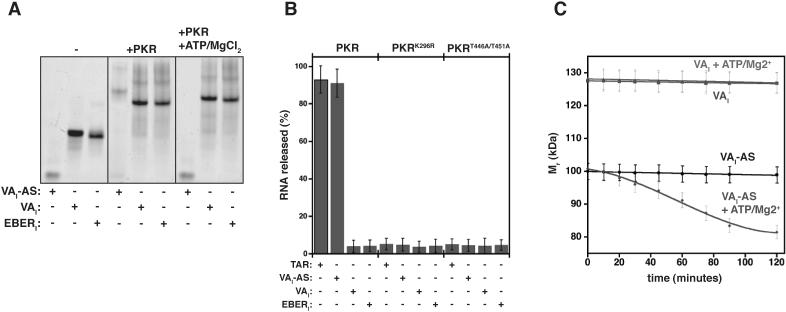
Inhibitory dsRNAs remain associated with PKR under activating conditions (A) Native gel mobility-shift for VAI, EBERI, or VAI-AS (200 nM) binding to PKR (400 nM) in reactions containing ATP (1 mM) and MgCl2 (2 mM) in certain cases. Samples were incubated at 30 °C for 90 minutes, resolved on 5% TBE gels, and visualized by SybrGreenII staining. (B) PKR-dsRNA complexes were pre-assembled (300 nM), and incubated at 30 °C for 90 minutes in the presence or absence of ATP (1 mM) and MgCl2 (1 mM). RNA release was quantified by resolving reaction components on native 5% TBE gels and dsRNA staining by SybrGreenII. Each data point represents a triplicate measurement. (C) Time-dependent molecular weight determination PKR-dsRNA complexes. Equimolar complexes of PKR and dsRNA (2 μM) were incubated in the cuvette at 30°C for the time specified prior to acquisition. ATP (1 mM) and MgCl2 (1 mM) were included in some cases. Each data point was repeated in triplicate.
To observe the release of RNA during the progress of PKR phosphorylation, apparent Mr of complexes was next determined by DLS at specific time intervals. A 1:1 complex of PKR-VAI-AS in the absence of reagents required for activation (ATP and MgCl2) does not change with respect to time (Fig. 5C, black). Addition of ATP and MgCl2 results in a time-dependent decrease in Mr, corresponding to a transition from the fully bound state to a mixture of dsRNA and PKR (Fig. 5C, blue). In contrast, an equimolar complex of VAI and PKR maintains a consistent Mr as a function of time (∼128 kDa) regardless of reaction conditions (Fig. 5C, red and green). Similar results were obtained when EBERI was employed (data not shown). Inhibitor RNAs do not release from PKR upon incubation under kinase activation conditions.
Inhibitors of PKR result in translational recovery
We next examined the effect of dsRNA activators and inhibitors on translation initiation. PKR activation by dsRNA leads to phosphorylation of eIf2α, resulting in attenuation of translation initiation, whereas inhibitors should have the opposite effect 45. To test this hypothesis, we quantitatively measured the synthesis of luciferase in HeLa S10 cell lysate that containing functional protein translation machinery, 5'-capped luciferase mRNA, and exogenous PKR. As expected, protein synthesis is significantly reduced upon inclusion of dsRNA activators in lysates (Fig. 6A). Addition of increasing amounts of inhibitory dsRNA (VAI) to extracts containing exogenously added PKR-activator complex results in the restoration of cap-dependent translational competency. Complete restoration is achieved when a 20-fold excess of inhibitor over activator is added; a result that is consistent with in vitro inhibition of autophosphorylation (Fig. 3B). As expected, restoring translational competency by addition of inhibitory dsRNA is accompanied by a decrease in eIF2α phosphorylation (Fig. 6B). Taken together, these results establish a causal link between in vitro phosphorylation of PKR and the inhibition of translation, with dsRNA inhibitors of PKR restoring translational competency.
Figure 6.

Inhibitors of PKR result in translational recovery. (A) HeLa S10 cell lysates containing the protein translation machinery, exogenous PKR (10 nM), and 5'-capped luciferase mRNA (50 nM) are incubated for 1 hour at 30 °C in the presence (grey) or absence (black) of HCV-IRES activator dsRNA (50 nM) and increasing amounts of VAI inhibitor (0−2 μM). Measurements were performed in triplicate and standardized relative to maximum Luciferase activity. (B) Western blot probed with an anti-(P-Ser51)-eIF2a antibody (ab4837−50, abcam) for selected reactions shown in (A).
DISCUSSSION
The mechanism by which dsRNA ligands modulate PKR kinase activity is central to understanding its function in innate immunity. The PKR response can be circumvented by viruses through transcription of highly-structured dsRNA inhibitors that bind to and inactivate PKR. Whereas the interaction between dsRNA inhibitors and PKR has been fairly well characterized, the mechanism of inhibition is poorly understood. Here we showed that dsRNA inhibitors interfere with the self association of PKR, thereby preventing PKR from acting as a substrate for trans-autophosphorylation. Numerous techniques were employed, including native gel shift mobility assays, isothermal titration calorimetry (ITC), autophosphorylation assays, dynamic light scattering, and in vitro translation assays.
Single stem-loops from both dsRNA activators and inhibitors mediate stable interaction with the dsRBDs of PKR 10. Here, we probed the interaction between dsRNA ligands (both activators and inhibitors) and PKR variants to delineate the requirements for binding using ITC and native gel shift mobility assays. The dsRBDs alone direct high-affinity interaction with dsRNA inhibitors, as no discernable interaction is observed with constructs of the kinase domain or interdomain linker alone. Furthermore, addition of the dsRBDs (residues 1−169) and the remainder of the protein (residues 170−551) in trans yields a complex containing only the dsRBDs and dsRNA; no high-affinity interaction with the interdomain linker or kinase domain is observed. Finally, mutations to critical regions in the kinase domain, including activation loop phosphorylation sites or ATP co-ordination site, have no effect on high-affinity interaction with dsRNA inhibitors.
dsRBDs of dephosphorylated PKR are sufficient and required for the interaction with either dsRNA activators or inhibitors, as previously shown. Phosphorylated PKR has minimal affinity for dsRNA inhibitors, indicating that once the phosphorylated state of the protein has been achieved, the inhibitory potential of these dsRNAs is attenuated. Kinetic studies further demonstrated that dsRNA inhibitors are preferentially effective at early points in the sigmoidal autophosphorylation profile (<15 minutes); as PKR becomes increasingly phosphorylated, the effectiveness of the inhibitors decreases. These observations are consistent with the hypothesis that dsRNA activators function to prime PKR for activation at low cellular PKR concentrations, but once activation is achieved, PKR is capable of trans-autophosphorylation of PKR in an RNA-independent manner 12. Furthermore, phosphorylation of the dsRBDs is not observed under these conditions 12, indicating that communication between the kinase and dsRBD domains is responsible for disrupting the interaction.
Dissociation of dsRNA activator from PKR is coincident with activation, presumably due to a reduced affinity with the phosphorylated form of the enzyme 12; 22. RNA release closely parallels activation, as almost complete dissociation is observed at the midpoint of the sigmoidal activation progress curve at all concentrations examined. Significant attenuation of RNA release is observed when inhibitor bound PKR complexes are examined, supporting the reciprocal link between phosphorylation state and PKR-dsRNA complex stability. Thus RNA inhibitors must operate at an early stage of the PKR response, since once PKR is phosphorylated, they are ineffective.
Activation of PKR proceeds through a bimolecular reaction mechanism, in which dsRNA activators and ultimately PKR phosphorylation serve to increase the propensity for PKR self-association 12. dsRNA inhibitors of PKR oppose these processes, since the inhibitors bind to PKR but prevent self-association. Therefore, dsRNA secondary structural elements not involved in the dsRBD interaction may block the ability of PKR to self-associate. Consistent with this hypothesis, truncations of dsRNA inhibitors, containing only the dsRBDs-binding stem-loop (i.e. VAI-AS or EBERI-CS), behave as activators of PKR 10. dsRNA inhibitors thus function to protect latent PKR from undergoing trans-autophosphorylation, and therefore activation. Trans- autophosphorylation assays indicate that inhibitor dsRNA-PKR complexes are not competent substrates for phosphorylation. This result is in stark contrast with activator dsRNA-PKR complexes, which are suitable substrates 12. Whether dsRNA inhibitors block substrate access to the kinase active site, or mask self-association interfaces must be further investigated.
Viral dsRNA inhibitors circumvent the PKR-mediated host-cell defense mechanism. To compliment our mechanistic data, we examined the effect of dsRNA ligands in cell extracts containing all of the necessary components of the protein translation machinery. As expected, addition of dsRNA activators of PKR, which result in phosphorylation of eIF2α, resulted in a significant decrease in protein synthesis. Conversely, translational competency is restored when sufficient viral dsRNA inhibitors are added to these cell extracts. Therefore, we have established a link between the in vitro mechanistic effects of inhibitors and their direct impact on the protein synthesis machinery.
The results presented here, combined with prior data from other groups, allows us to assess the functional importance of various secondary structural features of the dsRNA inhibitors. Both activators and inhibitors interact using a similar stem-loop, bind with similar affinities to PKR (KD ∼ 100nM), and have similar off-rates (personal communication, C.E. Aitken). The interaction is primarily mediated by the dsRBDs of PKR, indicating that the kinase domain and interdomain linker contribute negligible thermodynamic stability to these complexes. The additional secondary structural elements in VAI and EBERI must mediate inhibition of PKR. Mutations to the central stem of VAI are not tolerated 37; 41; 46, indicating that this region of the RNA, while not directly involved in binding PKR, plays an important role in inhibition. Footprinting studies have also suggested that PKR engages in a strong interaction with the minor groove of the apical stem and a weaker interaction with the central stem of VAI 40 . The dsRBDs alone protected only the apical stem from base-specific chemical probes, leaving the central stem exposed. Specific structural features in this region must be responsible, as increased length of the dsRNA inhibitors relative to activators is not sufficient to explain inhibition. Long, idealized dsRNA polymers serve as potent activators of PKR 36; 37; 47 and the hepatitis C virus internal ribosome entry site RNA (HCV IRES), a highly structured 374-nucleotide dsRNA molecule mediates potent autophosphorylation PKR in vitro (personal communication, T. Shimoike).
The physical basis for inhibition remains elusive, although a dynamic interaction between central stem of VAI and a specific region of PKR cannot be ruled out; further studies are required to explore this interaction. dsRNA inhibitors most likely prevent PKR self-association through direct steric blockage. However, an enticing possibility remains that PKR activation is mediated by the altering of the electrostatic landscape of the protein; inhibitory dsRNAs might prevent self-association through changing the electrostatic potential of the enzyme.
A simple model for the inactivation of PKR autophosphorylation by dsRNA inhibitors is consistent with the available data (Figure 7). PKR autophosphorylation proceeds through a bimolecular reaction mechanism in which trans-autophosphorylation occurs between PKR monomers. Interaction with dsRNA activators (and ultimately autophosphorylation) increase the propensity for PKR dimerization, thus increasing the rate of the bimolecular reaction 12. A single stem-loop from inhibitory dsRNAs interacts preferentially with the latent form of PKR via the dsRBDs; distal stem-loops of the RNA mediate the dimerization disruption. The net effect is a severe attenuation of PKR autophosphorylation and therefore inhibition of the enzyme. The data presented here provide a detailed mechanistic view of how dsRNA inhibitors function, and confirm previous insights into the mechanism of PKR activation. More detailed mechanistic and structural studies are clearly required to fully understand the function of this highly dynamic and intricately regulated enzyme.
Figure 7.

Model for the inhibition of PKR kinase activity. Model summarizing the framework for the inhibition of PKR; dsRBDs (R), kinase domain (K) and interdomain linker (L). Upon binding of activator dsRNA to the dsRBDs of PKR in the latent form, enhanced bimolecular interaction between two PKR molecules is observed. In this conformation, autophosphorylation occurs, leading to RNA release and activation competency of PKR. Subsequently, activated PKR may feed back to trans-phosphorylate remaining latent PKR. Addition of inhibitory dsRNA leads to interaction with the latent form of PKR; binding prevents dimerization and prevents PKR from acting as a substrate for trans-autophosphorylation. Structured stem-loops not required for interaction with the dsRBDs may be responsible for mediating inhibition.
MATERIALS AND METHODS
Sample preparation.
Expression and purification of full-length human PKR and domain constructs including dsRBD1/2 are as previously described 10; 12. Full length PKR mutant constructs (PKRK296R and PKRT446A/T451A) were generated using site-directed mutagenesis of the wild-type plasmid, and were expressed and purified identically to the wild-type protein. Phosphorylated PKR (PKRP) was generated by incubating wild-type full length human PKR (15 μM) in the presence of ATP (1 mM) and MgCl2 (2 mM) at 30 °C for 2 hours in a buffered solution containing Tris•HCl (pH 7.5), 100 mM NaCl, and 5 mM ß-mercaptoethanol (ß-Me), and purified as described elsewhere 12. RNA samples were prepared by in vitro transcription using T7 polymerase as described previously 10; 48 using a BsaI site for plasmid linearization.
Native Gel Shift Mobility Assay.
All samples were prepared in 50 mM Tris/Cl (pH 7.5), 100 mM NaCl, and incubated at room temperature for 10 minutes. Samples were mixed with non-denaturing load mix and loaded onto 5% non-denaturing TBE gels (Bio-Rad). Sample running buffer contained 0.5X TBE (50 mM Tris base, 41.5 mM boric acid, and 1 mM EDTA, final pH 8.3). Electrophoresis was performed at 60 V at 4 °C, and gels were stained with 1X SybrGreenII fluorescent dye (Molecular Probes Inc.) for quantitation. Fluorescently stained gels were scanned (GE Healthcare) and quantitated using the ImageJ software 49.
Isothermal Titration Calorimetry.
A VP-ITC microcalorimeter (Microcal) was used to analyze the stability of protein-RNA complexes. The sample cell contained the RNA ligand (10 μM RNA, 50 mM Tris•HCl, pH 7.5, 100 mM NaCl) and a concentrated protein solution (200 μM, 50 mM Tris•HCl, pH 7.5, 100 mM NaCl, 5 mM ß-Me) was placed in the syringe. Each titration was performed at 30 °C, and involved a single 2 μL injection, followed by twenty-four 10 μL injections of protein solution into the sample cell containing RNA. Titration curves were fit by a nonlinear least-squares method with a model for two binding sites using Microcal Origin (version 5.0) to extract thermodynamic parameters KA (association constant), ΔH (binding enthalpy), and N (stoichiometry). In each case, a single high-affinity site and a second weak affinity non-specific site were observed; reported results are from the high-affinity binding site. From these data, the changes in entropy (ΔS) and free energy (ΔG) were calculated using established equations 10; 32.
PKR Autophosphorylation Assays.
All phosphorylation assays for PKR were performed in 50 mM Tris/Cl (pH 7.5), 100 mM NaCl, 1 mM ATP, 2 mM MgCl2, and 1 μCi of 32P-γATP. Reactions were performed at 30 °C with the concentrations and incubation times as denoted in the figure legends. Reactions were quenched with SDS-PAGE load mix. Phosphorylated proteins were separated on a 4−20% SDS-PAGE gel (Bio-Rad), dried for 30 minutes at 80 °C, and autoradiographed (GE Healthcare) to trace the incorporation of 32P into PKR. Band intensities were quantitated using the ImageJ software 49.
Dynamic Light Scattering.
Protein samples were pre-filtered through a 0.22 μM Millex-GV syringe filter (Millipore) prior to analysis. Dynamic light scattering experiments were performed at 30 °C with a DynaPro-801 molecular sizing instrument (Protein Solutions Co.). A minimum of 50 data points at 10-second intervals was collected for each sample examined. The hydrodynamic radius, apparent molecular weight, and polydispersity were determined on the basis of an autocorrelation analysis of scattered light intensity data using the DYNAMICS (version 6.0) software package.
In vitro translation.
The basis for in vitro translation assays is a HeLa S10 cell lysate; preparation of cell extracts is discussed elsewhere 50. The lysate (50% vol.) is supplemented with 0.5 U/μl RNase inhibitor, SUPERase·In (Ambion), 25μM Amino Acid Mixture, Complete (Promega), 60mM KCl (for cap-dependent translation), and 1mM MgCl2 (required for activation of PKR). Modulation of cap-dependent translation by PKR was determined by adding purified PKR and RNA ligands to the lysate containing 5'-capped Renilla luciferase mRNA (reporter RNA) at concentrations denoted in the figure legend. Reporter RNA is preincubated at 65 °C for 3 min, and then immediately cooled in ice-cold water prior to addition. The translation reaction is incubated at 30 °C for 60 minutes prior to Luciferase activity assay. Using a reporter construct that contains cap-Luc, the activity (intensity of luminescence from substrate) of Renilla luciferase is determi ned by the luciferase assay system (P romega) and luminometer (Analytical Luminescence Laboratory).
ACKOWLEDGEMENTS
We thank M. Margaris for assistance, and other members of the Puglisi laboratory members for their help and advice. Supported by NIH AI47365 and GM078346. S.A. McKenna is supported by the Canadian Institutes of Health Research and the Alberta Heritage Foundation for Medical Research. D.A. Lindhout is supported by the Alberta Heritage Foundation for Medical Research. C.E. Aitken is supported by an NIH Molecular Biophysics training grant (T32 GM008294).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
REFERENCES
- 1.Malmgaard L. Induction and regulation of IFNs during viral infections. J Interferon Cytokine Res. 2004;24:439–54. doi: 10.1089/1079990041689665. [DOI] [PubMed] [Google Scholar]
- 2.Peel AL. PKR activation in neurodegenerative disease. J Neuropathol Exp Neurol. 2004;63:97–105. doi: 10.1093/jnen/63.2.97. [DOI] [PubMed] [Google Scholar]
- 3.Langland JO, Cameron JM, Heck MC, Jancovich JK, Jacobs BL. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006;119:100–10. doi: 10.1016/j.virusres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 4.Gale M,, Jr., Katze MG. Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol Ther. 1998;78:29–46. doi: 10.1016/s0163-7258(97)00165-4. [DOI] [PubMed] [Google Scholar]
- 5.Francois C, Duverlie G, Rebouillat D, Khorsi H, Castelain S, Blum HE, Gatignol A, Wychowski C, Moradpour D, Meurs EF. Expression of hepatitis C virus proteins interferes with the antiviral action of interferon independently of PKR-mediated control of protein synthesis. J Virol. 2000;74:5587–96. doi: 10.1128/jvi.74.12.5587-5596.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dever TE. Gene-specific regulation by general translation factors. Cell. 2002;108:545–56. doi: 10.1016/s0092-8674(02)00642-6. [DOI] [PubMed] [Google Scholar]
- 7.Clemens MJ. PKR--a protein kinase regulated by double-stranded RNA. Int J Biochem Cell Biol. 1997;29:945–9. doi: 10.1016/s1357-2725(96)00169-0. [DOI] [PubMed] [Google Scholar]
- 8.Clemens MJ, Elia A. The double-stranded RNA-dependent protein kinase PKR: structure and function. J Interferon Cytokine Res. 1997;17:503–24. doi: 10.1089/jir.1997.17.503. [DOI] [PubMed] [Google Scholar]
- 9.Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. Embo J. 1998;17:5458–65. doi: 10.1093/emboj/17.18.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKenna SA, Kim I, Liu CW, Puglisi JD. Uncoupling of RNA binding and PKR kinase activation by viral inhibitor RNAs. J Mol Biol. 2006;358:1270–85. doi: 10.1016/j.jmb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 11.McCormack SJ, Ortega LG, Doohan JP, Samuel CE. Mechanism of interferon action motif I of the interferon-induced, RNA-dependent protein kinase (PKR) is sufficient to mediate RNA-binding activity. Virology. 1994;198:92–9. doi: 10.1006/viro.1994.1011. [DOI] [PubMed] [Google Scholar]
- 12.McKenna SA, Lindhout DA, Kim I, Liu CW, Gelev VM, Wagner G, Puglisi JD. Molecular framework for the activaiton of PKR. J. Biol. Chem. 2007;285:11474–86. doi: 10.1074/jbc.M700301200. [DOI] [PubMed] [Google Scholar]
- 13.Dey M, Cao C, Dar AC, Tamura T, Ozato K, Sicheri F, Dever TE. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell. 2005;122:901–13. doi: 10.1016/j.cell.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 14.Taylor SS, Haste NM, Ghosh G. PKR and eIF2alpha: integration of kinase dimerization, activation, and substrate docking. Cell. 2005;122:823–5. doi: 10.1016/j.cell.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 15.Dar AC, Dever TE, Sicheri F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell. 2005;122:887–900. doi: 10.1016/j.cell.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 16.Lemaire PA, Tessmer I, Craig R, Erie DA, Cole JL. Unactivated PKR Exists in an Open Conformation Capable of Binding Nucleotides. Biochemistry. 2006;45:9074–9084. doi: 10.1021/bi060567d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romano PR, Garcia-Barrio MT, Zhang X, Wang Q, Taylor DR, Zhang F, Herring C, Mathews MB, Qin J, Hinnebusch AG. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2alpha kinases PKR and GCN2. Mol Cell Biol. 1998;18:2282–97. doi: 10.1128/mcb.18.4.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor DR, Lee SB, Romano PR, Marshak DR, Hinnebusch AG, Esteban M, Mathews MB. Autophosphorylation sites participate in the activation of the double-stranded-RNA-activated protein kinase PKR. Mol Cell Biol. 1996;16:6295–302. doi: 10.1128/mcb.16.11.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemaire PA, Lary J, Cole JL. Mechanism of PKR activation: dimerization and kinase activation in the absence of double-stranded RNA. J Mol Biol. 2005;345:81–90. doi: 10.1016/j.jmb.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 20.Vattem KM, Staschke KA, Wek RC. Mechanism of activation of the double-stranded-RNA-dependent protein kinase, PKR: role of dimerization and cellular localization in the stimulation of PKR phosphorylation of eukaryotic initiation factor-2 (eIF2) Eur J Biochem. 2001;268:3674–84. doi: 10.1046/j.1432-1327.2001.02273.x. [DOI] [PubMed] [Google Scholar]
- 21.Wu S, Kaufman RJ. A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J Biol Chem. 1997;272:1291–6. doi: 10.1074/jbc.272.2.1291. [DOI] [PubMed] [Google Scholar]
- 22.Jammi NV, Beal PA. Phosphorylation of the RNA-dependent protein kinase regulates its RNA-binding activity. Nucleic Acids Res. 2001;29:3020–9. doi: 10.1093/nar/29.14.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramos A, Grunert S, Adams J, Micklem DR, Proctor MR, Freund S, Bycroft M, St Johnston D, Varani G. RNA recognition by a Staufen double-stranded RNA-binding domain. Embo J. 2000;19:997–1009. doi: 10.1093/emboj/19.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu H, Henras A, Chanfreau G, Feigon J. Structural basis for recognition of the AGNN tetraloop RNA fold by the double-stranded RNA-binding domain of Rnt1p RNase III. Proc Natl Acad Sci U S A. 2004;101:8307–12. doi: 10.1073/pnas.0402627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryter JM, Schultz SC. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. Embo J. 1998;17:7505–13. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ben-Asouli Y, Banai Y, Pel-Or Y, Shir A, Kaempfer R. Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell. 2002;108:221–32. doi: 10.1016/s0092-8674(02)00616-5. [DOI] [PubMed] [Google Scholar]
- 27.Osman F, Jarrous N, Ben-Asouli Y, Kaempfer R. A cis-acting element in the 3'-untranslated region of human TNF-alpha mRNA renders splicing dependent on the activation of protein kinase PKR. Genes Dev. 1999;13:3280–93. doi: 10.1101/gad.13.24.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vuyisich M, Spanggord RJ, Beal PA. The binding site of the RNA-dependent protein kinase (PKR) on EBER1 RNA from Epstein-Barr virus. EMBO Rep. 2002;3:622–7. doi: 10.1093/embo-reports/kvf137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bommer UA, Borovjagin AV, Greagg MA, Jeffrey IW, Russell P, Laing KG, Lee M, Clemens MJ. The mRNA of the translationally controlled tumor protein P23/TCTP is a highly structured RNA, which activates the dsRNA-dependent protein kinase PKR. Rna. 2002;8:478–96. doi: 10.1017/s1355838202022586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spanggord RJ, Beal PA. Selective binding by the RNA binding domain of PKR revealed by affinity cleavage. Biochemistry. 2001;40:4272–80. doi: 10.1021/bi002512w. [DOI] [PubMed] [Google Scholar]
- 31.Maitra RK, McMillan NA, Desai S, McSwiggen J, Hovanessian AG, Sen G, Williams BR, Silverman RH. HIV-1 TAR RNA has an intrinsic ability to activate interferon-inducible enzymes. Virology. 1994;204:823–7. doi: 10.1006/viro.1994.1601. [DOI] [PubMed] [Google Scholar]
- 32.Kim I, Liu CW, Puglisi JD. Specific recognition of HIV TAR RNA by the dsRNA binding domains (dsRBD1-dsRBD2) of PKR. J Mol Biol. 2006;358:430–42. doi: 10.1016/j.jmb.2006.01.099. [DOI] [PubMed] [Google Scholar]
- 33.Bevilacqua PC, Cech TR. Minor-groove recognition of double-stranded RNA by the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR. Biochemistry. 1996;35:9983–94. doi: 10.1021/bi9607259. [DOI] [PubMed] [Google Scholar]
- 34.Robertson HD, Mathews MB. The regulation of the protein kinase PKR by RNA. Biochimie. 1996;78:909–14. doi: 10.1016/s0300-9084(97)86712-0. [DOI] [PubMed] [Google Scholar]
- 35.Kumar KU, Srivastava SP, Kaufman RJ. Double-stranded RNA-activated protein kinase (PKR) is negatively regulated by 60S ribosomal subunit protein L18. Mol Cell Biol. 1999;19:1116–25. doi: 10.1128/mcb.19.2.1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharp TV, Schwemmle M, Jeffrey I, Laing K, Mellor H, Proud CG, Hilse K, Clemens MJ. Comparative analysis of the regulation of the interferon-inducible protein kinase PKR by Epstein-Barr virus RNAs EBER-1 and EBER-2 and adenovirus VAI RNA. Nucleic Acids Res. 1993;21:4483–90. doi: 10.1093/nar/21.19.4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghadge GD, Malhotra P, Furtado MR, Dhar R, Thimmapaya B. In vitro analysis of virus-associated RNA I (VAI RNA): inhibition of the double-stranded RNA-activated protein kinase PKR by VAI RNA mutants correlates with the in vivo phenotype and the structural integrity of the central domain. J Virol. 1994;68:4137–51. doi: 10.1128/jvi.68.7.4137-4151.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clarke PA, Schwemmle M, Schickinger J, Hilse K, Clemens MJ. Binding of Epstein-Barr virus small RNA EBER-1 to the double-stranded RNA-activated protein kinase DAI. Nucleic Acids Res. 1991;19:243–8. doi: 10.1093/nar/19.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitajewski J, Schneider RJ, Safer B, Munemitsu SM, Samuel CE, Thimmappaya B, Shenk T. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell. 1986;45:195–200. doi: 10.1016/0092-8674(86)90383-1. [DOI] [PubMed] [Google Scholar]
- 40.Clarke PA, Mathews MB. Interactions between the double-stranded RNA binding motif and RNA: definition of the binding site for the interferon-induced protein kinase DAI (PKR) on adenovirus VA RNA. Rna. 1995;1:7–20. [PMC free article] [PubMed] [Google Scholar]
- 41.Mellits KH, Kostura M, Mathews MB. Interaction of adenovirus VA RNAl with the protein kinase DAI: nonequivalence of binding and function. Cell. 1990;61:843–52. doi: 10.1016/0092-8674(90)90194-j. [DOI] [PubMed] [Google Scholar]
- 42.Ma Y, Mathews MB. Structure, function, and evolution of adenovirus-associated RNA: a phylogenetic approach. J Virol. 1996;70:5083–99. doi: 10.1128/jvi.70.8.5083-5099.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gabel F, Wang D, Madern D, Sadler A, Dayie K, Daryoush MZ, Schwahn D, Zaccai G, Lee X, Williams BR. Dynamic flexibility of double-stranded RNA activated PKR in solution. J Mol Biol. 2006;359:610–23. doi: 10.1016/j.jmb.2006.03.049. [DOI] [PubMed] [Google Scholar]
- 44.Wu S, Kaufman RJ. trans-Autophosphorylation by the isolated kinase domain is not sufficient for dimerization or activation of the dsRNA-activated protein kinase PKR. Biochemistry. 2004;43:11027–34. doi: 10.1021/bi0360105. [DOI] [PubMed] [Google Scholar]
- 45.Sonenberg N, Dever TE. Eukaryotic translation initiation factors and regulators. Curr Opin Struct Biol. 2003;13:56–63. doi: 10.1016/s0959-440x(03)00009-5. [DOI] [PubMed] [Google Scholar]
- 46.Rahman A, Malhotra P, Dhar R, Kewalramani T, Thimmapaya B. Effect of single-base substitutions in the central domain of virus-associated RNA I on its function. J Virol. 1995;69:4299–307. doi: 10.1128/jvi.69.7.4299-4307.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mellits KH, Pe'ery T, Manche L, Robertson HD, Mathews MB. Removal of double-stranded contaminants from RNA transcripts: synthesis of adenovirus VA RNAI from a T7 vector. Nucleic Acids Res. 1990;18:5401–6. doi: 10.1093/nar/18.18.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim I, McKenna SA, Puglisi EV, Puglisi JD. Rapid purification of RNAs using Fast Performance Liquid Chromatography (FPLC) RNA. 2007;13:1–6. doi: 10.1261/rna.342607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abramoff MD, Magelhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- 50.Otto GA, Puglisi JD. The pathway of HCV IRES-mediated translation initiation. Cell. 2004;119:369–80. doi: 10.1016/j.cell.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 51.Clarke PA, Pe'ery T, Ma Y, Mathews MB. Structural features of adenovirus 2 virus-associated RNA required for binding to the protein kinase DAI. Nucleic Acids Res. 1994;22:4364–74. doi: 10.1093/nar/22.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Glickman JN, Howe JG, Steitz JA. Structural analyses of EBER1 and EBER2 ribonucleoprotein particles present in Epstein-Barr virus-infected cells. J Virol. 1988;62:902–11. doi: 10.1128/jvi.62.3.902-911.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muesing MA, Smith DH, Capon DJ. Regulation of mRNA accumulation by a human immunodeficiency virus trans-activator protein. Cell. 1987;48:691–701. doi: 10.1016/0092-8674(87)90247-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.