

Abstract
Systemic autoimmunity is characterized by widespread inflammation, autoantibody production and immune complex deposition. The regulatory mechanisms for the systemic autoimmunity are not fully understood. A paper by Li et al. revealed that Egr2 and Egr3, transcription factors required for T-cell anergy, are the regulators for systemic autoimmune disease. They showed evidence that Egr2 and Egr3 control cytokine productions and cell proliferation via SOCS and Batf regulation.
Keywords: Egr2, Egr3, SOCS1, SOCS3, Batf, Th17, autoimmunity
T-cell anergy is one important mechanism for immune tolerance. In CD4+ T cells, it was reported that Egr2 and Egr3 are required for anergy induction.1,2 Egr2 and Egr3 are the members of Cys2His2-type zinc finger transcription factor family, which consists of four members, Egr1, 2, 3 and 4. Egr2 plays an essential role in hindbrain development and myelination of the peripheral nervous system, and the null mutation of Egr2 resulted in perinatal or neonatal death due to respiratory and/or feeding deficits.3 Egr3-deficient mice show gait ataxia due to the lack of muscle spindles.4 Egr2 and Egr3 inhibit interferon (IFN)-γ and IL-2 secretion by T cells and enhance the expression of the E3 ligase Cbl-b, which is critical for the regulation of T cell tolerance and anergy.2
Previously, Zhu et al. reported Egr2 conditional knockout mice (Egr2-CKO), in which the Egr2 gene was deleted specifically in CD2+ lymphocytes.5 Egr2-CKO mice demonstrated massive infiltration of Th1 and Th17 cells in multiple organs and developed a lupus-like syndrome in later life. The Egr2-deficient T cells did not show alteration in primary activation, but highly proliferated in response to stimulation. Egr2-deficient CD4+ T cells produced higher amount of IFN-γ and IL-17 in response to TCR stimulation. CD44high T cells from Egr2-CKO mice exhibited defective expression of p21cip1 and Egr2 directly bind to the promoter of the cell cycle inhibitor p21cip1. These observations suggested the association between Egr2 and systemic autoimmunity via dysregulated T cell proliferation and cytokine production.
Recently, Li et al. reported the analysis of mice deficient for both Egr2 and Egr3 in B and T cells (CD2-Egr2−/−Egr3−/−).6 CD2-Egr2−/−Egr3−/− mice presented lethal and early-onset systemic autoimmunity. They developed lymphocytic infiltration in multiple organs and autoantibodies against nuclear antigens. On the other hand, the numbers of T and B lymphocytes of 5-week-old CD2-Egr2−/−Egr3−/− mice in the lymph node and spleen were similar to the age-matched control, and the number and suppressive function of CD4+CD25+ regulatory T cells (Treg) were not altered.
Surprisingly, IL-2 production and proliferation of B and T cells from CD2-Egr2−/−Egr3−/− mice were severely impaired upon antigen-receptor stimulation in vitro, suggesting the absence of reduced antigen receptor thresholds. Therefore, Egr2 and Egr3 are required for efficient proliferation of naïve B and T cells. This observation was unexpected, because Egr2 and Egr3 have been regarded as negative regulators of T cell receptor signaling via induction of E3 ubiquitin ligases related to T cell anergy.2
Production of inflammatory cytokines including IFN-γ, IL-4, IL-17 and IL-21 was significantly enhanced in Egr2- and Egr3-deficient CD4+ T cells. As antigen receptor signaling did not display hyperactivation in CD2-Egr2−/−Egr3−/− mice, enhancement of cytokine receptor signaling became another candidate pathway for the enhanced activation. In the stimulated Egr2- and Egr3-deficient B and T cells, both STAT1 and STAT3, not STAT5 were significantly activated, and expression of SOCS1 and SOCS3 was significantly reduced. Chromatin immunoprecipitation (ChIP) assay demonstrated direct binding of Egr2 to SOCS1 and SOCS3 promoters, and luciferase reporter assay showed that Egr2 induces SOCS1 and SOCS3 promoter activity. From these findings, it was concluded that Egr2 and/or Egr3 directly induce SOCS1 and SOCS3 expression, and it leads to the suppression of STAT1- and STAT3-mediated cytokine signaling (Fig. 1).
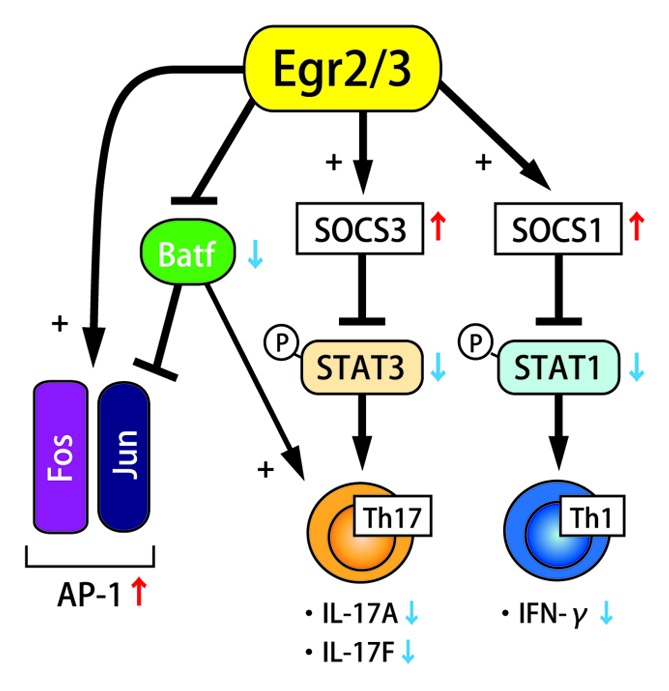
Figure 1. Overview of the function of Egr2/3.
SOCS1 and SOCS3 are suppressors of STAT1 and STAT3 activation, and they essentially regulate the STAT1- and STAT3-mediated cytokine expression and the differentiation of Th1 and Th17 cells.7 Although SOCS1-deficient T cells shift to Th1 cell differentiation and produce IFN-γ in the absence of TCR ligation,8 T cell-specific SOCS1 deficient mice do not develop spontaneous inflammatory pathologies, and their T cells are resistant to Th17 cell differentiation.9 On the other hand, SOCS3-deficient T cells are more susceptible to Th17 cell differentiation, but resistant to Th1 cell differentiation.10 In CD2-Egr2−/−Egr3−/− mice, systemic autoimmune disease was lethal and developed in their early life, and both Th1 and Th17 cell-derived cytokines excessively accumulated. The suppression of both SOCS1 and SOCS3 is supposed to be the cause of their severe inflammatory pathological phenotype. The direct regulation of both SOCS1 and SOCS3 expression by Egr2 suggests that the role of Egr2 and Egr3 is vital for preventing autoimmune diseases, and limiting immunopathology during productive immune responses.
In Egr2- and Egr3-deficient B and T cells, electrophoretic mobility shift assay (EMSA) analysis revealed the severe impairment of the activation of AP-1, an essential transcription factor for the production of IL-2 and cell cycle progression. Instead, an alternative molecular complex involving JunB, not c-Fos, was detected. Therefore, Batf, a member of the AP-1 complex that forms a heterodimer with c-Jun or JunB, was supposed to be a part of the alternative AP-1 complex.11 While Egr2 and Egr3 did not regulate Batf transcription, the binding of Egr2 to Batf blocked the association of Batf to AP-1 DNA probe. These findings indicated that Egr2 and Egr3 support AP-1 activation by the suppression of Batf function (Fig. 2). It was an unexpected observation that Egr2 and Egr3 positively regulate antigen receptor induced proliferation because Egr2 and Egr3 have been regarded as negative regulators of T cell receptor signaling.2 However, suppression of Batf function by Egr2 and Egr3 would not result in AP-1 enhancement in anergic T cells because AP-1, MAP kinase and NFκB are essentially suppressed in the partially stimulated anergic state.
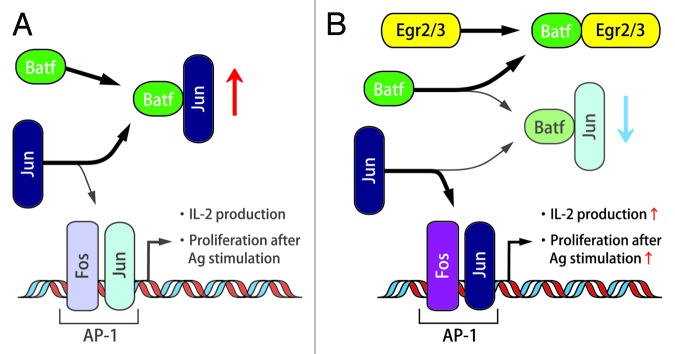
Figure 2. The relationship between Egr2/3 and Batf, Jun, AP-1. The function of Batf in the absence of Egr2/3 (A) and in the presence of Egr2/3 (B).
Recently, it has been reported that deletion of JunB in epithelial cells results in skin inflammation that is similar to systemic lupus erythematosus (SLE) with increased serum proinflammatory cytokines and a myeloproliferative disorder.12 Therefore, the fact that Egr2- and Egr-3 deficiency reduced Jun activity by the abrogation of direct Batf blocking implies the linkage between Egr-2 and SLE. Moreover, autoimmunity of Egr2- and Egr3-deficient B and T cells in the presence of impaired antigen receptor-induced proliferation resembles the findings from B and T cells in lupus patients, which show hyper-inflammatory activity in vivo and yet defective AP-1 activation and IL-2 expression in vitro.13 These observations indicate that Egr2 and Egr3 are key candidate transcription factors to control systemic autoimmunity. The linkage between Egr2 and lupus was also suggested by the observation that EGR2 is a risk factor for SLE in a case-control association study.14 Moreover, silencing of Batf in Egr2-and Egr3-deficient CD4+ T cells reduced expression of IL-17, consistent with previous findings on the role of Batf in IL-17 expression.11
This study is important in elucidating the role of Egr2 and Egr3 in systemic autoimmunity. Nevertheless, it may be difficult to explain autoantibody production leading to systemic autoimmunity by enhanced cytokine response, because enhanced cytokine production could be associated with autoimmune diseases other than systemic autoimmunity. The findings that Egr2 and Egr3 are not required for the function of CD4+CD25+ Treg is consistent with the fact that STAT5 is not affected in CD2-Egr2−/−Egr3−/− mice and the previous observation that Egr2-expression in CD4+CD25− T cells is not altered in Foxp3-mutated Scurfy mice.15 Egr2 and Egr3 may be associated with tolerogenic machinery that is different from CD4+CD25+Foxp3+ Treg and there is a possibility that Egr2 and Egr3 are important for the development of non-Foxp3 Tregs including Egr2-expressing CD4+CD25+LAG3+ Treg. IL-27 is an important factor inducing IL-10 secreting Treg, and we have recently found the requirement of Egr2 for the IL-10 production in IL-27 stimulated CD4+ T cells.16 Thus, Egr2 could be the regulatory transcription factor controlling multiple aspect of inflammatory response, and the precise contribution of Egr2 and Egr3 expressed in helper T cells, Tregs or B cells should be addressed in the future.
Disclosure of Potential Conflicts of Interest
K.F. has received research grant support from Bristol-Myers Squibb and Astellas Pharma.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/23952
References
- 1.Harris JE, Bishop KD, Phillips NE, Mordes JP, Greiner DL, Rossini AA, et al. Early growth response gene-2, a zinc-finger transcription factor, is required for full induction of clonal anergy in CD4+ T cells. J Immunol. 2004;173:7331–8. doi: 10.4049/jimmunol.173.12.7331. [DOI] [PubMed] [Google Scholar]
- 2.Safford M, Collins S, Lutz MA, Allen A, Huang CT, Kowalski J, et al. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat Immunol. 2005;6:472–80. doi: 10.1038/ni1193. [DOI] [PubMed] [Google Scholar]
- 3.Topilko P, Schneider-Maunoury S, Levi G, Baron-Van Evercooren A, Chennoufi AB, Seitanidou T, et al. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994;371:796–9. doi: 10.1038/371796a0. [DOI] [PubMed] [Google Scholar]
- 4.Tourtellotte WG, Milbrandt J. Sensory ataxia and muscle spindle agenesis in mice lacking the transcription factor Egr3. Nat Genet. 1998;20:87–91. doi: 10.1038/1757. [DOI] [PubMed] [Google Scholar]
- 5.Zhu B, Symonds AL, Martin JE, Kioussis D, Wraith DC, Li S, et al. Early growth response gene 2 (Egr-2) controls the self-tolerance of T cells and prevents the development of lupuslike autoimmune disease. J Exp Med. 2008;205:2295–307. doi: 10.1084/jem.20080187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li S, Miao T, Sebastian M, Bhullar P, Ghaffari E, Liu M, et al. The transcription factors Egr2 and Egr3 are essential for the control of inflammation and antigen-induced proliferation of B and T cells. Immunity. 2012;37:685–96. doi: 10.1016/j.immuni.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshimura A, Suzuki M, Sakaguchi R, Hanada T, Yasukawa H. SOCS, Inflammation, and Autoimmunity. Front Immunol. 2012;3:20. doi: 10.3389/fimmu.2012.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chong MM, Metcalf D, Jamieson E, Alexander WS, Kay TW. Suppressor of cytokine signaling-1 in T cells and macrophages is critical for preventing lethal inflammation. Blood. 2005;106:1668–75. doi: 10.1182/blood-2004-08-3049. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka K, Ichiyama K, Hashimoto M, Yoshida H, Takimoto T, Takaesu G, et al. Loss of suppressor of cytokine signaling 1 in helper T cells leads to defective Th17 differentiation by enhancing antagonistic effects of IFN-gamma on STAT3 and Smads. J Immunol. 2008;180:3746–56. doi: 10.4049/jimmunol.180.6.3746. [DOI] [PubMed] [Google Scholar]
- 10.Taleb S, Romain M, Ramkhelawon B, Uyttenhove C, Pasterkamp G, Herbin O, et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J Exp Med. 2009;206:2067–77. doi: 10.1084/jem.20090545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schraml BU, Hildner K, Ise W, Lee WL, Smith WA, Solomon B, et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature. 2009;460:405–9. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zenz R, Eferl R, Kenner L, Florin L, Hummerich L, Mehic D, et al. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. 2005;437:369–75. doi: 10.1038/nature03963. [DOI] [PubMed] [Google Scholar]
- 13.Rauen T, Benedyk K, Juang YT, Kerkhoff C, Kyttaris VC, Roth J, et al. A novel intronic cAMP response element modulator (CREM) promoter is regulated by activator protein-1 (AP-1) and accounts for altered activation-induced CREM expression in T cells from patients with systemic lupus erythematosus. J Biol Chem. 2011;286:32366–72. doi: 10.1074/jbc.M111.245811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myouzen K, Kochi Y, Shimane K, Fujio K, Okamura T, Okada Y, et al. Regulatory polymorphisms in EGR2 are associated with susceptibility to systemic lupus erythematosus. Hum Mol Genet. 2010;19:2313–20. doi: 10.1093/hmg/ddq092. [DOI] [PubMed] [Google Scholar]
- 15.Okamura T, Fujio K, Shibuya M, Sumitomo S, Shoda H, Sakaguchi S, et al. CD4+CD25-LAG3+ regulatory T cells controlled by the transcription factor Egr-2. Proc Natl Acad Sci U S A. 2009;106:13974–9. doi: 10.1073/pnas.0906872106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwasaki Y, Fujio K, Okamura T, Yanai A, Sumitomo S, Shoda H, et al. Egr-2 transcription factor is required for Blimp-1 mediated IL-10 production in IL-27 stimulated CD4(+) T cells. Eur J Immunol. 2013;43:1063–73. doi: 10.1002/eji.201242942. [DOI] [PubMed] [Google Scholar]