

Abstract
Murine macrophages are activated by interferon-γ (IFNγ) and/or TLR agonists such as bacterial endotoxin (LPS) to express an inflammatory (M1) phenotype characterized by expression of nitric oxide synthase-2 (iNOS) and inflammatory cytokines such as TNF-α and IL-12. In contrast, Th2 cytokines IL-4 and IL-13 activate macrophages by inducing expression of arginase-1 and the anti-inflammatory cytokine IL-10 in an IL-4 receptor-α (IL-4Rα) dependent manner. Macrophages activated in this way are designated as “alternatively activated” (M2a) macrophages. We have shown previously that adenosine A2A receptor (A2AR) agonists act synergistically with TLR2, 4, 7 and 9 agonists to switch macrophages into an “M2-like” phenotype that we have termed “M2d”. Adenosine signaling suppresses TLR-dependent expression of TNF-α, IL-12, IFN-γ and several other inflammatory cytokines by macrophages, and induces expression of VEGF and IL-10. We show here using mice lacking a functional IL-4Rα gene (IL-4Rα−/− mice) that this adenosine-mediated switch does not require IL-4Rα-dependent signaling. M2d macrophages express high levels of VEGF, IL-10 and iNOS, low levels of TNF-α and IL-12, and mildly elevated levels of arginase-1. In contrast, M2d macrophages do not express Ym1, Fizz1 (RELM-α) or CD206 at levels greater than those induced by LPS, and dectin-1 expression is suppressed. Use of these markers in vivo to identify “M2” macrophages thus provides an incomplete picture of macrophage functional status and should be viewed with caution.
Keywords: Macrophage, Alternative Activation, IL4Rα, Adenosine Receptor, TLR
Introduction
Macrophages play key roles in regulating inflammation, wound healing and innate and acquired immunity [1–4]. They are professional phagocytes equipped with cell surface and cytosolic receptors that enable them to recognize, phagocytize and destroy foreign organisms, as well as dead or damaged host cells and tissue components [4]. In response to a variety of stimuli, macrophages produce select arrays of products that mediate various aspects of inflammation, wound healing and innate and acquired immunity [5]. Expression of these mediators is closely regulated by micro-environmental influences [5, 6]. Macrophages are recruited from precursor monocytes in the circulation, which in turn derive from stem cells in the embryo and bone marrow [5, 7]. While distinct sub-populations of monocytes may exist, once recruited to a site of injury or infection, these monocytes differentiate into macrophages, and their expression of cell surface receptors, cytokines, growth and angiogenic factors and other mediators of inflammation and repair is regulated by micro-environmental signals at the site of recruitment [5–7].
Macrophage activation has been classified recently into “classical” and “alternative” activation pathways [5–7]. Classical activation is induced by IFN-γ with a variety of TLR agonists or by these agonists alone, and is canonically described as featuring strong expression of inflammatory cytokines such as TNF-α, IL-1β, IL-6 and IL-12, stimulation of the respiratory burst through activation of membrane oxidases, and induction of iNOS expression. Macrophages activated in this way have been termed M1 macrophages [5, 8]. “Alternative” activation is canonically defined as that induced by IL-4 and/or IL-13 through activation of the common IL4Rα subunit of the receptors for these cytokines [1]. These “alternatively activated” macrophages have been termed M2 macrophages, and are characterized in the mouse by elevated expression of anti-inflammatory cytokines and by elevated expression of CD206, Ym1, Fizz1 (RELM-α), dectin and arginase-1 [5, 8–11]. These markers, in conjunction with the expression of the pan-macrophage markers F4/80 and CD68, have been commonly used in mouse studies to identify M1 versus M2 macrophage populations. While this broad classification, which bears parallels with the Th1/Th2 paradigm for activation of the immune system, has some utility, it has become clear recently that there are other modes for regulation of macrophage phenotype, and the broad use of the M2 classification may no longer be adequate. Additional sub-classification of M2 macrophages into M2a, M2b and M2c categories has been proposed. M2a refers to IL4Rα;-dependent activation; M2b refers to activation by immune complexes with or without TLR agonists; M2c refers to activation by TGFβ [12]. We have described an activation paradigm that differs from these prototypical M1 and M2 pathways, that involves activation by TLR agonists in the presence of adenosine A2A receptor (A2AR) agonists [13]. This pathway switches macrophages from an M1 phenotype to an angiogenic “M2-like” phenotype that we have termed M2d [14]. The classical M2 (M2a) pathway is IL4Rα-dependent, and the IL-4Rα-dependent markers CD206, Ym1, Fizz1, dectin-1 and arginase-1 have been used in several studies to indicate the presence of M2 macrophages, In this study we examined the phenotype of M2d macrophages to determine 1) the expression levels of the canonical M2 markers CD206, Ym1, Fizz1, dectin-1 and arginase-1, and 2) whether the induction of the M2d phenotype requires signaling through the IL4Rα signaling pathway.
Materials and Methods
Preparation of Macrophages
Mouse peritoneal macrophages were harvested as previously described [13]. Briefly, 7–12 week old male mice were injected intraperitoneally with 2.5 ml of 4% thioglycolate broth (DIFCO, Detroit, MI). Four days post-injection mice were sacrificed by cervical dislocation and peritoneal macrophages were harvested and cultured as a monolayer in RPMI 1640 medium (Cellgro, Mediatech Inc., Herndon, VA), supplemented with 10% fetal bovine serum (Gemini Bio-Products, Calabasas, CA), 2 mM L-glutamine, 100 IU/ml Penicillin, and 100 μg/ml Streptomycin (Irvine Scientific, Santa Ana, CA). Monolayers were washed 4 hours after plating to remove any non-adherent cells. The cultures were shown to be >98% pure macrophages, as assessed by non-specific esterase staining and staining with the macrophage-specific F4/80 mAb. The following strains of mice were used for these experiments: IL4Rα mice homozygous for the Il4ra tm1Sz targeted mutation (IL-4Rα−/− mice) on the BALB/c background (BALB/c-IL4ratm1Sz/J) were from Jackson Labs (Bar Harbor, ME, USA). Wild-type BALB/c mice were also from Jackson Labs.
Extraction of RNA and Q-RT-PCR
Mouse peritoneal macrophages were plated in 60 mm dishes at a density of 5–10 × 106 cells per dish. Following overnight incubation, cells were treated with various reagents as indicated in the figure legends. RNA was isolated using Trizol reagent (Invitrogen Corp., Carlsbad, CA, USA), according to the manufacturer’s protocol for isolation of total RNA from animal cells. cDNA was synthesized from 1μg total RNA using TaqMan RT reagents (Applied Biosystems, Foster City, CA, USA) following the manufacturer’s instructions. Real-time quantitative PCRs were performed in an ABI Prism 7500 sequence detector using 1/10 vol of each cDNA reaction and TaqMan Universal PCR Master Mix. TaqMan gene expression assays for murine AR (Mm00802075_m1) and murine cyclophilin D (Mm00835365_g1) were purchased from Applied Biosystems. Real-time PCR reactions were carried out using the manufacturer’s protocol and mRNA levels were determined using both relative and absolute quantification. PCR reactions were performed in duplicate and expression levels for the transcripts of interest were normalized to that of endogenous cyclophilin D. Prior studies have indicated that cyclophilin D expression levels show minimal variation under the various activation conditions used in these experiments. Data were calculated as fold expression relative to the average of the untreated control group.
Flow cytometry analysis of macrophage marker expression
FACS analysis was used to determine the expression levels of various macrophage markers. Macrophages were incubated at 4°C for 20–30 minutes to allow detachment from UpCell™ culture dishes (Nunc). UpCell™ dishes are coated with a polymer that shifts from hydrophobic to hydrophilic below 32°C, thereby facilitating macrophage detachment without chemical or mechanical manipulation. Macrophages were pelleted at 400g at 4°C for 15 minutes, washed twice with sterile PBS(-Ca2+/Mg2+), and resuspended at a final concentration of 1 × 106 cells per 100μl of 5%FBS-PBS(-Ca2+/Mg2+). Resuspended macrophages were first incubated in darkness at 4°C for 30 minutes with 1μg anti-mouse CD16/32 (eBioscience), 4μl Pacific Blue-conjugated anti-mouse F4/80 (AbD Serotec), 4μl AlexaFluor 647-conjugated anti-mouse CD206 (AbD Serotec), 2.5 μl PE-conjugated anti-mouse CD124/IL4Rα (Becton Dickinson), and 4 μl RPE-conjugated anti-mouse Dectin-1 (AbD Serotec). Macrophages were washed three times with 1% FBS-PBS, resuspended in 100 μl fixation buffer (eBioscience), and incubated in darkness at room temperature for 20 minutes. After fixation, macrophages were washed three times with permeabilization buffer (eBioscience), resuspended in 100 μl permeabilization buffer, and incubated in darkness at room temperature for 20 minutes with 4 μl AlexaFluor 700-conjugated anti-mouse CD68 (AbD Serotec), 2.5 μl rabbit polyclonal anti-mouse Ym1 (Stem Cell Technologies), and 2.5 μl rabbit polyclonal anti-mouse RELM-α (Fizz1) (Abcam). Macrophages were washed three times with 1% FBS-PBS, resuspended in 100 μl permeabilization buffer and incubated in darkness at room temperature for 20 minutes with 1.5 μl AlexaFluor 488-conjugated anti-rabbit IgG (1:100 dilution, Invitrogen). Macrophages were washed three times with 1% FBS-PBS and resuspended in 1% FBS-PBS. Samples were analyzed using an LSR II flow cytometer (Becton Dickinson) and FlowJo software (Treestar Inc., Ashland, OR).
Cytokine Assays
Macrophages were plated in 12-well plates at a density of 0.5 × 106 cells per well. Post-treatment, macrophage-conditioned media was collected and levels of TNF-α (eBioscience), a typical M1 cytokine, and VEGF (R&D Systems), a typical M2d cytokine, were quantified by ELISA performed according to the manufacturers’ instructions. Samples were assayed in duplicate and normalized to relative cell number as determined by MTT assay. Levels of other cytokines in the conditioned media were analyzed in duplicate using the premixed 32-plex Milliplex™ MAP Mouse Cytokine/Chemokine Immunoassay kit (Millipore Corporation, Billerica, MA, USA) following the recommended protocol for tissue culture supernatant. Fluorescence was measured using the Luminex 100 xMAP technology system (Luminex Corporation, Austin, TX) and data were analyzed in Luminex 100 Integrated System Software 2.3 (Luminex Corporation, Austin, TX) using a four-parameter logistic curve fit.
Statistical Analyses
Values in the figures are expressed as means ± SD. Statistical analysis of the data were performed using Student’s t-test or one-way analysis of variance followed by Dunnett’s test as appropriate.
Results
IL4Rα signaling is not required for M2d macrophage polarization
To examine the contribution of IL-4Rα signaling to M2d macrophage polarization, we first examined cytokine and growth factor secretion by macrophages from wild-type and IL-4Rα−/− mice (Figures 1 and 2). Stimulation of macrophages by LPS strongly induced secretion of TNF-α, a typical pro-inflammatory cytokine, in both wild-type and IL-4Rα−/− mice (Figure 1A). In contrast, treatment of WT and IL-4Rα−/− macrophages concurrently with LPS and the non-specific A2A/A2BR agonist 5′-N-ethyl-carboxamido-adenosine (NECA) significantly reduced TNF-α secretion. IL-4 alone did not induce TNFα expression by WT or IL4Rα−/− macrophages; IL-4 treatment of WT macrophages together with LPS markedly suppressed TNFα expression by WT, but not by IL4Rα−/− macrophages. Treatment of macrophages from wild-type and IL-4Rα−/− mice with LPS alone had little effect on VEGF expression, but LPS together with NECA synergistically up-regulated VEGF expression in both cell types. These results clearly eliminate IL-4Rα signaling as a requirement for M2d macrophage polarization.
Figure 1. Comparison of M2a (IL-4) and M2d (LPS/NECA) activation on the production of (A) TNFα and (B) VEGF by murine peritoneal macrophages.

Thioglycollate-elicited macrophages from wild type (WT) BALB/c and IL-4Rα−/− mice were treated for 18 hours with LPS (100ng/ml), NECA (1μM), LPS/NECA, IL-4 (10ng/ml) and IL-4 with LPS, NECA and LPS/NECA. Cytokine levels were assayed using ELISA kits as described in Methods. Data indicate the mean ± SD of triplicate samples from a representative experiment. * P<0.01 for WT vs IL-4Rα−/− macrophages treated with IL-4/LPS; # P<0.05 for WT vs IL-4Rα−/− macrophages treated with IL-4/LPS/NECA.
Figure 2. Analysis of cytokine/chemokine production by wild type (WT) BALB/c and IL-4Rα−/− murine peritoneal macrophages.

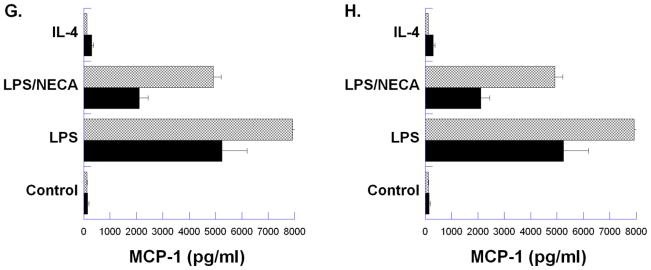
Thioglycollate-elicited macrophages from BALB/c and IL-4Rα−/− mice were treated for 18 hours with LPS (100ng/ml), LPS with NECA (1μM), or IL-4 (10ng/ml). Cytokine/chemokine levels were assayed from three replicate samples from each condition, and each sample was assayed in triplicate using Milliplex™ MAP immunoassays. Mean ± SD for each sample is presented.
Further analysis of cytokine production in wild-type and IL-4Rα−/− macrophages using Luminex technology (Figure 2) showed that several cytokines in addition to TNFα and VEGF were modulated by the adenosine receptor agonist NECA. Expression of IFN-γ, IL-12, IL-1α, IL-1β, MIP-1β and MCP-1, which are generally associated with a pro-inflammatory state, was markedly suppressed when LPS-treated macrophages were compared to macrophages treated in conjunction with NECA. IL-10 and G-CSF, on the other hand, that are generally considered to be anti-inflammatory, were increased. Overall, changes in cytokine release during induction of the M2d phenotype were similar in both WT and IL-4Rα−/− macrophages, further validating the independence of the M2d phenotype from the M2a IL-4Rα-dependent signaling. However, a notable difference in the expression of IFN-γ between WT and IL4Rα−/− macrophages was seen (Figure 2B). IFN-γ is generally considered a signature cytokine of Th1 cells. It is thus interesting that macrophages from IL-4Rα−/− mice expressed lower levels of IFN-γ in response to LPS than WT macrophages (Figure 2B). IFN-γ expression by WT macrophages was also strongly suppressed by NECA treatment.
Expression of M2a macrophage markers differs from that of M2d macrophages
Alternative activation of murine macrophages (IL-4Rα-dependent), M2a) has been shown in prior studies to enhance expression of several genes now considered canonical markers for the M2 phenotype, including arginase-1, Ym1, Fizz1, CD206 and dectin-1. Expression of these markers has been studied on murine macrophage populations in several in vivo models of inflammation and has been taken to indicate the presence of “alternatively activated” M2 macrophages. We have examined the in vitro expression of these markers, as well as of iNOS, a typical marker of M1 macrophage polarization, by peritoneal macrophages treated to induce M1, M2a, or M2d polarization (Figure 3–5).
Figure 3. Comparison of canonical M1 and M2 marker expression by M2a and M2d murine peritoneal macrophages.

Thioglycollate-elicited macrophages from BALB/c (WT) and IL-4Rα−/− mice were treated for 6 hours with LPS (100ng/ml), NECA (1μM) or IL-4 (10ng/ml), alone and in combination, as indicated. mRNA levels of the M1 marker iNOS (A) and the canonical murine M2 marker Arg1 (B) were determined using qRT-PCR. Data indicate the relative levels of mRNA from three representative experiments.
Figure 5. Analysis of the expression of M2 markers (Ym1, Fizz1 and CD206) by M2a and M2d murine peritoneal macrophages.

Thioglycollate-elicited macrophages from BALB/c (WT) and IL-4Rα−/− mice were treated for 6 hours with LPS (100ng/ml), NECA (1μM) or IL-4 (10ng/ml), alone and in combination, as indicated. mRNA levels of canonical M2 markers Ym1 (A), Fizz1 (B), and CD206 (C) were evaluated using qRT-PCR. Data indicate the mean relative levels of mRNA from three independent experiments. Each sample was analyzed in duplicate.
iNOS
Treatment with LPS alone dramatically elevated mRNA levels of the M1 macrophage marker iNOS and this level was augmented by concurrent treatment with NECA (Figure 3A). This pattern was also observed in IL-4Rα−/− macrophages, and was maintained in WT macrophages even with the presence of IL-4 in the external milieu.
Arginase
Increased arginase mRNA expression was observed in M2a macrophages compared to M2d-polarized macrophages (Figure 3B). Stimulation of adenosine receptors with NECA, in conjunction with IL-4 to induce the M2a phenotype, markedly increased arginase-1 expression over that induced by IL-4 alone, as we have reported previously [15]. This increase is unlike the expression patterns observed with Ym1, Fizz1 and CD206 (see below). Interestingly, activation of TLR receptor signaling by LPS in the presence of IL-4 abrogated the increased arginase mRNA levels seen with IL-4 treatment alone, Stimulation of IL-4/NECA-treated cells with LPS also suppressed arginase-1 expression, although to a lesser extent than that induced in cells treated with IL-4 alone. The effects observed in WT cells in the presence of IL-4R activation were absent in IL-4Rα−/− macrophages.
Ym1/Fizz1/CD206
Markedly different expression patterns of M2 markers were observed in M2a- and M2d-polarized peritoneal macrophages (Figures 4 and 5). Ym1 protein and mRNA expression was increased in WT but not IL-4Rα−/− macrophages upon induction of the M2a phenotype by IL-4; however, in response to M2d stimulation (LPS/NECA), Ym1 protein and mRNA levels remained unchanged in both WT and IL-4Rα−/− macrophages. While changes in Fizz1 and CD206 were not observable at the protein level by flow cytometry (data not shown), mRNA levels of these M2 markers demonstrated the same pattern as seen with Ym1 (Figure 5B and C). Furthermore, as observed with the cytokines TNF-α and VEGF, treatment with IL-4 had no effect on the protein and mRNA levels of these markers in M2d macrophages.
Figure 4. Flow Cytometry (FACS) comparison of the expression of Ym1, Dectin-1 and IL-4Rα by M2a (IL-4) and M2d (LPS/NECA) murine peritoneal macrophages.

A. Representative FACS analysis of thioglycollate-elicited macrophages from BALB/c (WT, left panels) and IL-4Rα−/− (IL-4Rα−/−, right panels) mice treated for 18 hours with LPS (100ng/ml) and NECA (1μM) (black), IL-4 (10ng/ml) (red), or a combination of all three (black dotted), as described in Methods. Cells were analyzed for expression of Ym1 (upper panels), Dectin-1 (middle panels), and IL-4Rα (lower panels). Shaded histograms represent marker expression in untreated controls. B. Analysis of Ym1, dectin-1 and IL-4Rα expression determined by FACS in treated vs untreated macrophages, Results indicate mean fold differences of median fluorescence values by treated macrophages vs controls from 3 independent experiments,
Dectin
Both wild-type and IL-4Rα−/− macrophages treated with LPS and NECA to induce the M2d phenotype exhibited significantly down-regulated surface expression of the M2 marker dectin-1 (Figure 4). In contrast, wild-type but not IL-4Rα−/− macrophages, polarized toward the M2a phenotype by exposure to IL-4, exhibited a small increase in dectin surface expression. Treatment of wild-type macrophages with a combination of LPS, NECA and IL-4 resulted in dectin-1 surface expression that mirrored that seen upon stimulation with LPS and NECA alone.
Regulation of IL-4Rα expression differs in M2a- and M2d-polarized macrophages
Flow cytometric analysis of IL-4Rα surface expression in M2a and M2d macrophages revealed yet another divergence in the protein expression profile of these distinctively polarized macrophages. While stimulation of WT macrophages with IL-4 led to a substantial abatement of IL-4Rα on the cell surface, induction of the M2d phenotype with LPS and NECA increased IL-4Rα surface expression (Figure 4). IL-4Rα surface expression in response to concurrent stimulation with LPS, NECA and IL-4 mimicked that observed in M2a polarization, i.e. down-regulation of surface dectin expression (Figure 4). As expected, IL-4Rα−− macrophages demonstrated no surface IL-4Rα expression (Figure 4).
Regulation of A2AR expression is similar in WT and IL-4Rα−/− macrophage activation
Previous studies have shown the up-regulation of A2ARs in macrophages activated by LPS stimulation [16–19]. This up-regulation is believed to sensitize macrophages to adenosine signaling by priming M1 macrophages to respond to adenosine and thus switching toward the M2d phenotype. As shown in Figure 6, LPS activation of both WT and IL-4Rα−/− macrophages strongly induced A2AR expression, demonstrating that the induction of A2ARs by LPS is independent of IL-4Rα signaling.
Figure 6. Analysis of adenosine A2A receptor (A2AR) expression in wild type (Balb-c) and IL-4Rα−/− mouse peritoneal macrophages.

Thioglycollate-elicited macrophages from BALB/c (WT) and IL4Rα;−/− mice were treated for 6 hours with LPS (100ng/ml), NECA (1 μM) and IL4 (10ng/ml) alone or in combination. mRNA levels of A2AR were determined using qRT-PCR. Data presented are the mean relative levels of A2AR mRNA from three independent experiments. Each sample was analyzed in duplicate.
Discussion
Macrophages comprise a diverse population of cells and demonstrate remarkable functional plasticity in response to dynamic micro-environmental cues. “Classical” (M1) activation of macrophages is induced by TLR agonists, either with or without the Th1 cytokine IFNγ, and results in an inflammatory phenotype characterized by expression of a series of inflammatory cytokines and chemokines, including TNFα, IL-1β, IL-6, MIP1α and IL-12 [5, 8]. “Alternatively activated” (M2) macrophages were originally described as induced by the Th2 cytokines IL-4 and IL-13 through the IL-4Rα subunit common to the receptors for these cytokines, leading to the expression of an anti-inflammatory and wound healing phenotype [1, 5, 8]. This IL-4Rα-dependent phenotype has been termed “M2a” [12]. Several recent studies have examined macrophage sub-populations in vivo using markers of the M2a phenotype to distinguish M1 from M2 macrophages [3, 20–24].
We have proposed a model for the induction of an M2-like macrophage phenotype that we have termed “M2d” [14]. This model involves the initial activation of macrophages by TLR agonists, which strongly induce expression of adenosine A2A and A2B receptors (A2ARs and A2BRs), thus priming these macrophages to respond to elevated levels of extracellular adenosine [16, 17, 19]. Adenosine is a metabolite produced by cells in response to stress by breakdown of ATP [25]. ATP is normally present at millimolar concentrations in cells, but in response to stress, including hypoxia and ischemia, ATP is broken down both intra-cellularly, and following release from cells by ectonucleotidases and ectonucleosidases (CD73 and CD39), to generate adenosine [25]. Intracellular and extracellular adenosine levels are equilibrated by membrane transporters, and extracellular steady state concentrations in response to stress can rise from low nano-molar to micro-molar [25]. Adenosine then stimulates cells through ligation of adenosine receptors, which are a family of four cell surface 7-transmembrane receptors (A1R, A2AR, A2BR, and A3) [26]. High extracellular adenosine levels can also be generated as a result of adenosine release from apoptotic and necrotic cells, which are a prominent component of the early wound healing environment [27, 28]. We have shown that adenosine stimulation of TLR-activated macrophages results in a switch from the production of inflammatory cytokines such as TNFα and IL-12, to the production of anti-inflammatory and angiogenic factors, including IL-10 and VEGF [13, 29]. This model thus provides a sequential pathway whereby macrophages initially mediate inflammation through TLR-dependent activation to an M1 phenoptype, but are then switched into an M2d anti-inflammatory and angiogenic phenotype by adenosine generated in response to hypoxia/ ischemia within the wound area. M2d macrophages, unlike previously described subsets of alternatively activated macrophages, are induced by “switching” from a classically activated inflammatory phenotype to an alternatively activated anti-inflammatory/pro-angiogenic phenotype.
We have also shown recently that adenosine signaling, through both A2ARs and A2BRs, promotes the effects of IL-4 and IL-13 in the induction of M2a activation, by augmenting the expression of alternatively activated macrophage markers including arginase-1 and MGL1/CD301[15]. The studies presented here indicate that the M2d phenotypic switch mediated by TLRs with adenosine receptors is activated independently of the canonical IL-4Rα/STAT6 “alternative activation” pathway, and that macrophages activated in this way do not display the same marker profile reported previously for “alternatively activated” M2a macrophages. M2a activation is induced by signaling through the IL-4Rα/STAT6 pathway, does not involve TLR signaling, and is promoted by adenosine signaling [1, 6, 15, 30]. Here we show that macrophages from mice lacking IL-4Rα (IL-4Rα−/− mice) respond to the TLR4 agonist LPS in a similar manner to those from wild type mice, in terms of their expression of TNFα and several other inflammatory cytokines, as well as in their strong up-regulation of expression of A2ARs 19, [31, 32]. IL-4Rα−/− macrophages also respond to adenosine receptor stimulation by switching from the expression of TNFα and IL-12 to expressing VEGF and IL-10 in a manner akin to that of wild type macrophages. Previous studies from our lab have shown that this pathway is MyD88, IRAK4 and TRAF6-dependent, and involves regulation of NF-κB [29, 33, 34]. The adenosine signaling involved in this pathway modulates PLCβ2 expression and signaling rather than Gsα-dependent regulation of cAMP, and involves induction of HIF1α expression and stabilization in a hypoxia-independent manner [34, 35]. IL-4 and IFN-γ are the canonical cytokines of Th2 and Th1 responses respectively. LPS-stimulated macrophages from IL-4Rα−/− mice express lower levels of IFN-γ than WT macrophages, indicative of their potential role in Th2 responses. In addition, IFN-γ expression by LPS-treated WT macrophages is suppressed by NECA (Figure 2B), also suggestive of a switch to a Th2 phenotype.
Since the adenosine-dependent switch of murine macrophages to the M2d phenotype is independent of IL-4Rα signaling, we investigated whether the expression of the canonical M2a markers Ym1, Fizz1, CD206, dectin-1 and arginase-1 is similar in M2a and M2d activation. Our results clearly indicate that while these markers might indicate the presence of the IL-4Rα-dependent M2a macrophages in vivo, lack of expression of these markers does not indicate that populations of macrophages with an M2-like anti-inflammatory and angiogenic phenotype are absent. Arginase, Ym1, Fizz1, CD206, dectin and arginase-1 are markers of murine M2a macrophages, and M2d macrophages are not identified on the basis of this marker set [5, 8–11]. In contrast, expression of A2ARs and expression of VEGF and IL-10 are features of M2d macrophages [13, 29, 31, 32]. Expression of adenosine receptors has been demonstrated in macrophages in vitro using both ligand binding assays and Q-RT-PCR [16, 29]. Unfortunately, currently available antibodies to these adenosine receptors are not adequate for the identification of murine M2d macrophages either by FACS or immunohistochemistry, as they lack the required sensitivity. Further analysis of the expression of cellular markers to enable the identification of M2d macrophages in vivo is clearly required. These studies are currently underway.
Macrophage function has been shown to be integral to proper tissue repair in a number of injury models, and temporal changes in macrophage phenotype have been documented during this process [3, 36–38]. The functional phenotype of macrophages changes from the initial stage of wound debridement, through the proliferative phase of repair involving fibroblast migration, proliferation, and angiogenesis, to the later stages involving resolution of inflammation, maturation of connective tissue, regression of neo-vascular blood vessels, and scar formation [3, 36–39] At the outset of the healing process, classically activated (M1) macrophages predominate, suggesting a role in the clearance of apoptotic and necrotic tissue from the injured area. As healing progresses, the population shifts towards alternatively activated (M2), macrophages, suggesting a switch to the promotion of granulation tissue formation as well as to the attenuation of the inflammatory phase. In particular, macrophages are central to the development and stabilization of new blood vessels in the injured area [39]. Macrophage polarization is largely determined by local environmental signals, and dysregulation of this process severely compromises tissue repair in vivo. While the micro-environmental signals governing this process have remained largely elusive, TLR signaling has been linked to initiation of the inflammatory response in numerous injury models. Both endogenous and exogenous TLR agonists induce “classical” M1 activation of macrophages, and may play a role in wound healing [40]. The presence of “alternatively activated” M2 macrophages has been observed during the repair process; the signals involved in the induction of this phenotype have however, not been defined [41, 42]. Significantly, macrophages expressing an M2 phenotype have been observed even in the absence of IL-4Rα/STAT6 signaling [42]. This finding strongly suggests that factors that are independent of IL-4Rα signaling play a role in the induction of multiple M2 phenotypes during tissue repair.
Acknowledgments
Supported by grants from the U.S. Public Health Service (RO1-GM068636) to SJL and RO1-GM066189 to GH.
References
- 1.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 2.Leibovich SJ, Ross R. The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol. 1975;78:71–100. [PMC free article] [PubMed] [Google Scholar]
- 3.Lucas T, et al. Differential roles of macrophages in diverse phases of skin repair. J Immunol. 2010;184:3964–3977. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 4.Galli SJ, et al. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol. 2011;12:1035–1044. doi: 10.1038/ni.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 7.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 8.Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 9.Stein M, et al. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–292. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Willment JA, et al. Dectin-1 expression and function are enhanced on alternatively activated and GM-CSF-treated macrophages and are negatively regulated by IL-10, dexamethasone, and lipopolysaccharide. J Immunol. 2003;171:4569–4573. doi: 10.4049/jimmunol.171.9.4569. [DOI] [PubMed] [Google Scholar]
- 11.Kreider T, et al. Alternatively activated macrophages in helminth infections. Curr Opin Immunol. 2007;19:448–453. doi: 10.1016/j.coi.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez FO, et al. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 13.Pinhal-Enfield G, et al. An angiogenic switch in macrophages involving synergy between Toll-like receptors 2, 4, 7, and 9 and adenosine A(2A) receptors. Am J Pathol. 2003;163:711–721. doi: 10.1016/S0002-9440(10)63698-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrante CJ, Leibovich SJ. Regulation of macrophage polarization and wound healing. Advances in wound care. 2012;1:10–16. doi: 10.1089/wound.2011.0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Csoka B, et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012;26:376–386. doi: 10.1096/fj.11-190934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphree LJ, et al. Lipopolysaccharide rapidly modifies adenosine receptor transcripts in murine and human macrophages: role of NF-kappaB in A(2A) adenosine receptor induction. Biochem J. 2005;391:575–580. doi: 10.1042/BJ20050888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramanathan M, et al. Differential regulation of HIF-1alpha isoforms in murine macrophages by TLR4 and adenosine A(2A) receptor agonists. J Leukoc Biol. 2009;86:681–689. doi: 10.1189/jlb.0109021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramanathan M, et al. Synergistic up-regulation of vascular endothelial growth factor (VEGF) expression in macrophages by adenosine A2A receptor agonists and endotoxin involves transcriptional regulation via the hypoxia response element in the VEGF promoter. Mol Biol Cell. 2007;18:14–23. doi: 10.1091/mbc.E06-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elson G, et al. Induction of murine adenosine A(2A) receptor expression by LPS: analysis of the 5′ upstream promoter. Genes Immunology. 2013 doi: 10.1038/gene.2012.60. [DOI] [PubMed] [Google Scholar]
- 20.Komohara Y, et al. M2 macrophage/microglial cells induce activation of Stat3 in primary central nervous system lymphoma. J Clin Exp Hematop. 2011;51:93–99. doi: 10.3960/jslrt.51.93. [DOI] [PubMed] [Google Scholar]
- 21.Kurahara H, et al. M2-Polarized Tumor-Associated Macrophage Infiltration of Regional Lymph Nodes Is Associated With Nodal Lymphangiogenesis and Occult Nodal Involvement in pN0 Pancreatic Cancer. Pancreas. 2012 doi: 10.1097/MPA.0b013e318254f2d1. [DOI] [PubMed] [Google Scholar]
- 22.Niino D, et al. Ratio of M2 macrophage expression is closely associated with poor prognosis for Angioimmunoblastic T-cell lymphoma (AITL) Pathol Int. 2010;60:278–283. doi: 10.1111/j.1440-1827.2010.02514.x. [DOI] [PubMed] [Google Scholar]
- 23.Prokop S, et al. M2 polarized macrophages and giant cells contribute to myofibrosis in neuromuscular sarcoidosis. Am J Pathol. 2011;178:1279–1286. doi: 10.1016/j.ajpath.2010.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruffell D, et al. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci U S A. 2009;106:17475–17480. doi: 10.1073/pnas.0908641106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hasko G, et al. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol. 2001;41:775–787. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- 27.Greenhalgh DG. The role of apoptosis in wound healing. Int J Biochem Cell Biol. 1998;30:1019–1030. doi: 10.1016/s1357-2725(98)00058-2. [DOI] [PubMed] [Google Scholar]
- 28.Rai NK, et al. Apoptosis: a basic physiologic process in wound healing. Int J Low Extrem Wounds. 2005;4:138–144. doi: 10.1177/1534734605280018. [DOI] [PubMed] [Google Scholar]
- 29.Ramanathan M, et al. Differential regulation of HIF-1alpha isoforms in murine macrophages by TLR4 and adenosine A(2A) receptor agonists. J Leukoc Biol. 2009;86:681–689. doi: 10.1189/jlb.0109021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mylonas KJ, et al. Alternative activation of macrophages by filarial nematodes is MyD88-independent. [10.1016/j.imbio.2012.07.006];Immunobiology. 2012 doi: 10.1016/j.imbio.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Csoka B, et al. A2A adenosine receptors and C/EBPbeta are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood. 2007;110:2685–2695. doi: 10.1182/blood-2007-01-065870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nemeth ZH, et al. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J Immunol. 2005;175:8260–8270. doi: 10.4049/jimmunol.175.12.8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macedo L, et al. Wound healing is impaired in MyD88-deficient mice: a role for MyD88 in the regulation of wound healing by adenosine A2A receptors. Am J Pathol. 2007;171:1774–1788. doi: 10.2353/ajpath.2007.061048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramanathan M, et al. Synergistic up-regulation of vascular endothelial growth factor (VEGF) expression in macrophages by adenosine A2A receptor agonists and endotoxin involves transcriptional regulation via the hypoxia response element in the VEGF promoter. Mol Biol Cell. 2007;18:14–23. doi: 10.1091/mbc.E06-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grinberg S, et al. Suppression of PLCbeta2 by endotoxin plays a role in the adenosine A(2A) receptor-mediated switch of macrophages from an inflammatory to an angiogenic phenotype. Am J Pathol. 2009;175:2439–2453. doi: 10.2353/ajpath.2009.090290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnold L, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. 2007;204:1057–1069. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ricardo SD, et al. Macrophage diversity in renal injury and repair. J Clin Invest. 2008;118:3522–3530. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Willenborg S, et al. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood. 2012;120:613–625. doi: 10.1182/blood-2012-01-403386. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X, Mosser DM. Macrophage activation by endogenous danger signals. J Pathol. 2008;214:161–178. doi: 10.1002/path.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brancato SK, Albina JE. Wound macrophages as key regulators of repair: origin, phenotype, and function. Am J Pathol. 2011;178:19–25. doi: 10.1016/j.ajpath.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daley JM, et al. The phenotype of murine wound macrophages. J Leukoc Biol. 2010;87:59–67. doi: 10.1189/jlb.0409236. [DOI] [PMC free article] [PubMed] [Google Scholar]