

Abstract
Frontotemporal lobar degeneration (FTLD) with ubiquitin-positive, tau-negative inclusions, and linkage to chromosome 17 was recently found to be caused by mutations in the progranulin (PGRN) gene. In this study, we screened a group of 51 FTLD patients for PGRN mutations and identified a novel exon 6 splice donor site deletion (IVS6+5_8delGTGA) in 2 unrelated patients. This mutation displayed an altered splicing pattern generating 2 aberrant transcripts and causing frameshifts of the coding sequence, premature termination codons, and a near absence of PGRN mRNA from the mutated alleles most likely through nonsense-mediated decay. The subsequent PGRN haploinsufficiency is consistent with previously described PGRN mutations. We present a molecular characterization of the IVS6+5_8delGTGA mutation and also describe clinical and neuropathologic features from the 2 patients carrying this PGRN mutation. From the screening of these 51 FTLD patients, we could also identify the earlier reported mutation Gln130fs, and several coding sequence variants that are most likely nonpathogenic.
Keywords: frontotemporal lobar degeneration, frontotemporal dementia, progranulin, ubiquitin, TDP-43
Frontotemporal lobar degeneration (FTLD) is the third most common cause of dementia accounting for approximately 10% of all dementia cases.1–3 Clinically, FTLD is characterized by a progressive change in personality, behavior, and language with memory impairment being less frequent.4 Gross examination of the FTLD brain typically shows severe atrophy of the frontal and temporal cortices.5 Microscopically, 2 main pathologic subtypes can be identified. Many cases display intraneuronal inclusions of the microtubule-associated protein tau (MAPT). However, the majority of cases show ubiquitin and TAR DNA binding protein-43 (TDP-43)-positive intraneuronal inclusions.6–8
Up to 40% of FTLD patients have a positive family history of the disease1,9,10 and in 10% to 20% of the familial cases the disease can be explained by mutations in the MAPT gene.11–13 Neuropathologically, these patients are characterized by intraneuronal accumulation of hyperphosphorylated tau. Mutations in the charged multivesicular body protein 2B14 and valosin-containing protein15 genes have also been found in rare familial FTLD cases. Recently, mutations in the gene encoding progranulin (PGRN) were identified as a major cause for FTLD associated with ubiquitin and TDP-43-positive pathology. 16,17 To date, more than 60 different PGRN mutations have been identified and studies suggest that PGRN mutations account for up to 20% of familial FTLD.18,19 A majority of the identified PGRN mutations introduce premature termination codons in the PGRN mRNA sequence owing to nonsense, splice-site, or frameshift mutations. The mutant mRNA transcripts are most likely degraded by nonsense-mediated decay (NMD), resulting in a loss of functional PGRN protein.16,17 Recently, hemizygous deletions of the PGRN gene were reported in patients with FTLD, in further support of haploinsufficiency as the pathogenic mechanism in these patients.20,21
In this study, we have conducted a mutation screen of the PGRN gene in 51 FTLD patients. We present a molecular, clinical, and neuropathologic characterization of a novel deletion mutation in the splice donor site of PGRN exon 6, IVS6+5_8delGTGA.
MATERIALS AND METHODS
Patients
The PGRN mutation screen included 51 FTLD patients. Thirty-two were clinically diagnosed FTLD cases from Sweden, and 12 of these had a known positive family history of dementia. The remaining 19 patients originated from the United States and had been neuropathologically verified at Massachusetts General Hospital. Of these, eight were TDP-43-positive and 11 were tau-positive. At least 3 of the patients had a known positive family history of the disease. All patients investigated had earlier been analyzed and found negative for mutations in the MAPT gene. The study was approved by the Ethical Committees of Uppsala University, Uppsala, Sweden and Massachusetts General Hospital, Boston, USA. Written informed consent was obtained from all individuals.
PGRN Genomic Analyses
Genomic DNA was prepared from blood or fresh frozen brain tissue using a standard protocol. All coding exons with flanking intronic sequences and the noncoding exon 0 of the PGRN gene were amplified by polymerase chain reaction (PCR) and sequenced according to the manufacturer’s instructions (Big Dye terminator v3.1, Applied Biosystems, Foster City, CA). Primers used for PGRN mutation screening16 were positioned so that the entire exons and the expected splice sites within the introns could be examined. The sequence variants identified in this study were analyzed in 165 unrelated control individuals. The PCR products were separated on ABI 3700 (Applied Biosystems) and analyzed using the GeneMapper program (Applied Biosystems).
PGRN mRNA Analyses
Frontal lobe brain tissue was homogenized and total RNA was isolated using Trizol Reagent (Invitrogen, Paisley, UK). The RNA samples were treated with DNase 1 (Invitrogen) before first-strand cDNA synthesis with the superscript first-strand synthesis system for RT-PCR (Invitrogen). To show degradation of the mutant allele in a patient carrying the IVS6+5_8delGTGA mutation, sequence analysis was done on a synonymous polymorphism (rs25646) located in exon 4. Exons 3–4 of PGRN were amplified from brain cDNA and the resulting PCR products were sequenced using Big Dye terminator v3.1 sequencing chemistry (Applied Biosystems). To assess mRNA levels of PGRN, quantitative PCR (qPCR) using Power SYBR green PCR Mastermix (Applied Biosystems) was conducted on brain cDNA from one of the patients carrying the IVS6+5_8delGTGA mutation, the patient carrying the Gln130fs mutation and a healthy control. Samples were analyzed in triplicates and normalized against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA levels.
Analysis of IVS6+5_8delGTGA Mutation Splicing Effects
PCR amplification was conducted on brain cDNA of a patient carrying the IVS6+5_8delGTGA mutation and a healthy control, using a cDNA-specific forward primer located across the exon 4–5 border (5′-atgccccaggcttcc tgctgtg) and a reverse primer located across the exon 7 to 8 border (5′-acatcccccactgtgtgcgcag). A genomic DNA sample was included in the analysis to confirm that no genomic DNA was amplified. The PCR products were subsequently used in a nested PCR using a forward exon 5 primer (5′-ttcctgctgtgaagacagggtgca) and a reverse exon 7 primer (5′-tagtgaggaggtccgtggtagcgt) to investigate RNA transcripts resulting from the IVS6+5_8delGTGA mutation. To examine the possible retention of intron 6 in the RNA transcript, a forward exon 5 primer (5′-ttcctgctgtgaagacagggtgca) and reverse intron 6 primer (5′-tgagtggggacaggggtgggtctt) were used in a nested PCR. The resulting PCR fragments were resolved on a 1.2% agarose gel to verify product length (Flashgel DNA system, Lonza, Basel, Switzerland). The PCR products were sequenced using Big Dye terminator v3.1 sequencing chemistry (Applied Biosystems) to confirm their identity.
Founder Effect Analysis
Genotyping of the 2 patients carrying the IVS6+5_8-delGTGA mutation for microsatellite markers D17S1814, D17S1793, D17S932, D17S1789, D17S930, D17S1804, D17S931, and D17S958 was carried out by PCR amplification using genomic DNA and fluorescently labeled primers. Fragments were separated on an ABI 3700 (Applied Biosystems) and the results were analyzed using the GeneMapper program (Applied Biosystems).
Immunohistochemical Analyses
Immunohistochemistry was carried out on 5-μm formalin-fixed and paraffin-embedded brain sections from the hippocampus, amygdala, temporal, and frontal cortices, using antibodies directed against ubiquitin (rabbit anti-ubiquitin, 1:200; Dako, Glostrup, Denmark), TDP-43 (rabbit anti-TDP-43, 1:500; ProteinTech Group, Inc., Chicago, IL), amyloid-β (anti-amyloid-β, 10D5, 1:200, Elan Pharmaceuticals, San Francisco, CA), tau (rabbit anti-human tau, 1:3000, Dako), and α-synuclein (anti-α-synuclein–Syn-1, BD Transduction Laboratories, San Jose, CA). Sections were subsequently incubated with a secondary biotinylated anti-mouse or anti-rabbit immunoglobulin-G antibody (1:200; Invitrogen) followed by treatment with avidin-biotin-conjugated enzyme complex (Vector Laboratories, Burlingame, CA). The staining was visualized with diaminobenzidine (DAB, Vector Laboratories) and counterstained with hematoxylin.
RESULTS
Mutation Screening of the PGRN Gene
Genetic analysis of the PGRN gene in this collection of patients with FTLD showed one novel deletion of 4 nucleotides, IVS6+5_8delGTGA, which was identified in the splice donor site of PGRN exon 6 in 2 patients (Table 1, Fig. 1A). In addition an earlier reported deletion of 4 nucleotides, Gln130fs, was identified in PGRN exon 4 in another patient (Table 1).16,18 The genetic analysis of PGRN also showed 3 coding sequence variants (2 missense and 1 silent mutation) and 2 intronic sequence changes located in introns 2 and 7 (Table 1). Two of the coding sequence variants (Asp128Asp and Arg433Trp) and the 2 intronic sequence changes were present in healthy controls or in healthy family members. However, one coding sequence variant, the novel Leu53Pro (found in a clinical case with familial FTD), was not identified in 165 healthy controls (Table 1). As there was no available tissue sample for cDNA analysis from the Leu53Pro patient or DNA samples from additional family members for segregation analysis of the mutation, we were not able to further investigate the pathogenic relevance of this PGRN mutation. However, in silico predictions based on evolutionary conservation (SIFT v.2),22 predicted the Leu53Pro mutation not to significantly affect protein function (data not shown).
TABLE 1.
Progranulin (PGRN) Coding Sequence Changes Identified in 51 Frontotemporal Lobar Degeneration (FTLD) Cases
| Region | Mutation Genomic* | Predicted cDNA Alteration | Predicted Protein Alteration | rs Number | Frequency Patients % | Frequency Controls % | Notes |
|---|---|---|---|---|---|---|---|
| EX 2 | g.100347T>C | c.158T>C | Leu53Pro | 2.0 | 0 | ||
| IVS 2 | g.100474G>A | rs9897526 | 21.6 | 16.3 | |||
| EX 4 | g.101164T>C | c.384T>C | Asp128Asp | rs25646 | 7.8 | — | Not segregating with disease |
| EX 4 | g.101168_101171 delCAGT | c.388_391del CAGT | Gln130SerfsX125 | 2.0 | 0 | ||
| IVS 6 | g.101707_101710 delGTGA | c.599_708del | Val200GlyfsX18 | 3.9 | 0 | ||
| c.708_709ins708 +1_708+236 | Ala237ValfsX17 | ||||||
| IVS 7 | g.102270G>A | 17.6 | 10.4 | ||||
| EX 10 | g.103034C>T | c.1297C>T | Arg433Trp | 2.0 | 1.9 |
Numbering relative to reverse complement of GenBank accession number AC003043.1 and starting at nucleotide 1.
FIGURE 1.

A, Genomic DNA sequence of the PGRN exon 6 splice donor site of 1 patient showing the 4 base pair deletion that causes the IVS6+5_8delGTGA mutation. B, Genomic DNA sequence of PGRN exon 4 from one IVS6+5_8delGTGA patient shows the synonymous polymorphism, rs25646 (upper panel). Sequencing of brain cDNA of PGRN exon 4 from the IVS6+5_8-delGTGA patient shows a reduction of the C allele of the rs25646 polymorphism (lower panel). C, Quantitative PCR analysis shows a 17% reduction of PGRN mRNA levels in brain from the IVS6+5_8delGTGA patient and a 40% reduction in the Gln130fs patient, when compared with a healthy control. Data are shown as the average of triplicate samples.
Analysis of PGRN Wild-type and Mutant Alleles
Sequence analysis of genomic DNA of the IVS6+5_8-delGTGA patient showed a synonymous polymorphism in exon 4 (rs25646), with the patient carrying one C and one T allele (Fig. 1B). Sequence analysis of brain cDNA from the same IVS6+5_8delGTGA patient showed a substantial reduction of the C allele, supporting the expected degradation of the mutant PGRN transcript (Fig. 1B). There was no tissue sample available for RNA extraction from the second patient with the IVS6+5_8delGTGA mutation and hence no cDNA analysis could be done in this individual.
Analysis of PGRN mRNA Levels
Quantitative PCR analysis of PGRN brain mRNA levels showed a 17% reduction in one of the patients carrying the IVS6+5_8delGTGA mutation and a 40% reduction in the patient carrying the Gln130fs mutation, when compared with a healthy control (Fig. 1C).
Analysis of IVS6+5_8delGTGA Mutation Splicing Effects
To examine the effect of the IVS6+5_8delGTGA deletion on PGRN mRNA splicing, PCR amplification of brain cDNA from PGRN exons 5 to 7 was conducted. The analysis showed a PCR product of 353 base pairs in the patient carrying the IVS6+5_8delGTGA mutation and in a healthy control sample (Fig. 2A), confirmed by sequence analysis to represent the wild-type allele containing exons 5, 6, and 7 (data not shown). The sample from the patient carrying the IVS6+5_8delGTGA mutation also displayed a faint PCR product of 243 base pairs, which corresponds to the size of a transcript in which exon 6 is excluded (Fig. 2A). Sequence analysis confirmed this PCR product to be exon 5 spliced onto exon 7 (Fig. 2B). Moreover, the IVS6+5_8delGTGA sample showed an additional faint band of 589 base pairs on gel electrophoresis (Fig 2A). To investigate if this band represented a PGRN splice form caused by the retention of the intron following exon 6, a PCR using primers located in PGRN exon 5 and the intron following exon 6 was condutced. Sequence analysis of this PCR fragment showed a sequence containing exon 5, followed by exon 6 and the mutated intron 6 sequence with the IVS6+5_8delGTGA deletion (Fig. 2B). The absence of intronic sequence following exon 5 excludes the possibility of genomic DNA contamination of this sample.
FIGURE 2.

A, Agarose gel electrophoresis of PCR products amplified from brain cDNA shows the wild-type fragment (353 bp) containing exon 5, 6, and 7 in the IVS6+5_8delGTGA patient (lane 2) and in a healthy control (lane 3). Lane 2 also shows a fragment representing the exclusion of exon 6 (243 bp) and a fragment caused by the retention of intron 6 (589 bp) in the IVS6+5_8delGTGA patient. No PCR amplification product is seen in a genomic DNA sample (lane 4). B, Schematic representation of the different splice forms produced as a result of the IVS6+5_8delGTGA mutation. Sequence chromatograms show the retention of intron 6 (upper panel) and the skipping of exon 6 (lower panel) in the IVS6+5_8delGTGA patient.
Founder Effect Analysis
To determine whether the 2 patients carrying the IVS6+5_8delGTGA mutation shared a common genetic founder, haplotype analysis was done using microsatellite markers flanking the PGRN gene. However, the haplotype analysis did not support a common genetic origin for the 2 patients as they did not seem to share a haplotype across the PGRN locus (Table 2). Also, sequence analysis of one of the IVS6+5_8delGTGA patients, heterozygous for a synonymous polymorphism in exon 4 (rs25646), suggested that the mutated allele in this patient was represented by the C allele, which was almost absent in the cDNA sequence (Fig. 1B), whereas the other IVS6+5_8delGTGA patient was homozygous for the T allele (data not shown). Hence, the 2 patients carrying the IVS6+5_8delGTGA mutation were most likely not related, indicating that this mutation has originated independently in different individuals.
TABLE 2.
Haplotype Analysis of the IVS6+5_8delGTGA Patients
| Marker | Position (Mb) | Patient 1 Genotypes | Patient 2 Genotypes |
|---|---|---|---|
| D17S1814 | 38.1 | 145/157 | 145/151 |
| D17S1793 | 40.4 | 193/193 | 193/197 |
| D17S932 | 41.2 | 193/193 | 187/197 |
| D17S1789 | 41.7 | 179/194 | 194/194 |
| rs25646 | 42.4 | C/T | T/T |
| D17S930 | 42.7 | 101/105 | 101/103 |
| D17S1804 | 43.0 | 236/236 | 230/232 |
| D17S931 | 45.0 | 89/97 | 95/97 |
| D17S958 | 46.2 | 94/94 | 98/98 |
Clinical and Neuropathologic Features
The first patient with the IVS6+5_8delGTGA deletion was a female who at the age of 73 years presented with memory disturbances. Analyses with single photon emission-computed tomography and magnetic resonance imaging (MRI) showed decreased perfusion, especially of the right frontal and parietal lobes, and also atrophy of the same regions. Within 2 years the patient developed Parkinsonian symptoms such as gait impairment and tremor. After 4 years the patient was suffering from a severe dementia with muteness and severe Parkinsonism. The disease duration was approximately 7 years. The family disease history was positive, with both the patient’s mother and 1 of her siblings having suffered from a late-onset type of dementia. Neuropathologically this case displayed a marked atrophy of the frontal lobe with lesser involvement of temporal and parietal lobes. There was a moderate to advanced neuronal loss and severe gliosis in affected areas. Immunostaining for ubiquitin and TDP-43 showed numerous cells, primarily in the dentate gyrus and the superficial layers of the parahippocampal gyrus, displaying both cytoplasmic and intranuclear deposits (Fig. 3A). Virtually no tau-immunoreactivity and no amyloid-β plaques were detected.
FIGURE 3.
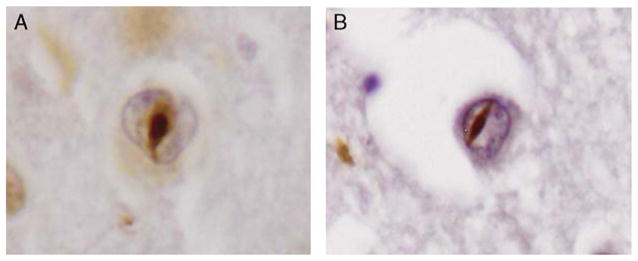
A, TDP-43-positive neuronal intranuclear inclusion in the superficial temporal cortex of the first case with the IVS6+5_8delGTGA mutation and (B) the second case carrying the IVS6+5_8delGTGA mutation.
The second patient with the IVS6+5_8delGTGA deletion was a female who at the age of 65 years began to have word finding difficulties and spatial disorientation. A computed tomography and a subsequent MRI displayed asymmetric left cortical atrophy. With increased disease duration, the patient started to show increasing irritability, a flattened affect in addition to other personality changes. After 4 years the patient became incontinent, mute, and suffered from increased muscle tone but no clear Parkinsonian features. The disease duration was 9 years. There was no reported family history of the disease. Neuropathologically, this case described an advanced atrophy of the frontal and temporal lobes. Neuronal loss and gliosis was accompanied by cortical spongiosis and white matter degeneration. Also this case displayed numerous ubiquitin and TDP-43-positive inclusions, both in the cytoplasm and in the nuclei of neurons in the dentate gyrus and the superficial layers of the parahippocampal gyrus (Fig. 3B). Only very sparse neurofibrillary pathology and rare amyloid-β plaques could be visualized.
DISCUSSION
In this study, we set out to examine the prevalence of PGRN mutations in a group of 51 FTLD patients found negative for MAPT mutations. The genetic analysis of PGRN showed a novel frameshift mutation, IVS6+5_8-delGTGA mutation that was identified in 2 FTD patients. In addition, the earlier reported Gln130fs mutation was found in another FTD patient. The identification of 3 pathogenic mutations in 51 samples corresponds to a PGRN mutation frequency of 5.9%, which is comparable to earlier studies.17,18,23,24 Most PGRN mutations described create premature termination codons in the mRNA sequence. Mutant PGRN mRNA is most likely subsequently degraded by NMD causing reduced levels of PGRN transcripts and protein.16,17 Both the PGRN mutations identified in the present investigation create premature termination codons, which are predicted to result in NMD of mutant mRNA and ultimately reduced levels of PGRN protein.
The IVS6+5_8delGTGA deletion is located in the splice donor site of exon 6. Altering this splice site could result in skipping of exon 6 and a premature termination codon in exon 7 when spliced onto exon 5 (Val200-GlyfsX18). Retention of the intron following exon 6 is another possible outcome that would create a premature termination codon in the retained intron (Ala237ValfsX17). We see evidence of both these transcripts in a brain cDNA sample from a mutation carrier although only in minor amounts, as a majority of these transcripts have most likely been subjected to NMD. The reduction of mutant PGRN brain mRNA was supported by qPCR although the difference in total PGRN levels between the IVS6+5_8-delGTGA deletion carrier and a healthy control was quite modest. However, the detection of decreased levels of PGRN brain mRNA owing to NMD could be complicated by inflammatory processes in the brain and an upregulation of PGRN expression in activated microglia.16,25 A cDNA sequence analysis of a synonymous polymorphism within the PGRN exon 4 in the mutation carrier confirms, however, that the wild-type allele represents the major transcript and that the mutated transcript levels are very low. Owing to the lack of additional tissue samples or cell lines from the IVS6+5_8delGTGA mutation cases, we were unable to investigate the stability of the aberrant transcripts or the potential presence of low levels of truncated protein. It is therefore at the moment difficult to determine the significance, if any, of these splice variant transcripts. As there was no tissue sample available for RNA extraction from the second IVS6+5_8delGTGA mutation carrier, we were unable to conduct any mRNA analysis in this individual. We could therefore not confirm the reduced levels of the mutated transcript or the alternative splicing effects seen in the first patient. However, the lower levels of PGRN transcripts observed in the first individual are consistent with observations from a number of earlier described PGRN mutations.
The mechanism by which a loss of PGRN leads to neurodegeneration is still unclear. In the central nervous system, PGRN is expressed in cerebellar Purkinje cells, pyramidal cells of the hippocampus, and to a certain extent also in neurons of the cerebral cortex.26 However, little is known about the normal function of PGRN in the central nervous system. A recent study suggests PGRN to function as a neurotrophic factor important for neuronal survival.27 PGRN has also been suggested to play a role in neuroinflammation and microglial activation.16,25,28,29 A loss of functional PGRN could therefore affect neuronal survival and glial inflammatory processes in the brain.
In conclusion, our genetic analysis of 51 FTLD patients showed a novel PGRN frameshift mutation, the IVS6+5_8delGTGA deletion in addition to the earlier reported Gln130fs mutation. Our analyses show that both mutations lead to a reduction of PGRN mRNA, most likely through NMD of the mutant mRNA and resulting PGRN haploinsufficiency, as earlier shown for other pathogenic PGRN mutations.
Acknowledgments
This work was supported by the Swedish Research Council, the Swedish Brain Foundation, Stohne’s Foundation, the Swedish Alzheimer Foundation, the Swedish Dementia Association, and the Swedish Society of Medicine.
References
- 1.Ratnavalli E, Brayne C, Dawson K, et al. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. doi: 10.1212/wnl.58.11.1615. [DOI] [PubMed] [Google Scholar]
- 2.Harvey RJ, Skelton-Robinson M, Rossor MN. The prevalence and causes of dementia in people under the age of 65 years. J Neurol Neurosurg Psychiatry. 2003;74:1206–1209. doi: 10.1136/jnnp.74.9.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ikeda M, Ishikawa T, Tanabe H. Epidemiology of frontotemporal lobar degeneration. Dement Geriatr Cogn Disord. 2004;17:265–268. doi: 10.1159/000077151. [DOI] [PubMed] [Google Scholar]
- 4.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 5.Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathologica. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taniguchi S, McDonagh AM, Pickering-Brown SM, et al. The neuropathology of frontotemporal lobar degeneration with respect to the cytological and biochemical characteristics of tau protein. Neuropathol Appl Neurobiol. 2004;30:1–18. doi: 10.1046/j.0305-1846.2003.00481.x. [DOI] [PubMed] [Google Scholar]
- 7.Lipton AM, White CL, III, Bigio EH. Frontotemporal lobar degeneration with motor neuron disease-type inclusions pre-dominates in 76 cases of frontotemporal degeneration. Acta Neuropathol (Berl) 2004;108:379–385. doi: 10.1007/s00401-004-0900-9. [DOI] [PubMed] [Google Scholar]
- 8.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 9.Stevens M, van Duijn CM, Kamphorst W, et al. Familial aggregation in frontotemporal dementia. Neurology. 1998;50:1541–1545. doi: 10.1212/wnl.50.6.1541. [DOI] [PubMed] [Google Scholar]
- 10.Rosso SM, Donker Kaat L, Baks T, et al. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126(Pt 9):2016–2022. doi: 10.1093/brain/awg204. [DOI] [PubMed] [Google Scholar]
- 11.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 12.Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 13.Spillantini MG, Murrell JR, Goedert M, et al. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skibinski G, Parkinson NJ, Brown JM, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37:806–808. doi: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- 15.Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 16.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 17.Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 18.Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- 19.Gijselinck I, Van Broeckhoven C, Cruts M. Granulin mutations associated with frontotemporal lobar degeneration and related disorders: an update. Human Mutation. 2008;29:1373–1386. doi: 10.1002/humu.20785. [DOI] [PubMed] [Google Scholar]
- 20.Gijselinck I, van der Zee J, Engelborghs S, et al. Progranulin locus deletion in frontotemporal dementia. Hum Mutat. 2008;29:53–58. doi: 10.1002/humu.20651. [DOI] [PubMed] [Google Scholar]
- 21.Rovelet-Lecrux A, Deramecourt V, Legallic S, et al. Deletion of the progranulin gene in patients with frontotemporal lobar degeneration or Parkinson disease. Neurobiology of Disease. 2008;31:41–45. doi: 10.1016/j.nbd.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 22.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Research. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le Ber I, van der Zee J, Hannequin D, et al. Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat. 2007;28:846–855. doi: 10.1002/humu.20520. [DOI] [PubMed] [Google Scholar]
- 24.Pickering-Brown SM, Rollinson S, Du Plessis D, et al. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain. 2008;131(Pt 3):721–731. doi: 10.1093/brain/awm331. [DOI] [PubMed] [Google Scholar]
- 25.Mukherjee O, Pastor P, Cairns NJ, et al. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol. 2006;60:314–322. doi: 10.1002/ana.20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daniel R, He Z, Carmichael KP, et al. Cellular localization of gene expression for progranulin. J Histochem Cytochem. 2000;48:999–1009. doi: 10.1177/002215540004800713. [DOI] [PubMed] [Google Scholar]
- 27.Van Damme P, Van Hoecke A, Lambrechts D, et al. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41. doi: 10.1083/jcb.200712039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker CA, Manuelidis L. Unique inflammatory RNA profiles of microglia in Creutzfeldt-Jakob disease. Proc Natl Acad Sci U S A. 2003;100:675–679. doi: 10.1073/pnas.0237313100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malaspina A, Kaushik N, de Belleroche J. Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J Neurochem. 2001;77:132–145. doi: 10.1046/j.1471-4159.2001.t01-1-00231.x. [DOI] [PubMed] [Google Scholar]