

Abstract
Local anesthetics and related drugs block ionic currents of Na+, K+ and Ca2+ conducted across the cell membrane by voltage-dependent ion channels. Many of these drugs bind in the permeation pathway, occlude the pore and stop ion movement. However channel-blocking drugs have also been associated with decreased membrane stability of certain tetrameric K+ channels, similar to the destabilization of channel function observed at low extracellular K+ concentration. Such drug-dependent stability may result from electrostatic repulsion of K+ from the selectivity filter by a cationic drug molecule bound in the central cavity of the channel. In this study we used the pore domain of the KcsA K+ channel protein to test this hypothesis experimentally with a biochemical assay of tetramer stability and theoretically by computational simulation of local anesthetic docking to the central cavity. We find that two common local anesthetics, lidocaine and tetracaine, promote thermal dissociation of the KcsA tetramer in a K+-dependent fashion. Docking simulations of these drugs with open, open-inactivated and closed crystal structures of KcsA yield many energetically favorable drug-channel complexes characterized by nonbonded attraction to pore-lining residues and electrostatic repulsion of K+. The results suggest that binding of cationic drugs to the inner cavity can reduce tetramer stability of K+ channels.
Keywords: local anesthetics, K+ channels, lidocaine, tetracaine, membrane protein, ion channels
Introduction
Voltage-gated Na+ channels (Nav channels) are recognized as the primary target of a large number of diverse local anesthetic (LA) drugs including tertiary amines such as lidocaine and tetracaine that suppress firing of action potentials.1 Likewise, LA drugs are important anti-arrhythmic, anti-epileptic and anti-myotonic agents used to stabilize abnormal conditions of hyperexcitability by blocking particular protein products(s) of the nine gene isoforms of human Nav channels.2,3 Many tetrameric K+ channels are also blocked by hydrophobic organic cations such as alkyl derivatives of tetraethylammonium (TEA+) and amine derivatives with LA activity.4-6 Block of certain drug-sensitive K+ channels may induce or suppress abnormal symptoms of electrical excitability. For example, a form of potentially fatal ventricular fibrillation known as acquired long-QT syndrome is triggered by promiscuous drug block of the human hERG (Kv11.1) cardiac channel that mediates repolarization of the ventricular action potential.7,8 In contrast, drugs that block the human Kv1.5 channel may be useful in suppressing atrial fibrillation.9,10
In addition to acute channel block by simple occlusion of the ion conduction pathway, drug molecules also affect long-term stability of certain K+ channels in the cell membrane. For example, some blockers, including TEA+ and anti-arrhythmic drugs (e.g., quinidine), destabilize certain K+ channels, such as the squid axon delayed rectifier, the Drosophila Shab channel and the human Kv1.5 channel, by promoting irreversible loss of K+ conductance.10-12 In contrast, other drugs (e.g., quinidine, hydrophobic TEA+ derivatives and astemizole) stabilize the activity of certain K+ channels such as naturally occurring hERG mutants associated with long-QT Syndrome Type-2 (LQT2).13 Over 290 human genetic defects in the hERG channel are linked to LQT2, a condition characterized by susceptibility to cardiac arrhythmia, ventricular fibrillation and sudden death. These mutations may alter hERG gating; however, most LQT2 mutations affect biogenesis of the channel protein due to structural instability, effects on protein folding and disruption of membrane trafficking.8,14,15 The related phenomenon of drug-acquired LQT-syndrome7,8,16 is due to intrinsic susceptibility of hERG current to blockade by many clinically useful drugs (e.g., antihistamine terfenadine/Seldane and antidepressant fluoxetine/Prozac). Promiscuous drug-sensitivity of hERG has been attributed to high affinity of the hydrophobic inner cavity of the channel for diverse drugs, partly determined by two aromatic residues (Y652, F656) of the S6 inner-pore helix that enhance drug-binding affinity.7,17-20
Insight to the mechanism of hERG channel instability is revealed by results showing that low extracellular K+ results in rapid conversion of the native channel to a non-conducting state followed by a decline in surface membrane density of the channel protein.21 Permeant inorganic cations (e.g., Rb+ and Cs+) preserve hERG expression and function in the absence of K+, whereas weakly permeant Na+ and Li+ are ineffective.21 Studies of hERG pore mutations support the conclusion that K+ occupancy of the hERG channel is required to maintain function and membrane stability.21 Correspondingly, low-K+ diet and serum hypokalemia (<3.5 mM K+) promote long-QT cardiac arrhythmia by decreasing hERG surface density in ventricular myocytes.22,23 Interestingly, some hERG-blocking drugs such as astemizole act as chemical chaperones to enhance trafficking of misfolded LQT2 hERG mutants to the cell membrane.13,24 The chaperone mechanism appears to involve stabilizing drug interactions with the inner pore of hERG, since TEA+ derivatives such as C10-TEA (decyl-triethylammonium) rescue trafficking of mutant hERG with an efficacy correlated with their blocking affinity.13
Attention has been drawn to the possibility of using specific blockers of Kv1.5 to control symptoms of atrial fibrillation, the most common form of cardiac arrhythmia, since this K+ channel gene is selectively expressed in human atria.9,25-27 Kv1.5 is also a candidate for rational drug design by virtue of high sequence identity to Kv1.2, a voltage-gated K+ channel of known crystal structure.28-30 Block of Kv1.5 by LAs and related drugs has been studied extensively by traditional electrophysiological approaches;25,31-33 however, recent work revealed a new mode of action of the anti-arrhythmic drug, quinidine. Block of Kv1.5 by quinidine with a half-inhibition constant, K0.5 = 1 μM, in atrial myocytes induces rapid internalization (~10 min) of the channel protein.10 Quinidine-induced internalization of Kv1.5 is stereospecific and sensitive to mutations of the channel pore.10 Drug-induced internalization of Kv1.5 and drug- and K+-dependent stability of hERG described above suggests that chronic treatment with certain drugs is likely to result in long-term changes in cellular expression of functional K+ channels. Remodeling of K+ channel expression linked to drug-induced membrane instability may be more important than acute block of ionic current in determining long-term effects of therapeutic drug treatment and may be relevant to the failure of new drugs due to cardiotoxicity.23
Effects of LA drugs on long-term functional activity of K+ channels described above are likely to be related to a general phenomenon involving loss of channel function in the absence of K+. First described by Almers and Armstrong in squid axons,34 loss of K+ current upon exposure to zero or low K+ concentration on one or both sides of the membrane has been observed for the Ca2+-activated channel (KCa3.1) in human erythrocytes,35 the BKCa (KCa1.1) channel,36 the Drosophila Shaker (Kv1) and Shab (Kv2) voltage-gated K+ channels37-39 and the human hERG K+ channel.21,40 We have pursued the structural basis for this phenomenon in thermal stability studies of KcsA, a membrane protein of Streptomyces lividans that is a good experimental model for the K+ channel pore domain.41,42 These studies showed that permeant inorganic ions (e.g., K+, Rb+, Tl+, NH4+) and blockers (Cs+, Ba2+) that bind in the selectivity filter43,44 protect KcsA tetramer from thermal dissociation. In contrast, thermal stability of KcsA tetramer is much weaker in the presence of impermeant cations, such as Na+, Li+ and choline+.41 These results confirm that K+ channel quaternary structure is K+-dependent and presumably linked to structural deformation of the selectivity filter observed in low K+.45,46 In support of this interpretation, mutations of the innermost K+ site (S4) of the KcsA selectivity filter formed by four Thr75 residues perturb inorganic cation binding and also affect tetramer stability.42 Such evidence for KcsA and other K+ channels47-49 suggests that destabilization of tetrameric structure may be an important aspect of K+-dependent regulation of K+ channel expression and drug-dependent changes in expression of human Kv1.5 and hERG.5 channels described above.
To pursue this hypothesis in the present work, we investigated the effect of two well-known LA drugs, lidocaine and tetracaine, on tetramer stability of KcsA. Since KcsA is amenable to structural analysis by X-ray crystallography, it may be a useful model for investigating the interaction of LAs with K+ channels such as Kv1.5 and hERG (Kv11.1). We also explored this possibility by in silico docking simulations of lidocaine and tetracaine to various conformations (closed, open, inactivated) of KcsA using the Monte Carlo energy minimization method. Our results from these two approaches show that LA drugs diminish tetramer stability of KcsA in a K+-dependent fashion, likely by binding to residues that line the inner pore and repelling K+ from the selectivity filter.
Results
Effects of local anesthetics on tetramer stability of KcsA
Classical electrophysiological studies of neuronal preparations suggested that KV channels are less sensitive to block by LA drugs than Nav channels. For example, Taylor50 found that 0.1% (w/v) procaine (4.2 mM) blocked the peak Na+ conductance of voltage-clamped squid axon by 60%; whereas outward K+ conductance (delayed rectifier current) was only blocked by 20%. Assuming a one-site model for the concentration dependence of procaine block, these data correspond to IC50 values of ~3 mM and ~17 mM for procaine block of squid Nav and Kv current respectively. However, various other K+ channels have since been found to exhibit higher affinity for LA drugs. For example, mammalian Kv1.5 and Kv11.1 (hERG) are blocked by the LA drug, bupivacaine, with IC50 values of 9 μM and 20 μM, respectively.6 To investigate whether LA drugs may generally affect tetramer stability of the K+ channel pore domain, we studied the effect of lidocaine and tetracaine using our previously described assay of thermal stability of the KcsA tetramer as monitored by SDS-PAGE.41,42
The results indicate that 20 mM lidocaine has a significant effect on thermal stability as indicated by loss of KcsA tetramer content upon 10-min exposure to temperatures above 80 °C in the presence of 5 mM KCl (Fig. 1A). As previously described,41,42 KcsA tetramer is completely stable in the presence of 5 mM KCl, and absence of LA drug for the temperature range tested up to 99 °C (Fig. 1A). In the absence of K+ the mid-point temperature for KcsA tetramer dissociation is lowered significantly to ~46 °C (Fig. 1B). However, under these latter conditions of zero K+, 20 mM lidocaine did not significantly affect the temperature dependence of tetramer stability (Fig. 1B).
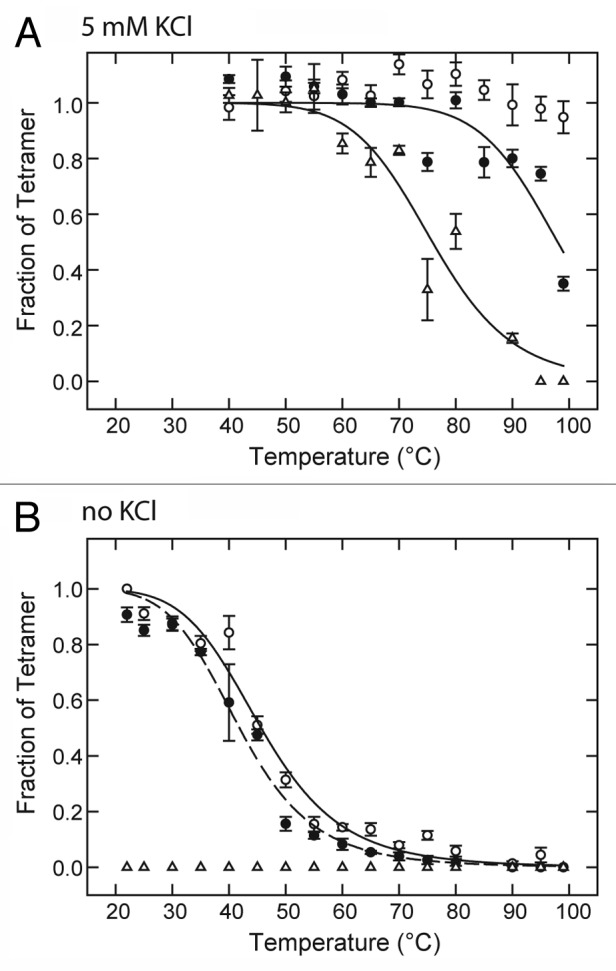
Figure 1. Effect of LAs on temperature dependence of KcsA tetramer stability in the presence and absence of K+. KcsA tetramer was measured by SDS-PAGE analysis of a series of identical samples incubated at various temperatures for 10 min before addition of SDS-PAGE sample buffer. The sample assay mixture contained 10 mM Hepes-Tris, pH 7.4, 100 mM cholineCl, either 5 mM KCl (A) or no added KCl (B) and either no LA (○), 20 mM lidocaine (●) or 5 mM tetracaine (△). Solid lines indicate nonlinear regression fits to a logistic function of temperature described in Materials and Methods. Fit parameters: (A) 20 mM lidocaine (●) : T0.5 = 98.0 ± 2.1 °C, n = 14.1 ± 4.8; (A) 5 mM tetracaine (△): T0.5 = 75.9 ± 1.7 °C, n = 10.7 ± 2.3; (B) no LA (○): T0.5 = 45.7 ± 0.9 °C, n = 6.4 ± 0.7; (B) 20 mM lidocaine (●) : T0.5 = 41.9 ± 0.7 °C, n = 6.4 ± 0.6.
In contrast to lidocaine, 5 mM tetracaine decreases the mid-point temperature of tetramer dissociation to ~76 °C in the presence of 5 mM KCl (Fig. 1A) and promotes the complete dissociation of tetramer at 22 °C or above in the absence of K+ (Fig. 1B). These results show that KcsA tetramer is sensitive to destabilization by LA drugs, and the effect is highly dependent on the particular LA drug molecule and the presence or absence of K+.
The dependence of tetramer content on LA concentration was characterized for both drugs in the presence of 5 mM KCl and a 10-min exposure to 90 °C. Under these conditions, the native KcsA tetramer is completely stable in the absence of drug (Fig. 1A); however, the tetramer dissociates as a function of drug concentration with IC50 values of 25 mM for lidocaine and 4.2 mM for tetracaine (Fig. 2). For sake of comparison, these latter values are similar to the range of LA concentration observed for acute block of Na+ and K+ conductance by procaine in the squid giant axon.50 In the absence of K+ and at room temperature (22 °C), the dependence of tetramer content on the concentration of tetracaine is shifted to a lower range of LA concentration with an IC50 value of 1.2 mM (Fig. 2). These results are consistent with the hypothesis that both lidocaine and tetracaine promote ligand-dependent dissociation of KcsA tetramer at elevated temperature.
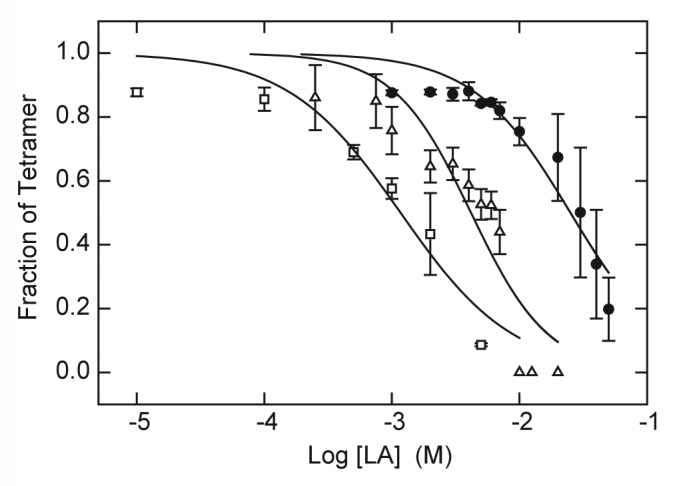
Figure 2. Destabilization of KcsA tetramer as function of LA concentration. KcsA tetramer was measured by SDS-PAGE analysis of samples titrated with increasing concentrations of lidocaine (●) or tetracaine (△, □ ). The sample assay mixture contained 10 mM Hepes-Tris, pH 7.5, 100 mM cholineCl and either 5 mM KCl (● , △) or no added KCl (□ ). Samples were incubated either at 90°C (● , △) or 22 °C (□ ) for 10 min before addition of SDS-PAGE sample buffer. Solid lines indicate nonlinear regression fits to a logistic function of LA concentration as described in Materials and Methods. Fit parameters: lidocaine, 5 mM KCl, 90°C (● ): IC50 = 25.1 ± 2.6 mM, n = 1.14 ± 0.13; tetracaine, 5 mM KCl, 90 °C (△): IC50 = 4.2 ± 0.6 mM, n = 1.43 ± 0.33; tetracaine, 0 KCl, 22 °C (□ ): 1.2 ± 0.2 mM, n = 0.98 ± 0.22.
If the higher apparent affinity of KcsA for tetracaine observed in the absence of K+ is due to an antagonistic interaction between the LA molecule and K+, the K+-concentration dependence of tetramer stability would be expected to shift to higher K+ concentration in the presence of LA. As shown in Figure 3, this appears to be the case. In the absence of LA, K+ promotes the stability of KscA tetramer with a concentration dependence characterized by midpoint value (K0.5) of 1.48 ± 0.04 mM K+ and an apparent slope factor of n = 3.19 ± 0.26. In the presence of 5 mM tetracaine this relationship is shifted to higher K+ with K0.5 = 5.03 ± 0.90 mM K+ and smaller slope factor of n = 1.04 ± 0.19. A similar shift to higher K+ concentration is also observed in the presence of 20 mM lidocaine (Fig. 3).

Figure 3. Stabilization of KcsA tetramer as a function of K+ concentration in the absence and presence of LA. KcsA tetramer was measured by SDS-PAGE analysis of samples titrated with increasing K+ concentration in the absence of LA (○) or in the presence of 20 mM lidocaine (● ) or 5 mM tetracaine (△). The sample assay mixture contained ~500 ng KcsA, 10 mM Hepes-Tris, pH 7.4, 100 mM choline Cl, indicated concentrations of KCl and either no LA, 20 mM lidocaine or 5 mM tetracaine. All samples were incubated at 90 °C for 10 min before addition of SDS-PAGE sample buffer. Solid lines indicate nonlinear regression fits to a logistic function of K+ concentration as described in Materials and Methods. Fit parameters: no LA (○): K0.5 = 1.48 ± 0.04 mM, n = 3.19 ± 0.26; 20 mM lidocaine (● ): K0.5 = 6.65 ± 0.8 mM, n = 1.05 ± 0.13; 5 mM tetracaine (△): K0.5 = 5.03 ± 0.90, n = 1.04 ± 0.19.
Molecular simulations of lidocaine and tetracaine binding within the inner cavity of KcsA
The preceding results suggest that the KcsA tetramer, which is a structural homolog of the pore domain of different K+ channel proteins, binds two different prototypical LA drugs. LA binding to KcsA appears to be accompanied by destabilization of the structural integrity of the tetramer in a temperature- and K+-dependent fashion; i.e., K+ occupancy of the selectivity filter inhibits LA binding and vice versa. However, the different behavior of lidocaine and tetracaine implies that the relative efficacy of various LAs in tetramer destabilization is likely to be quite dependent on the structure of the drug molecule.
To investigate this hypothesis from a theoretical perspective, we performed computational simulations of the binding of lidocaine and tetracaine to available crystal structures of KcsA. Lidocaine and tetracaine were docked to models built using X-ray structures of KcsA in the open, closed and inactivated states. In the open KcsA structure (3PJS),51 the large cytoplasmic C-terminal portion was removed. We imported crystallographic coordinates of atoms to the ZMM program and the model was Monte Carlo-minimized. The selectivity filter was populated with two K+ ions and two water molecules43 in two different two-ion occupancy configurations of the selectivity filter. In the 1/3 configuration (Fig. 4), K+ ions were placed in sites S1 and S3, and water molecules were placed in sites S2 and S4. In the 2/4 configuration (Fig. 5), K+ ions were placed in sites S2 and S4, and water molecules were placed in sites S1 and S3. In the open-inactivated KcsA conformation (Fig. 6) with the distance of ~32 Å between Cα atoms of diagonally opposed Thr112 residues (3F5W),52 we removed the Fab antibody, used the symmetry module of ZMM program53 to generate and optimize the symmetric structure and used it to dock ligands. In the X-ray structure of this crystal form, the selectivity filter is populated with two K+ ions at sites S1 and S4. Since this configuration is proposed to be an important feature of the open-inactivated state, we did not test other configurations. In this structure two water molecules are seen at the inner pore axis. One molecule is located at site S5 of the central pore cavity, at the focus of the pore helices. Another water molecule is present at 8.8 Å from site S5 in the cytoplasmic direction. We removed both water molecules. In the closed KcsA-tetrabutylammonium complex (1J95),54 we removed tetrabutylammonium (TBA+) from the inner cavity and K+ ions from sites S1 and S3, and retained K+ ions in sites S2 and S4 (Fig. 7). Intensive Monte Carlo minimizations yielded many low-energy binding modes for each ligand-channel complex. Representative low-energy complexes of both LAs docked into the models of the open, open-inactivated, and closed KcsA are shown in Figures 4–7 and their calculated energy components are given in Table 1.

Figure 4. Example of energy-minimized structure simulation of lidocaine (A) and tetracaine (B) docked in the open KcsA structure (PDB entry 3PJS) with S1/S3 K+ occupancy of the selectivity filter. Front subunit is removed for clarity. Right panel is an enlargement of the docked LA molecule zoomed from the left panel. In this and subsequent structure figures, the side chains of Thr75, Ile100 and Phe103 residues as well as the backbone carbonyls of Thr75 are shown as thin sticks. LA molecules and water molecules in the selectivity filter are shown as thick sticks. Potassium ions are shown as yellow spheres.

Figure 5. Examples of energy minimized complexes of lidocaine (A) and tetracaine (B and C) with the open KcsA (PDB entry 3PJS) and S2/S4 K+ occupancy of the selectivity filter. Two different docked locations of tetracaine are shown in (B) and (C).

Figure 6. Examples of energy-minimized complexes of lidocaine (A) and tetracaine (B) with the open-inactivated KcsA structure (PDB entry 3F5W) and S1/S4 K+ occupancy of the selectivity filter. Note that the apparently different crossing angles of inner helices in the right panels of (A) vs. (B) are due to different views of the ligand-channel complex chosen for an optimal display of the LA molecule.

Figure 7. Examples of energy-minimized complexes of lidocaine (A) and tetracaine (B) with closed KcsA (PDB entry 1J95) and K+ ions at sites S2 and S4. Right panel of (B) shows detailed enlargement of tetracaine, which forms H-bonds with Thr75 and Thr107.
Table 1. Ligand-channel energy (E, kcal/mol) and top contributions to ita.
| State | Ligand | K ions | E | K+ | F103 | T75 | T74 | I100 |
|---|---|---|---|---|---|---|---|---|
| Open |
Lidocaine |
S1/S3 |
−23.4 |
0.37 |
−8.20 |
−4.15 |
−0.73 |
−2.52 |
| S2/S4 |
−15.1 |
2.42 |
−6.25 |
−1.61 |
−0.11 |
−2.12 |
||
| Tetracaine |
S1/S3 |
−20.6b |
0.74 |
−2.02 |
−1.36 |
−0.72 |
−1.09 |
|
| S2/S4 |
−13.4c |
6.72 |
−9.17 |
−2.03 |
0.17 |
−0.75 |
||
| Open- inactivated |
Lidocaine |
S1/S4 |
−17.5 |
0.58 |
−1.38 |
−6.64 |
−0.22 |
−2.17 |
| Tetracaine |
S1/S4 |
−20.2 |
−0.38 |
−1.97 |
−2.99 |
−0.01 |
−2.57 |
|
| Closed | Lidocaine |
S2/S4 |
−19.4d |
0.62 |
−2.3 |
−3.64 |
0.04 |
−3.79 |
| Tetracaine | S2/S4 | −23.1e | 3.51 | −2.45 | −3.54 | −0.16 | −2.86 |
a Summed contributions from the sidechains of the four same-number residues and from both K ions. Contributions from backbones may be large, especially for Thr74 and Gly99, which may be tested by synthetic replacement with unnatural amino acids. bOther top contributors are Ser102 (−0.67), Leu36 (−0.86), Val97 (−0.61), Thr33 (−0.35), H2O at site S4 (−0.57), Val106 (−0.36) and Met96 (−0.50). cAnother top contributor is Thr107 (−0.68). dOther top contributors are Val106 (−0.93) and Thr107 (−0.37). eAnother top contributor is Thr107 (−3.53).
In the open-KcsA model, configuration 1/3 is most favorable for both lidocaine and tetracaine (Table 1). Lidocaine fits neatly in the central cavity. Its ammonium group binds close to site S5 at the focus of P-helices (Fig. 4A). Such a location of the ammonium group was proposed for LAs in the Na+-deficient models of the Nav1.4 Na+ channel in the open55 and closed56 states. Lidocaine adopts a “horizontal” binding mode with the aromatic ring directed toward the subunit interface. The lidocaine carbonyl group approaches the selectivity filter where it has favorable dipole-dipole interactions with the water molecule at site S4 and hydroxyl groups of Thr75. Phenylalanines Phe103 provide the largest contributions to lidocaine-channel energy, in part due to cation-pi interactions, which are sensed by the AMBER force field.56 Other strong contributors to lidocaine binding energy are Ile100 and Thr74. The K+ ion at site S3 experiences a weak repulsion from lidocaine. While “vertical” orientation of tetracaine, which was proposed for the open Nav channel55 was found among low-energy binding modes (not shown), in the energetically most preferable binding mode the ligand extends its “tail” in the subunit interface (Fig. 4B). The ammonium group of tetracaine binds closer to the selectivity filter than that of lidocaine. Due to this latter interaction tetracaine repels the S3 K+ ion twice as strong as lidocaine. The top contributors to tetracaine-KcsA energy are Phe103, Thr75, Ile100, as well as Ser102 and Leu36.
In the 2/4 configuration of the open KcsA, the binding energies for both LAs are substantially weaker than in the 1/3 configuration (Table 1). In the lowest-energy complex (Fig. 5A), lidocaine and a K+ ion in site S4 electrostatically repel each other. Despite this repulsion, the ammonium group of lidocaine remains close to site S5 at the cavity center due to favorable interactions with threonine Thr75 and interaction of the aromatic moiety of the LA with the side chains of Phe103 and Ile100. Low-energy tetracaine-KcsA complexes are diverse. In some complexes, tetracaine extends along the cytoplasmic part of the open pore, far below the selectivity filter (e.g., Fig. 5B). In other complexes, it also extends along the pore but approaches the selectivity filter (Fig. 5C). The tetracaine-channel energies in both complexes only differ by <0.5 kcal/mol. Our computations are not precise enough to favor a particular docked tetracaine complex. In the latter complex, a strong electrostatic repulsion between a K+ ion at site S4 and the ammonium group is compensated by attraction of the drug to Phe103 and Thr75.
In the open-inactivated KcsA, the lowest-energy binding mode shows lidocaine positioned with its ammonium group 8.6 Å away from the K+ ion at site S4 (Fig. 6A). The complex is stabilized by two H-bonds between the amide nitrogen of lidocaine and the hydroxyl side chains of Thr75 residues. The carbonyl group of tetracaine approaches the K+ ion (Fig. 6B), which contributes a small favorable energy to ligand-channel energy. When compared with the open KcsA in configuration 2/4, the ligands in the open-inactivated KcsA experience weaker attraction to Phe103, much stronger attractions to Thr75 residues and weaker repulsion from the K+ ion. The latter is explained by the fact that the inner pore of the open-inactivated channel is wider than that in the open channel, and drug ligands have more room for maneuver to avoid electrostatic repulsion with the K+ ion.
The closed KcsA conformation can readily accommodate lidocaine or tetracaine in the central cavity (Fig. 7). Ligand interactions with the closed KcsA conformation are at least as strong as those predicted for the open-inactivated channel due to favorable interactions with Phe103, Thr75 and Ile100. This suggests the possibility of trapping LA in the central cavity upon channel closure as previously suggested by crystal structures of complexes of KcsA and TBA+.54,57-59 Both LA ligands experience electrostatic repulsion with the K+ ion in site S4 of the closed channel.
Discussion
The purpose of this work was to explore whether the well-studied and readily manipulated KcsA K+ channel protein might provide structural insights into complex interactions of LAs and other drug molecules with voltage-gated ion channels. In terms of function, KcsA can replace the pore domain of a eukaryotic Kv channel with conservation of normal gating activity.60 It is also blocked by organic cations, TEA+ and TBA+, from the intracellular side of membrane in a fashion similar to other K+ channels.58,61 The fact that drug sensitivity of K+ channels can be modified by mutation of residues of the M2/S6 transmembrane helix that lines the inner cavity of the channel17-20,25 implies that LA pharmacology of KcsA is amenable to molecular engineering. Thus, KcsA may be useful per se or in chimera strategies for pursuing the structural basis of drug interactions with the K+ channel pore domain. In particular, effects of LAs and similar drugs on tetramer stability of KcsA may involve molecular events analogous to those that play a role in long-term drug effects at the cellular level; i.e., drug-dependent loss of functional Kv1.5 channels from the cell membrane10 or drug-dependent rescue of membrane trafficking defects of dysfunctional LQT2 mutants of the hERG13,24 as discussed in the Introduction.
Our results generally support this possibility based on effects of two generic tertiary amine LA drugs on thermal stability of the KcsA tetramer and computational results from docking simulations. Lidocaine has a weak but reproducible effect on thermal stability of the KcsA tetramer observed as enhanced tetramer dissociation in the presence of K+ at high temperature (Fig. 1A), an effect corresponding to a K0.5 value of 25 mM lidocaine for tetramer dissociation at 90 °C (Fig. 2). The effect of the ester-linked tetracaine molecule is considerably more pronounced than that of amide-linked lidocaine with K0.5 values of 4.2 and 1.2 mM tetracaine in the presence and absence of 5 mM K+, respectively (Fig. 2). The low affinity of lidocaine (K0.5 = 25 mM) in the KcsA tetramer stability assay may be reasonably compared with the low affinity of procaine (IC50 ~17 mM) in blocking delayed rectifier K+ current of squid giant axon.50 The 5.9-fold higher apparent affinity of tetracaine vs. lidocaine in the present tetramer stability assay in the presence of 5 mM K+ is comparable to the 6.7-fold higher potency of tetracaine vs. lidocaine observed for in vivo animal studies of local anesthesia.62 The relative anesthetic potency of a series of different LA molecules is known to be highly correlated with the bulk hydrophobicity of the molecule as measured by the octanol/water partition coefficient (Q7.4) of the drug.62 Values of Q7.4 based on total drug concentrations at equilibrium in octanol/water at 25 °C and pH 7.4 are reported as 221 for tetracaine and 43 for lidocaine corresponding to a hydrophobicity ratio of 5.1 for tetracaine/lidocaine.63 It is important to recognize that such comparisons may be purely coincidental but warrant further investigation.
Additional insight to the mechanism of LA-induced destabilization of KcsA tetramer may be found in the mutually antagonistic interaction between LAs and K+. Approximately 4-fold higher concentrations of tetracaine are required to destabilize KcsA tetramer in the presence of 5 mM K+ than in the absence of K+ (Fig. 2). Similarly, a ~4-fold increase in the concentration range of K+ needed to promote stabilization of KcsA tetramer is observed in the presence of LAs as compared with K+ titration in the absence of drug (Fig. 3). These results obtained with the KcsA channel are reminiscent of many electrophysiological observations on the K+-dependence of K+ current stability in cellular assays and antagonistic interactions between K+ and organic cations that block diverse K+ channels. For example, the Drosophila Shab channel, a homolog of the mammalian Kv2 subfamily, is reversibly blocked by the anti-arrhythmic drug quinidine with a Kd of 11 μM in the presence of K+.12 However, Shab K+-current is irreversibly lost when quinidine blocker is added in the absence of external K+.12 These latter studies support the conclusion that occupancy of a pore-blocking site in the channel by quinidine cation antagonizes K+ binding to the selectivity filter, which leads to structural collapse of the channel to a non-conducting conformation. A possible scenario for such a mechanism is that drug occupancy first induces a non-conducting tetramer that subsequently irreversibly dissociates to non-functional monomers in the cell membrane or at a later stage of intracellular protein turnover. Such dissociation of K+ channel tetramers to monomers may accompany the loss of hERG channels from the cell membrane under conditions of serum hypokalemia21 and similar membrane disappearance and internalization of KV1.5 channels triggered by quinidine.10 Tetramer stability of KcsA is considered to be unusually robust in comparison to quaternary subunit interactions of many oligomeric proteins, which readily dissociate under conditions of SDS-PAGE and elevated temperature. The fact that KcsA tetramer exhibits K+-dependent destabilization in the presence of two generic low-affinity LA drugs such as lidocaine and tetracaine implies that similar phenomena are likely to occur for other K+ channels of weaker tetramer stability and for drug blockers of higher affinity.
We investigated the hypothesis of electrostatic repulsion between LA drugs and K+ from a theoretical standpoint in molecular docking simulations of lidocaine and tetracaine for four models of KcsA: open state with K+ ions bound to the selectivity filter in either the S1/S3 or S2/S4 configurations,45,51 the open-inactivated state with K+ ions in the S1/S4 configuration52 and the closed state with K+ ions in the S2/S4 configuration.54 In the closed and open KcsA crystal structures, only two K+ ions bind simultaneously in the outer pore.45,51 In both the non-conducting C-type inactivated state52 and closed state at low K+ concentration,46 only two K+ ions are observed in the selectivity filter, and one of them binds at site S4. To address the question of state-dependent binding, the low-energy binding modes and major ligand energy contributions to LA binding were compared for all states of the channel (Table 1).
For the open channel, the predicted interaction energy of both lidocaine and tetracaine is 7–8 kcal/mol lower (higher affinity) for the S1/S3 state vs. the S2/S4 K+ occupancy state. The lower affinity of the drugs for the S2/S4 ion configuration is partially due to the higher repulsive interaction between K+ and the LA drug in the S2/S4 vs. the S1/S3 state as expected for closer proximity of the S4 K+ ion to the protonated amine group of the LA located in the central cavity. Differences in K+ repulsion between tetracaine vs. lidocaine are similarly due to closer proximity of the ammonium group of docked tetracaine vs. lidocaine to the K+-binding filter. The wider central cavity of the open-inactivated channel lowers the K+ repulsion energy by allowing the LA molecules to move farther away from the filter resulting in a larger distance of separation between the S4 K+ and the drug ammonium group. In the S2/S4 state of the closed channel a weaker K+ repulsion is observed for lidocaine vs. tetracaine due to the particular favored orientations of the drug molecules in which the ammonium group of lidocaine is positioned farther away from the S4 K+.
Our calculations are consistent with the proposal that electrostatic repulsion occurs between K+ ions in the selectivity filter and the positively charged ammonium group of an LA molecule or other drug that binds in the inner pore, below the selectivity filter. The computational results also show that the bound LA molecules have strong attractive interactions to certain residues in the inner cavity such as Thr75, Ile100 and Phe103. Theoretical calculations of the absolute interaction energy of K+ ions with cation binding sites of the K+ channel selectivity filter are subject to considerable uncertainty; however estimates suggest that the free energy of K+ binding is weaker than LA interactions with the cavity residues.64,65 Under such circumstances LA-K+ repulsion would tend to favor displacement of K+ ions from the selectivity filter. In the absence of K+, the carbonyl groups that line the filter repel each other and destabilize inter-subunit interactions.46 Such displacement of K+ from the S4 site by the LA molecule would account for destabilization of the tetrameric KcsA structure observed in our experiments. Interaction of cationic drug ligands with permeant cations has previously been proposed to contribute to state-dependent action of certain drugs (e.g, ref. 55).
The simulated docking of lidocaine and tetracaine into the open, C-type inactivated, and closed states of KcsA described here is an attempt to address the atomistic basis of state-dependent drug binding to a potassium channel. This problem is relevant to the use-dependent action of LAs proposed in the classic “modulated receptor hypothesis,” which proposes that higher-affinity drug binding to open and inactivated states vs. closed, resting states of the Nav channel gives rise to enhanced block of current with repetitive depolarization.1,63,66,67
Regarding the computational results, some words of caution are appropriate for proper perspective on the molecular simulations. Energy characteristics of the representative structures shown in Table 1 and Figures 4–7 are obtained using the AMBER force field, with the distance- and environment-dependent dielectric permittivity function developed to maximize the success rate of correct predictions of water-soluble ligand-protein complexes of known X-ray structures.68 The absolute values of the KcsA-LA energies should be treated with caution because the entropy was not calculated and electrostatic interactions within the ion channel may differ from those in water-soluble proteins. The structures shown are the lowest-energy representatives of many possible binding modes, which may differ from the representative structure up to few kcal/mol. Visual inspection shows that in many, but not all structures accumulated in respective Monte Carlo-minimization trajectories, the LA binding modes are rather similar to those shown in Figure 4.
It is interesting to compare our docking simulations of LA molecules in KcsA with previous results of mutational analysis of drug block of the Kv1.5 and hERG K+ channels. The sequence alignment of Figure 8 illustrates considerable amino acid conservation of K+ channel pore structure from the N-terminal pore helix to the C-terminal inner helix that lines the inner cavity which forms a binding site for TBA+57–59 and organic molecules similar to LA drugs. Such a sequence alignment has previously been used to construct molecular models of the drug binding sites of Kv1.5 and hERG based on known crystal structures of K+ channel proteins such as KcsA and Kv1.2.17,19,25,28-30 Mutational scanning of this region conducted to identify potential drug-binding residues of Kv1.5 found the greatest loss in drug blocking affinity for mutation of the following residues: Thr479, Thr480, Ile 502, Val505, Leu 506, Ile508 and Val512.10,25,33 Similarly, mutation of the following residues of hERG produced the greatest loss in drug blocking affinity: Thr623, Ser624, Val625, Gly648, Tyr652 and Phe656.17,19,20 These latter residues of Kv1.5 and hERG highlighted in Figure 8 coincide well with KcsA residue locations that contribute the greatest interaction energy with docked lidocaine and tetracaine as identified in our simulations: Thr74, Thr75, Ile100, Phe103 and Thr107 (Table 1). Detailed examination of the LA docking results shows that such residue correlations vary for the particular drug and the particular binding mode of the drug. However, this comparison further supports the notion that K+ channel residues whose side chains face the inner pore are available for favorable chemical interactions with drugs bound within the inner conduction pathway.

Figure 8. Sequence alignment of residues corresponding to the inner core pore domain of KcsA and related K+ channels. Range of residue numbers listed for aligned sequences corresponds to NCBI protein database entries for KcsA (Acc. P0A334), human Kv1.2 (Acc. NP_004965), human Kv1.5 (Acc. NP_002225) and hERG/Kv11.1 (Acc. Q12809). Residues identical to KcsA are colored red. Asterisks (*) correspond to residues of KcsA located at monomer-monomer interfaces of closed (PDB entry 1K4C) or open (PDB entry 3FB7) conformations of KcsA crystal structures as determined by the EMBL-EBI PISA (Protein Interfaces, Surfaces and Assemblies) analysis program (www.ebi.ac.uk/msd-srv/prot_int/pistart.html).71,72 Drug-binding residues of Kv1.510,25,33 and hERG17,19,20 identified by mutational analysis are highlighted in yellow. Residues of KcsA found to exhibit the highest interaction energy with lidocaine and tetracaine in docking simulations performed in this study are also highlighted in yellow.
The absolute blocking affinity of different K+ channels for LA molecules and other clinical drugs varies considerably. For example, various drugs block the hERG K+ channel with sub-micromolar affinity19,20 in comparison to millimolar affinity for block of squid K+ current by procaine50 In this regard, apparent millimolar affinity of KcsA for lidocaine and tetracaine is likely to reflect the chemical structure of these drugs and the polarity of side chain residues lining the inner cavity, such as Thr107 of KcsA vs. Phe565 of hERG. Mutational analysis may provide further insight to this hypothesis and allow construction of a chimeric version of KcsA, which exhibits hERG-like drug sensitivity.
Figure 8 also shows the location of residues that form the monomer-monomer contact regions in both closed (1K4C) and open (3FB7) conformations of KcsA crystal structures as identified by the PISA analysis program.69,70 The fact that the majority of all residues (~70%) in this region contribute to the interfacial contact regions of the tetramer is consistent with the finding that that LA binding to the central cavity formed by this domain perturbs tetramer stability. In regard to findings that some drugs such as alkyl derivatives of TEA+ actually stabilize trafficking mutants of hERG,13,24 we have observed that TBA+ exhibits a tetramer stabilizing effect similar to K+ when tested in our SDS-PAGE assay of KcsA exposed to high temperature in the absence of K+ (unpublished results). More work is clearly needed to understand such structure-dependent differences in tetramer stability that depend on the particular drug and K+ channel isoform. As seen in some of our simulations, certain drugs may partially insert into the tetramer interface and weaken inter-subunit contacts while other drugs may form bridging interactions between subunits that help to hold the tetramer together. In summary, we have shown that KcsA can be used to investigate the phenomenon of drug-dependent tetramer stability of K+ channels in a simple experimental assay of the purified protein and in computational experiments performed by molecular simulation techniques.
Materials and Methods
Expression and purification of KcsA
A synthetic gene for native KcsA with a (His)6 sequence inserted after Met1 cloned into the pASK90 plasmid was used to express the His-tagged KscA protein in E. coli strain BL21.71 Expression and purification of KcsA for use in tetramer stability assays were performed essentially as described.41,42 One milliliter of pure KscA eluted from a Ni-NTA column (BioRad) in phosphate buffer [10 mM KCl, 95 mM NaH2PO4, 400 mM imidazole, pH 7.0 and 1 mM dodecylmaltoside (C12M)] was concentrated to ~100 μl in an Amicon Ultra-4 (10 kDa filter cutoff) centrifugal concentration device (Millipore). The concentrate was diluted to 4 ml with choline buffer (100 mM cholineCl, 10 mM Hepes-Tris, pH 7.4, 0.75 mM C12M) and concentrated again to ~100 μl. This dilution/concentration procedure was repeated an additional 4 times and the final concentrate was diluted to 500 μl using choline buffer minus C12M for all steps. The latter filtration-exchange procedure is expected to reduce residual contaminant concentration of K+ and Na+ to less than 1 μM in the final KcsA preparation by simple dilution-exchange into buffer containing 100 mM cholineCl, 10 mM Hepes-Tris, pH 7.4 and approximately 8 mM C12M. KcsA protein concentration was determined by the BCA assay (Pierce Chemical Co.)
Assay of KcsA tetramer stability
Assay samples in a final volume of 10 μl in PCR tubes were prepared to contain ~500 ng KcsA, 100 mM cholineCl, 10 mM Hepes-Tris, pH 7.4, ~6 mM C12M and variable concentrations of KCl, lidocaine or tetracaine. Individual Samples were equilibrated for 1 h at 22 °C and transferred to individual wells of a thermal cycler machine set to a constant temperature in the range of 25–98 °C. Samples were incubated at elevated temperature for 10 min, placed on ice for 1 min and briefly spun in a mini-centrifuge. Concentrated 6X SDS-PAGE sample buffer (2 μl) was added to each 10 μl sample to yield final concentrations of 10% glycerol, 62.5 mM TRIS-HCl, pH 6.8, 2% SDS and 0.01% bromphenol blue. Ten milliliters of each sample was pipetted into wells of precast 12% acrylamide TRIS-HCl ready SDS-PAGE Gels (Bio-Rad) and subjected to electrophoresis at 200V for ~35 min in running buffer (25 mM Tris base, 192 mM glycine, pH 8.3, 0.1% SDS). The gel was rinsed in deionized H2O for 7 min and stained with GelCode Blue Stain Reagent for 1 h, rinsed for 1 h and destained overnight in deionized H2O. Stock solutions of 250 mM lidocaine or tetracaine were prepared by dissolving the HCl salt of each compound (purchased from Sigma) in water adjusted to pH ~6.5 with Tris base. Tetracaine solutions were freshly prepared before each use.
Data analysis
Images of scanned gels were analyzed for relative amount of KcsA tetramer using ImageJ software to measure the relative area of the stained band as described previously.41 Measured peak area of the tetramer band was normalized to that of unheated control samples for each experiment. Summary data for the fraction of tetramer relative to control are plotted as the mean ± SE (n = 3 to 5). For experiments (Fig. 1) where the fraction of tetramer (F) decreases as a function of temperature (T), data points were fit to the following logistic function using the nonlinear regression wizard of Sigma-Plot 11 software (SysStat):
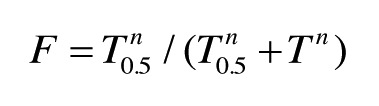 |
where T0.5 is the temperature at f = 0.5 and n is an integer. For experiments where F decreases as a function of [LA] (Fig. 2), data points were fit to
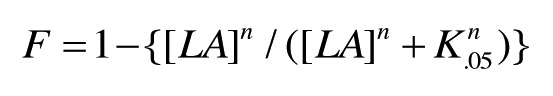 |
where K0.5 is the concentration of LA at 50% inhibition. For experiments (Fig. 3) where F increases as a function of [K+], data points were fit to
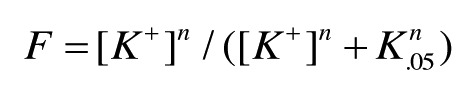 |
where K0.5 is the concentration of K+ at 50% of maximal tetramer.
Energy calculations and computational protocol for docking lidocaine and tetracaine to various conformations of KcsA
Energy was calculated using the AMBER force field.72,73 Atomic charges at ligands were calculated using the MOPAC program.74 Electrostatic energy was calculated using the environment- and distance-dependent dielectric without desolvation energy.68 All calculations were performed using the ZMM program, which optimizes the energy in the space of internal (generalized) coordinates.75,76 Bond angles were kept rigid in the protein but were allowed to vary in the ligands. The energy optimization was performed by the Monte Carlo-minimization (MCM) protocol.77
Docking of each ligand was performed in two stages. At the first stage, 2048 MCM trajectories were submitted with random ligand starting torsional angles, positions and orientation within the inner pore. Each energy minimization was 20 iterations long, and each trajectory was terminated when 20 consecutive energy minimizations did not decrease the energy of the apparent global minimum found in the trajectory. The KcsA backbone was fixed. The ligand torsion angles and KcsA sidechains that had at least one atom within 6 Å from the ligand were randomly sampled. The lowest energy structures were accumulated in a stack. A new structure was added in the stack if the ligand generalized coordinates did not have an approximate match there. If a match was found, the new structure replaced it only if it had a lower energy. At the second stage, a hundred of the top structures in the stack were refined in longer MCM trajectories. The backbone torsions of KcsA were allowed to vary. To prevent large deviations of the backbone from the X-ray template, α carbons were constrained to the template by pins. A pin is a flat-bottom parabolic energy function that allows atoms to deviate penalty-free up to 1 Å from the template and imposes a penalty of 10 kcal mol−1 Å−1 for larger deviations. Each energy minimization was 200 iterations long, and each trajectory was terminated when 2,000 consecutive energy minimizations did not decrease the energy of the apparent global minimum found in the trajectory.
Acknowledgments
N.W.G. was supported by a graduate teaching fellowship in the Biology Department at Clarkson University. E.G.M. is supported by an Early Career LDRD award from Sandia National Laboratories. Sandia National Laboratories is a multi-program laboratory managed and operated by Sandia Corporation, a wholly owned subsidiary of Lockheed Martin Corporation, for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-AC04-94AL85000.
Computations were made possible by the facilities of the Shared Hierarchical Academic Research Computing Network (SHARCNET:www.sharcnet.ca). This part of the work was supported by the grant GRPIN/238773-2009 from the Natural Sciences and Engineering Research Council of Canada to B.S.Z.
Glossary
Abbreviations:
- hERG (Kv11.1)
human ether-a-go-go related channel
- Kv
voltage-gated K+ channel
- LA
local anesthetic
- Nav
voltage-gated Na+ channel
- TBA+
tetrabutylammonium
- TEA+
tetraethylammonium
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/24455
References
- 1.Nau C, Wang GK. Interactions of local anesthetics with voltage-gated Na+ channels. J Membr Biol. 2004;201:1–8. doi: 10.1007/s00232-004-0702-y. [DOI] [PubMed] [Google Scholar]
- 2.Catterall WA. Common modes of drug action on Na+ channels: local anesthetics, antiarrhythmics and anticonvulsants. Trends Pharmacol Sci. 1987;8:57–65. doi: 10.1016/0165-6147(87)90011-3. [DOI] [Google Scholar]
- 3.Catterall WA. Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol. 2012;590:2577–89. doi: 10.1113/jphysiol.2011.224204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armstrong CM. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J Gen Physiol. 1971;58:413–37. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.French RJ, Shoukimas JJ. Blockage of squid axon potassium conductance by internal tetra-N-alkylammonium ions of various sizes. Biophys J. 1981;34:271–91. doi: 10.1016/S0006-3495(81)84849-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kindler CH, Yost CS. Two-pore domain potassium channels: new sites of local anesthetic action and toxicity. Reg Anesth Pain Med. 2005;30:260–74. doi: 10.1016/j.rapm.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Sanguinetti MC, Mitcheson JS. Predicting drug-hERG channel interactions that cause acquired long QT syndrome. Trends Pharmacol Sci. 2005;26:119–24. doi: 10.1016/j.tips.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–9. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 9.Carlsson L, Duker G, Jacobson I. New pharmacological targets and treatments for atrial fibrillation. Trends Pharmacol Sci. 2010;31:364–71. doi: 10.1016/j.tips.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Schumacher SM, McEwen DP, Zhang L, Arendt KL, Van Genderen KM, Martens JR. Antiarrhythmic drug-induced internalization of the atrial-specific k+ channel kv1.5. Circ Res. 2009;104:1390–8. doi: 10.1161/CIRCRESAHA.108.192773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khodakhah K, Melishchuk A, Armstrong CM. Killing K channels with TEA+ Proc Natl Acad Sci U S A. 1997;94:13335–8. doi: 10.1073/pnas.94.24.13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gomez-Lagunas F. Quinidine interaction with Shab K+ channels: pore block and irreversible collapse of the K+ conductance. J Physiol. 2010;588:2691–706. doi: 10.1113/jphysiol.2010.193128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ficker E, Obejero-Paz CA, Zhao S, Brown AM. The binding site for channel blockers that rescue misprocessed human long QT syndrome type 2 ether-a-gogo-related gene (HERG) mutations. J Biol Chem. 2002;277:4989–98. doi: 10.1074/jbc.M107345200. [DOI] [PubMed] [Google Scholar]
- 14.Ficker E, Dennis A, Kuryshev Y, Wible BA, Brown AM. HERG channel trafficking. Novartis Found Symp. 2005;266:57–69, discussion 70-4, 95-9. doi: 10.1002/047002142X.ch6. [DOI] [PubMed] [Google Scholar]
- 15.Sanguinetti MC. HERG1 channelopathies. Pflugers Arch. 2010;460:265–76. doi: 10.1007/s00424-009-0758-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajamani S, Eckhardt LL, Valdivia CR, Klemens CA, Gillman BM, Anderson CL, et al. Drug-induced long QT syndrome: hERG K+ channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br J Pharmacol. 2006;149:481–9. doi: 10.1038/sj.bjp.0706892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci U S A. 2000;97:12329–33. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, Seebohm G, Sanguinetti MC. Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc Natl Acad Sci U S A. 2002;99:12461–6. doi: 10.1073/pnas.192367299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sánchez-Chapula JA, Navarro-Polanco RA, Culberson C, Chen J, Sanguinetti MC. Molecular determinants of voltage-dependent human ether-a-go-go related gene (HERG) K+ channel block. J Biol Chem. 2002;277:23587–95. doi: 10.1074/jbc.M200448200. [DOI] [PubMed] [Google Scholar]
- 20.Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC. Molecular determinants of HERG channel block. Mol Pharmacol. 2006;69:1709–16. doi: 10.1124/mol.105.020990. [DOI] [PubMed] [Google Scholar]
- 21.Massaeli H, Guo J, Xu J, Zhang S. Extracellular K+ is a prerequisite for the function and plasma membrane stability of HERG channels. Circ Res. 2010;106:1072–82. doi: 10.1161/CIRCRESAHA.109.215970. [DOI] [PubMed] [Google Scholar]
- 22.Guo J, Massaeli H, Xu J, Jia Z, Wigle JT, Mesaeli N, et al. Extracellular K+ concentration controls cell surface density of IKr in rabbit hearts and of the HERG channel in human cell lines. J Clin Invest. 2009;119:2745–57. doi: 10.1172/JCI39027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robertson GA. Endocytic control of ion channel density as a target for cardiovascular disease. J Clin Invest. 2009;119:2531–4. doi: 10.1172/JCI40427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wible BA, Hawryluk P, Ficker E, Kuryshev YA, Kirsch G, Brown AM. HERG-Lite: a novel comprehensive high-throughput screen for drug-induced hERG risk. J Pharmacol Toxicol Methods. 2005;52:136–45. doi: 10.1016/j.vascn.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Decher N, Pirard B, Bundis F, Peukert S, Baringhaus K-H, Busch AE, et al. Molecular basis for Kv1.5 channel block: conservation of drug binding sites among voltage-gated K+ channels. J Biol Chem. 2004;279:394–400. doi: 10.1074/jbc.M307411200. [DOI] [PubMed] [Google Scholar]
- 26.Tamargo J, Caballero R, Gómez R, Delpón EI. I(Kur)/Kv1.5 channel blockers for the treatment of atrial fibrillation. Expert Opin Investig Drugs. 2009;18:399–416. doi: 10.1517/13543780902762850. [DOI] [PubMed] [Google Scholar]
- 27.Beshore DC, Liverton NJ, McIntyre CJ, Claiborne CF, Libby B, Culberson JC, et al. Discovery of triarylethanolamine inhibitors of the Kv1.5 potassium channel. Bioorg Med Chem Lett. 2010;20:2493–6. doi: 10.1016/j.bmcl.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 28.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 29.Andér M, Luzhkov VB, Aqvist J. Ligand binding to the voltage-gated Kv1.5 potassium channel in the open state--docking and computer simulations of a homology model. Biophys J. 2008;94:820–31. doi: 10.1529/biophysj.107.112045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Q, Du L, Wang X, Li M, You Q. Modeling the binding modes of Kv1.5 potassium channel and blockers. J Mol Graph Model. 2008;27:178–87. doi: 10.1016/j.jmgm.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 31.Valenzuela C, Delpón E, Tamkun MM, Tamargo J, Snyders DJ. Stereoselective block of a human cardiac potassium channel (Kv1.5) by bupivacaine enantiomers. Biophys J. 1995;69:418–27. doi: 10.1016/S0006-3495(95)79914-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steidl JV, Yool AJ. Distinct mechanisms of block of Kv1.5 channels by tertiary and quaternary amine clofilium compounds. Biophys J. 2001;81:2606–13. doi: 10.1016/S0006-3495(01)75904-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aréchiga IA, Barrio-Echavarria GF, Rodríguez-Menchaca AA, Moreno-Galindo EG, Decher N, Tristani-Firouzi M, et al. Kv1.5 open channel block by the antiarrhythmic drug disopyramide: molecular determinants of block. J Pharmacol Sci. 2008;108:49–55. doi: 10.1254/jphs.08084FP. [DOI] [PubMed] [Google Scholar]
- 34.Almers W, Armstrong CM. Survival of K+ permeability and gating currents in squid axons perfused with K+-free media. J Gen Physiol. 1980;75:61–78. doi: 10.1085/jgp.75.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heinz A, Passow H. Role of external potassium in the calcium-induced potassium efflux from human red blood cell ghosts. J Membr Biol. 1980;57:119–31. doi: 10.1007/BF01868998. [DOI] [PubMed] [Google Scholar]
- 36.Vergara C, Alvarez O, Latorre R. Localization of the K+ lock-In and the Ba2+ binding sites in a voltage-gated calcium-modulated channel. Implications for survival of K+ permeability. J Gen Physiol. 1999;114:365–76. doi: 10.1085/jgp.114.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ambriz-Rivas M, Islas LD, Gomez-Lagunas F. K+-dependent stability and ion conduction of Shab K+ channels: a comparison with Shaker channels. Pflugers Arch. 2005;450:255–61. doi: 10.1007/s00424-005-1411-9. [DOI] [PubMed] [Google Scholar]
- 38.Gómez-Lagunas F. Shaker B K+ conductance in Na+ solutions lacking K+ ions: a remarkably stable non-conducting state produced by membrane depolarizations. J Physiol. 1997;499:3–15. doi: 10.1113/jphysiol.1997.sp021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gómez-Lagunas F. Na(+) interaction with the pore of Shaker B K(+) channels: zero and low K(+) conditions. J Gen Physiol. 2001;118:639–48. doi: 10.1085/jgp.118.6.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L, Dennis AT, Trieu P, Charron F, Ethier N, Hebert TE, et al. Intracellular potassium stabilizes human ether-à-go-go-related gene channels for export from endoplasmic reticulum. Mol Pharmacol. 2009;75:927–37. doi: 10.1124/mol.108.053793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krishnan MN, Bingham J-P, Lee SH, Trombley P, Moczydlowski EG. Functional role and affinity of inorganic cations in stabilizing the tetrameric structure of the KcsA K+ channel. J Gen Physiol. 2005;126:271–83. doi: 10.1085/jgp.200509323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krishnan MN, Trombley P, Moczydlowski EG. Thermal stability of the K+ channel tetramer: cation interactions and the conserved threonine residue at the innermost site (S4) of the KcsA selectivity filter. Biochemistry. 2008;47:5354–67. doi: 10.1021/bi702281p. [DOI] [PubMed] [Google Scholar]
- 43.Zhou Y, MacKinnon R. The occupancy of ions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J Mol Biol. 2003;333:965–75. doi: 10.1016/j.jmb.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 44.Jiang Y, MacKinnon R. The barium site in a potassium channel by x-ray crystallography. J Gen Physiol. 2000;115:269–72. doi: 10.1085/jgp.115.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morais-Cabral JH, Zhou Y, MacKinnon R. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 2001;414:37–42. doi: 10.1038/35102000. [DOI] [PubMed] [Google Scholar]
- 46.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001;414:43–8. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 47.Pagliuca C, Goetze TA, Wagner R, Thiel G, Moroni A, Parcej D. Molecular properties of Kcv, a virus encoded K+ channel. Biochemistry. 2007;46:1079–90. doi: 10.1021/bi061530w. [DOI] [PubMed] [Google Scholar]
- 48.Chatelain FC, Gazzarrini S, Fujiwara Y, Arrigoni C, Domigan C, Ferrara G, et al. Selection of inhibitor-resistant viral potassium channels identifies a selectivity filter site that affects barium and amantadine block. PLoS One. 2009;4:e7496. doi: 10.1371/journal.pone.0007496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang S, Alimi Y, Tong A, Nichols CG, Enkvetchakul D. Differential roles of blocking ions in KirBac1.1 tetramer stability. J Biol Chem. 2009;284:2854–60. doi: 10.1074/jbc.M807474200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taylor RE. Effect of procaine on electrical properties of squid axon membrane. Am J Physiol. 1959;196:1071–8. doi: 10.1152/ajplegacy.1959.196.5.1071. [DOI] [PubMed] [Google Scholar]
- 51.Uysal S, Cuello LG, Cortes DM, Koide S, Kossiakoff AA, Perozo E. Mechanism of activation gating in the full-length KcsA K+ channel. Proc Natl Acad Sci U S A. 2011;108:11896–9. doi: 10.1073/pnas.1105112108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cuello LG, Jogini V, Cortes DM, Perozo E. Structural mechanism of C-type inactivation in K(+) channels. Nature. 2010;466:203–8. doi: 10.1038/nature09153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bruhova I, Zhorov BS. KvAP-based model of the pore region of shaker potassium channel is consistent with cadmium- and ligand-binding experiments. Biophys J. 2005;89:1020–9. doi: 10.1529/biophysj.105.062240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou M, Morais-Cabral JH, Mann S, MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001;411:657–61. doi: 10.1038/35079500. [DOI] [PubMed] [Google Scholar]
- 55.Tikhonov DB, Zhorov BS. Sodium channels: ionic model of slow inactivation and state-dependent drug binding. Biophys J. 2007;93:1557–70. doi: 10.1529/biophysj.106.100248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bruhova I, Tikhonov DB, Zhorov BS. Access and binding of local anesthetics in the closed sodium channel. Mol Pharmacol. 2008;74:1033–45. doi: 10.1124/mol.108.049759. [DOI] [PubMed] [Google Scholar]
- 57.Lenaeus MJ, Vamvouka M, Focia PJ, Gross A. Structural basis of TEA blockade in a model potassium channel. Nat Struct Mol Biol. 2005;12:454–9. doi: 10.1038/nsmb929. [DOI] [PubMed] [Google Scholar]
- 58.Faraldo-Gómez JD, Kutluay E, Jogini V, Zhao Y, Heginbotham L, Roux B. Mechanism of intracellular block of the KcsA K+ channel by tetrabutylammonium: insights from X-ray crystallography, electrophysiology and replica-exchange molecular dynamics simulations. J Mol Biol. 2007;365:649–62. doi: 10.1016/j.jmb.2006.09.069. [DOI] [PubMed] [Google Scholar]
- 59.Yohannan S, Hu Y, Zhou Y. Crystallographic study of the tetrabutylammonium block to the KcsA K+ channel. J Mol Biol. 2007;366:806–14. doi: 10.1016/j.jmb.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 60.Lu Z, Klem AM, Ramu Y. Ion conduction pore is conserved among potassium channels. Nature. 2001;413:809–13. doi: 10.1038/35101535. [DOI] [PubMed] [Google Scholar]
- 61.Kutluay E, Roux B, Heginbotham L. Rapid intracellular TEA block of the KcsA potassium channel. Biophys J. 2005;88:1018–29. doi: 10.1529/biophysj.104.052043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Langerman L, Bansinath M, Grant GJ. The partition coefficient as a predictor of local anesthetic potency for spinal anesthesia: evaluation of five local anesthetics in a mouse model. Anesth Analg. 1994;79:490–4. doi: 10.1213/00000539-199409000-00015. [DOI] [PubMed] [Google Scholar]
- 63.Butterworth JF, 4th, Strichartz GR. Molecular mechanisms of local anesthesia: a review. Anesthesiology. 1990;72:711–34. doi: 10.1097/00000542-199004000-00022. [DOI] [PubMed] [Google Scholar]
- 64.Åqvist J, Luzhkov V. Ion permeation mechanism of the potassium channel. Nature. 2000;404:881–4. doi: 10.1038/35009114. [DOI] [PubMed] [Google Scholar]
- 65.Thompson AN, Kim I, Panosian TD, Iverson TM, Allen TW, Nimigean CM. Mechanism of potassium-channel selectivity revealed by Na(+) and Li(+) binding sites within the KcsA pore. Nat Struct Mol Biol. 2009;16:1317–24. doi: 10.1038/nsmb.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Strichartz GR, Ritchie JM. The action of local anesthetics on ion channels of excitable tissues. In: Strichartz GR, ed. Handbook of Experimental Pharmacology, Volume 81. New York: Springer-Verlag, 1987:21-52. [Google Scholar]
- 68.Garden DP, Zhorov BS. Docking flexible ligands in proteins with a solvent exposure- and distance-dependent dielectric function. J Comput Aided Mol Des. 2010;24:91–105. doi: 10.1007/s10822-009-9317-9. [DOI] [PubMed] [Google Scholar]
- 69.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–97. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 70.Krissinel E. Crystal contacts as nature’s docking solutions. J Comput Chem. 2010;31:133–43. doi: 10.1002/jcc.21303. [DOI] [PubMed] [Google Scholar]
- 71.Heginbotham L, Odessey E, Miller C. Tetrameric stoichiometry of a prokaryotic K+ channel. Biochemistry. 1997;36:10335–42. doi: 10.1021/bi970988i. [DOI] [PubMed] [Google Scholar]
- 72.Weiner SJ, Kollman PA, Case DA, Singh UC, Chio C, Alagona G, et al. A new force field for molecular mechanical simulation of nucleic acids and proteins. J Am Chem Soc. 1984;106:765–84. doi: 10.1021/ja00315a051. [DOI] [Google Scholar]
- 73.Weiner SJ, Kollman PA, Nguyen DT, Case DA. An all atom force field for simulations of proteins and nucleic acids. J Comput Chem. 1986;7:230–52. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- 74.Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP. AM1: A new general purpose quantum mechanical model. J Am Chem Soc. 1985;107:3902–9. doi: 10.1021/ja00299a024. [DOI] [Google Scholar]
- 75.Zhorov BS. Vector method for calculating derivatives of energy of atom-atom interactions of complex molecules according to generalized coordinates. J Struct Chem. 1981;22:4–8. doi: 10.1007/BF00745970. [DOI] [Google Scholar]
- 76.Zhorov BS. Vector method for calculating derivatives of the energy deformation of valence angles and torsion energy of complex molecules according to generalized coordinates. J Struct Chem. 1983;23:649–55. doi: 10.1007/BF00746185. [DOI] [Google Scholar]
- 77.Li Z, Scheraga HA. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc Natl Acad Sci U S A. 1987;84:6611–5. doi: 10.1073/pnas.84.19.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]