

Abstract
Use of second generation sequencing (SGS) technologies for transcriptional profiling (RNA-Seq) has revolutionized transcriptomics, enabling measurement of RNA abundances with unprecedented specificity and sensitivity and the discovery of novel RNA species. Preparation of RNA-Seq libraries requires conversion of the RNA starting material into cDNA flanked by platform-specific adaptor sequences. Each of the published methods and commercial kits currently available for RNA-Seq library preparation suffers from at least one major drawback, including long processing times, large starting material requirements, uneven coverage, loss of strand information and high cost. We report the development of a new RNA-Seq library preparation technique that produces representative, strand-specific RNA-Seq libraries from small amounts of starting material in a fast, simple and cost-effective manner. Additionally, we have developed a new quantitative PCR-based assay for precisely determining the number of PCR cycles to perform for optimal enrichment of the final library, a key step in all SGS library preparation workflows.
Keywords: Next generation sequencing (NGS), second generation sequencing (SGS), RNA-Seq, library preparation, quantitative PCR (qPCR)
Introduction
Transcriptional profiling using second generation sequencing (SGS) (i.e., RNA-Seq) is a powerful tool for quantitatively characterizing the transcriptome of any organism or cell type.1,2 RNA-Seq enables measurement of relative abundances of transcripts, discovery of novel coding and non-coding transcripts, identification of splice junction sites and alternative splicing events and identification of sequence polymorphisms that differentiate RNA samples.3,4 The utility of RNA-Seq depends upon preparation of representative cDNA libraries for sequencing. Production of strand-specific RNA-Seq libraries involves reverse transcription of RNA to generate complementary single-stranded cDNA, synthesis of the second cDNA strand by PCR and addition of SGS platform-specific adaptors to the ends of the cDNA molecules either during cDNA synthesis or afterwards via ligation. Strand specificity is maintained either through addition of directional non-identical sequence tags at the 5′ and 3′ ends during cDNA synthesis, or by incorporating dUTP (instead of dTTP) into the second cDNA strand prior to tag ligation and then degrading this stand with uracil-DNA glycosylase (UDG).5,6 Alternatively, sequence adaptors can be ligated onto RNA molecules for direct RNA sequencing, but this leads to inherent biases toward shorter insert lengths and higher sequence error rates.7 Given that rRNA (rRNA) typically comprise > 90% of the total RNA population, strategies to deplete it have been implemented in most RNA-Seq library preparation protocols, providing more efficient access to the full diversity of transcriptome constituents.8-10 Each of the published methods and commercially available products for RNA-Seq library preparation suffers from at least one of the following disadvantages: Requirement for large amounts of RNA starting material, long processing times, labor intensive processing and high cost.5,6
We have developed a simple and cost-effective technique for preparing representative, strand-specific RNA-Seq libraries. In this technique, called “Peregrine,” short non-identical tag sequences are incorporated at the 5′ and 3′ ends of the first strand of cDNA during its synthesis. These tags preserve strand specificity information, and are used as primer binding sites for incorporation of full-length SGS adaptors and barcodes during second strand synthesis. In the initial cDNA synthesis step, Peregrine produces cDNA bearing short tag sequences (~20 bp) that do not interfere with downstream annealing reactions, such as those central to duplex-specific nuclease (DSN) mediated normalization11,12 and other molecular suppression techniques for depletion of unwanted high-abundance sequences, including rRNA.9 The much longer SGS-compatible adaptors (~60 bp), which can interfere with annealing reactions, are added at a subsequent step.
Additionally, we have developed a qPCR-based assay for precisely determining the number of PCR cycles to perform for optimal enrichment of the final RNA-Seq library. In all other library preparation techniques, the number of PCR cycles used for library enrichment is chosen on the basis of the input amount (a rough estimate), or empirically determined by subjecting replicate libraries to different cycling regimes (labor-intensive and costly). Our qPCR-based assay is a more direct and precise means of maximizing amplification of the library while minimizing biases imposed by over-cycling. The assay is simple and fast, and can be easily integrated into any RNA-Seq library preparation protocol to maximize the quality and yield of final product.
In this study, we demonstrate the capabilities of the Peregrine RNA-Seq library preparation technique, as well as our qPCR-based assay for optimizing the final enrichment step in library preparation. To test its performance in supporting analysis of transcriptomes of different complexity, we used Peregrine to generate RNA-Seq libraries from total RNA extracted from bacterial cells as well as from human primary cells. To evaluate its compatibility with molecular suppression methods, we used Peregrine to generate libraries from total RNA extracts that had been depleted of rRNA via Ribo-Zero (Epicentre), and in other experiments libraries generated from total RNA were depleted of rRNA-derived cDNA via DSN-mediated normalization. To determine the minimum amount of starting material required for preparation of representative RNA-Seq libraries, we used Peregrine to generate libraries from varying amounts (1‒100 ng) of total RNA extracted from a human cell line. We also directly compared the performance of Peregrine to that of ScriptSeq (Epicentre), a commercially available product that is commonly used for preparation of strand-specific RNA-Seq libraries.10,13-16 We found that our Peregrine RNA-Seq library preparation method produced representative, strand-specific cDNA libraries from ≥ 10 ng of RNA starting material, in ≤ 5 h, at a per-sample cost (~$5) significantly lower than offered by commercially available products.5,6
Results
Peregrine RNA-Seq library preparation method: Design considerations and mechanics
The Peregrine method was designed to minimize the time, cost, and number of sample manipulations required for preparation of representative RNA-Seq libraries. As illustrated in Figure 1, the RNA starting material is first chemically fragmented, to ensure production of cDNA that is of appropriate size for SGS. The RNA fragments are then used as templates for synthesis of the first cDNA strand by the Moloney murine leukemia virus (MMLV) reverse transcriptase. First strand cDNA synthesis is primed using PP_RT, a random hexamer sequence preceded by a 22 nt tag. As the MMLV reverse transcriptase reaches the 5′ end of the RNA template, the enzyme's terminal transferase activity adds three cytosine deoxyribonucleotides (5′-CCC-3′) to the 3′ end of the first strand of cDNA, and this sequence serves as the binding site for PP_TS, a primer comprised of a complementary sequence of three guanine ribonucleotides (5′-ggg-3′) preceded by a 19 nt tag. The resulting DNA:RNA hybrid facilitates a template switching reaction,17 in which the reverse transcriptase further extends the first strand of cDNA to incorporate the entire sequence complementary to primer PP_TS. This generates a first-strand cDNA product that reflects the full length of its RNA template, and which is flanked by short, non-identical tags in known orientation relative to the 5′ and 3′ ends of its template; the latter feature preserves strand specificity information. The tags also serve as priming sites for second strand synthesis and PCR amplification of the double-stranded (ds) cDNA. The tag-flanked ds cDNA can be depleted of highly abundant species through DSN-mediated normalization. In the final step of RNA-Seq library preparation, primers PP_A and PP_I are used to incorporate the full-length terminal adaptors necessary for Illumina sequencing.

Figure 1. Overview of the Peregrine RNA-Seq library preparation technique. Chemically fragmented RNA anneals with the random hexamer end of the primer PP_RT, and MMLV reverse transcriptase polymerizes the first cDNA strand. Upon reaching the end of the RNA template, the enzyme adds the untemplated sequence CCC to the 3′ end of the new cDNA strand. This serves as the annealing site for the complementary ribonucleotide sequence (ggg) of oligonucleotide PP_TS. At this point the enzyme switches templates, extending the first cDNA strand to incorporate sequence complementary to the PP_TS template. A qPCR assay can be performed to determine the number of PCR cycles required for optimal second strand synthesis and library amplification. During second strand synthesis, Illumina sequencing adaptors (one including a barcode) are incorporated at the ends of the cDNA. Depletion of rRNA-derived sequences may be performed prior to RNA fragmentation (Ribo-Zero) or following PCR (DSN-mediated normalization).
qPCR assay for optimization of second strand cDNA synthesis and library enrichment
A critical step in any SGS library preparation is the selective PCR enrichment of DNA molecules that bear the correct adaptor sequences at their 5′ and 3′ ends.18 Under-amplification at this stage leads to low yields, whereas over-amplification can lead to primer concatemers and non-representative libraries. To address this problem, we have developed a qPCR assay for determining the number of PCR cycles required for optimal amplification of a library. In this method, first strand cDNA synthesis products are combined with primers complementary to the short tags incorporated at their ends, and an EvaGreen-based qPCR19 is performed for 25 cycles (~35 min). Results from a representative experiment are shown in Figure 2. As expected, with increasing numbers of PCR cycles the overall yield of library products increased (Fig. 2A). Optimal library enrichment was achieved at cycle 11, the determined threshold cycle [C(t)]. Additional cycling was counterproductive: By cycle 13, unusually large products (≥ 1 kb) were generated with greater frequency, and by cycle 15, the majority of products were ≥ 200 bp larger than those observed at cycle 11 (Fig. 2B). The over-cycled libraries contained high levels of primer concatemers, and yielded greater numbers of reads that failed to pass the quality filter (data not shown). Results from a second, independent experiment (Fig. S1) show that cycling for the determined C(t) (in this case, 12 cycles) generated a library in which long inserts predominate (79% of reads included ≥ 30 bp of insert sequence). While less cycling (eight cycles) generated a library with a higher proportion of long inserts (94% ≥ 30 bp), there was ~15-fold less product than generated in cycling to the C(t) (data not shown). Additional cycling (16 cycles) yielded a library with a markedly lower proportion of long inserts (65% ≥ 30 bp) and high levels of primer concatemers (9% of reads consisted of only primer sequence). The results from these and similar experiments indicate that PCR amplification for the number of cycles corresponding to the C(t) in the qPCR assay ensures optimal library enrichment, as it generates maximum yield without imposing over-amplification biases. This qPCR strategy can be used to optimize enrichment of libraries prepared by any method, for analysis on any SGS platform.

Figure 2. qPCR assay for optimizing second-strand synthesis and library enrichment. The final PCR step of library preparation must generate sufficient yield for SGS, yet avoid biases associated with over-amplification. (A) Amplification curves from qPCR test performed in quadruplicate. First-strand cDNA products generated from human PBMC total RNA served as template in qPCR reactions. The horizontal line indicates the detection threshold. The green vertical bar indicates the cycle number at which fluorescence intensity exceeded the detection threshold [i.e., the cycle threshold, C(t)]; in this experiment, the C(t) is cycle 11. The gray vertical lines and horizontal arrow indicate the range within which the C(t) typically falls. The colored vertical bars [including the green one marking C(t)] indicate the cycle numbers selected for testing in the Figure 2B experiment. (B) Second strand synthesis yields and product sizes. The first-strand cDNA products described above served as template in PCR reactions, the products of which were assessed using a DNA High Sensitivity chip on a Bioanalyzer machine. The colored traces indicate the quantity and size of reaction products generated through use of cycle numbers selected as described above. Note that as cycle number surpassed C(t), product sizes increased (particularly evident at ≥ 1,000 bp).
Peregrine library preparation reproducibility and compatibility with rRNA depletion methods
To evaluate the performance of the Peregrine library preparation technique, we first used it for analysis of transcripts expressed by the well-characterized E. coli K-12 strain.20,21 In these experiments, we assessed Peregrine's compatibility with two methods for depleting rRNA. Since total RNA extracts are dominated by rRNA species, it is a widely adopted practice to deplete rRNA, or their cDNA products, during library preparation, in order to increase the coverage of other, more informative constituents of the transcriptome. For instance, Yi et al. demonstrated the use of DSN-mediated normalization to deplete rRNA-derived cDNA species for analysis of the E. coli transcriptome.22 Additionally, Epicentre’s Ribo-Zero kit is designed to remove rRNA prior to library preparation. Thus, in these experiments we prepared E. coli K-12 cDNA libraries using Peregrine, alone or in combination with an rRNA depletion step (DSN-mediated normalization, or Ribo-Zero), for transcriptome analysis. These libraries were directly compared with ones generated from the same starting material using ScriptSeq (Epicentre), a commercially available product commonly used for preparation of strand-specific RNA-Seq libraries.10,13-16
Our results demonstrate that the majority of reads from Peregrine-prepared cDNA libraries could be mapped with high confidence to the reference E. coli genome (Table S1). In absence of an rRNA depletion step, ~96% of the mapped reads were assigned to rRNA, consistent with observations made in other studies (e.g., ref. 22). As expected, the use of DSN or Ribo-Zero to deplete rRNA reduced the proportion of mapped reads assigned to rRNA (to ~40% or ~23% of mapped reads, respectively), and increased those of reads mapping to transcriptome constituents other than rRNA (Fig. 3; Fig. S2A and Table S1). Aside from these differences, the untreated and rRNA-depleted libraries strongly resembled one another, as evident from their high coefficient of determination (R2) and Spearman rank correlation coefficient (ρ) values (Fig. S2A). Technical replicates of Peregrine library preparation, with or without rRNA depletion, showed a high degree of similarity, as evident in small standard deviation values (≤ 6%) in read mapping statistics (Table S1) and high coefficient of determination (R2) values (≥ 0.92) in scatter plots (Fig. S3A). By these standards, the reproducibility of Peregrine library preparation was comparable to that of ScriptSeq (Fig. S3 and Table S1). In summary, our results indicate that the Peregrine library preparation method consistently generated high-quality RNA-Seq libraries from E. coli K-12 total RNA, and was compatible with two different methods for rRNA depletion.
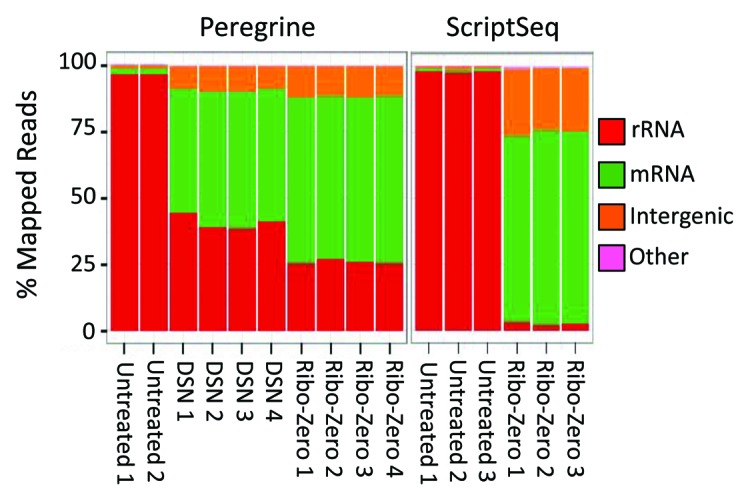
Figure 3. Categorization of Peregrine and ScriptSeq library reads mapping to different RNA species comprising the E. coli K-12 transcriptome. Libraries were prepared from E. coli K-12 total RNA. Each library was sequenced, and reads aligning to the reference E. coli genome were categorized according to the type of RNA species to which they mapped. Additional read statistics for the libraries are included in Table 1; Tables S1 and S2. Left panel: Peregrine libraries were untreated (two replicates), normalized via DSN treatment (four replicates) or prepared from Ribo-Zero treated RNA (four replicates). Right panel: ScriptSeq libraries were untreated (three replicates) or prepared from Ribo-Zero treated RNA (three replicates).
Strand specificity of Peregrine RNA-Seq libraries
Studies of prokaryotic and eukaryotic transcriptomes have revealed a wide variety of RNA species, including regulatory noncoding RNA that are antisense to mRNA.23-30 Proper characterization of transcriptome diversity requires methods that reliably distinguish sense from antisense RNA molecules. The Peregrine library preparation method is designed to support accurate strand assignment, as each cDNA molecule that it generates is flanked by non-identical tags in known orientation relative to the 5′ and 3′ ends of its RNA template (Fig. 1). To verify that Peregrine preserves strand specificity information, we analyzed strand assignment for reads derived from libraries generated from E. coli K-12 RNA. In libraries that were not depleted of rRNA, we found that > 99% of the reads that mapped to rRNA genes were assigned to the sense strand (as defined by the reference genome annotation) (Table 1). This result is consistent with expectations, as there is no evidence of antisense transcription from bacterial rRNA genes. Approximately 95% of the reads that mapped to CDS were assigned to the sense strand. The level of antisense assignment (~5%) is comparable to those seen in libraries prepared by other strand-specific methods,5 and is thought to reflect the fact that many CDS in E. coli are transcribed from both the sense and antisense strands.23,24 Consistent with this interpretation, we found that for 90 CDS previously shown to support robust antisense transcription (ref. 24, see their Table S3), only ~87% of the mapping reads were assigned to the sense strand (Table 1). ScriptSeq libraries that were not depleted of rRNA showed similar trends in strand specificity. Taken together, these results indicate that the Peregrine library preparation method successfully preserves strand specificity information.
Table 1. Strand specificity of Peregrine and ScriptSeq libraries prepared from E. coli RNA.
| Prep & Treatment | rRNA | CDS | CDS w/known asRNA |
|---|---|---|---|
| Peregrine Untreated |
99.77 ± 0.01 |
94.77 ± 0.04 |
86.97 ± 0.35 |
| Peregrine DSN |
99.86 ± 0.01 |
94.20 ± 0.23 |
87.17 ± 0.19 |
| Peregrine Ribo-Zero |
2.21 ± 0.09 |
87.15 ± 1.28 |
81.72 ± 1.51 |
| ScriptSeq Untreated |
99.53 ± 0.10 |
97.85 ± 0.19 |
94.04 ± 1.24 |
| ScriptSeq Ribo-Zero | 22.33 ± 4.27 | 97.87 ± 0.30 | 93.11 ± 0.92 |
Peregrine and ScriptSeq libraries were prepared from the same sample of E. coli K-12 total RNA, sequenced and aligned to the reference E. coli genome. Read statistics for the libraries analyzed are included in Tables S1 and S2. Reads mapping to rRNA genes, CDS, or a subset of 90 CDS from which robust antisense transcription has been observed (ref.e 24, see their Table S3) were assessed for strand specificity. Values indicate the proportion of reads mapping to the sense strand (mean ± standard deviation).
We had anticipated that inclusion of an rRNA depletion step would not appreciably affect strand specificity in Peregrine library preparation, and indeed we found this to be the case for DSN-mediated normalization (Table 1). On the other hand, inclusion of Ribo-Zero treatment generated libraries in which the vast majority of reads mapping to rRNA were assigned to the antisense strand. Presumably these reads derived from the Ribo-Zero probe (sequences complementary to rRNA) rather than from endogenous rRNA, as reads assigned to antisense rRNA were extremely rare in untreated and DSN-normalized libraries. Ribo-Zero treatment also affected strand assignment for reads mapping to CDS (only ~87% assigned to the sense strand, as compared with 94–95% for untreated and DSN-normalized libraries). Libraries prepared using ScriptSeq showed similar trends, including a dramatic increase in antisense rRNA assignments upon inclusion of Ribo-Zero treatment. However, Ribo-Zero had no detectable effect on strand assignment for ScriptSeq reads mapping to CDS. In summary, our results indicate that Peregrine library preparation successfully preserved stand specificity information when used in isolation or in combination with DSN-mediated normalization, but suffered noticeable loss of strand specificity when combined with Ribo-Zero treatment.
Uniformity of transcriptome sampling in Peregrine RNA-Seq libraries
Effective RNA-Seq library preparation methods provide access to the broad diversity of RNA species comprising the transcriptome, and preserve the relative abundances of those species in representative libraries. Both of these functions depend upon the uniformity with which transcriptome constituents are sampled during library preparation. To assess transcriptome sampling in Peregrine library preparation, we first compared the transcriptional profiles represented in Peregrine libraries to those represented in ScriptSeq libraries that were generated from the same starting material (E. coli K-12 RNA depleted of rRNA via Ribo-Zero). We found that while the transcriptional profiles represented in the libraries roughly resembled one another at a gross level (R2 = 0.61, Spearman rank correlation coefficient = 0.82), the Peregrine libraries supported sequencing of a greater number of transcriptome constituents (163 detected in Peregrine but not ScriptSeq libraries, vs. seven detected in ScriptSeq but not Peregrine libraries) (Fig. 4A). Similarly, the Peregrine libraries yielded fewer reads that mapped to CDS (1.2 M, compared with 1.5 M for ScriptSeq), yet these were distributed across a greater fraction of CDS in the genome (99%, compared with 97% for ScriptSeq) (Table S3). Moreover, the distributions of mean coverage levels for operons represented in the libraries indicate that Peregrine produced a narrower range of coverage levels across operons (mean ± standard deviation of the Log10 distribution: 1.16 ± 0.43 and 0.82 ± 0.79 for Peregrine and ScriptSeq, respectively) (Fig. 4B). These observations suggest that Peregrine sampled the transcriptome constituents at least as uniformly as did ScriptSeq.
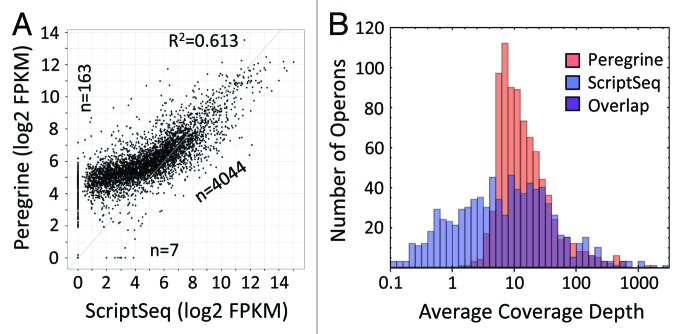
Figure 4.E. coli K-12 operon coverage provided by reads from Peregrine and ScriptSeq libraries. Read statistics for the library sub-samples analyzed are included in Tables S3 and S4. (A) Scatter plot of Log10 of FPKM values for each operon. Points on the axes represent transcripts with zero coverage in one of the two libraries. The number of data points in the diagonal cloud and on the axes is indicated. The coefficient of determination (R2) value, as calculated for all transcripts represented in the libraries, is indicated as well. The coverage levels provided by Peregrine and ScriptSeq libraries are correlated, but ScriptSeq has lower coverage values for low-expression operons and somewhat higher values for high-expression operons. The Spearman rank correlation coefficient between the two methods is 0.819 for operons with mean coverage > 1 in both sets of libraries. (B) Histograms of Log10 of mean coverage levels of operons, with Peregrine in red and ScriptSeq in blue. Mean (standard deviation) of each distribution is 1.16 (0.43) and 0.82 (0.79). Quartile divisions for mean coverage (not Log10) are (7.02128, 11.2367, 22.5935) and (1.67284, 7.17042, 23.0521), indicating a disparity of coverage for low-expression operons that is compensated by high coverage of a small number of high-expression operons.
Continuity and uniformity of transcript coverage in Peregrine RNA-Seq libraries
RNA-Seq library preparation methods that provide uniform, full-length sequence coverage of expressed transcripts are particularly useful when characterizing transcription start sites,31 5′-/3′-untranslated regions,32,33 splicing variants,34,35 operon organization36 and pathogen transcriptomes.37-39 Thus, in evaluating the performance of Peregrine library preparation it was important to characterize the sequencing coverage provided in terms of its distribution along the length of individual transcripts. Additionally, it was important to determine whether uniformity of coverage across operons (i.e., uniformity in transcriptome sampling) is consistently associated with uniformity of coverage within operons. For these analyses, we once again directly compared Peregrine vs. ScriptSeq libraries prepared from the same starting material (E. coli K-12 RNA depleted of rRNA via Ribo-Zero).
To assess continuity of sequence coverage along transcript length, we counted the number of coverage gaps (≥ five contiguous bases without coverage) for each operon represented in the library, normalized the counts for operon length, and plotted them as a function of each operon's average coverage depth. As expected, operons with low average coverage depth generally showed many gaps in coverage, whereas those with high average coverage depth showed greater continuity of coverage (Fig. 5). The trend was more pronounced in Peregrine libraries than in ScriptSeq libraries, as indicated by empirical linear fits of the data. Indeed, the slope of the Peregrine fit (-1.15) was almost twice that of the ScriptSeq fit (-0.65), despite the fact that the libraries yielded similar numbers of reads mapping to operons (1.7 M and 1.8 M for Peregrine and ScriptSeq, respectively). Additionally, we determined that the coverage-weighted average number of segments into which each operon's coverage was broken (calculated for each operon as one plus the number of gaps) was 1.55 for Peregrine, and 2.27 for ScriptSeq. These values compare well to those reported for other methods (ref. 5, see their Fig. 5A), such that by this measure Peregrine ranks as the top performing method, and ScriptSeq as among the better ones. In summary, our results indicate greater continuity of coverage, and imply greater uniformity in distribution of coverage depth, along transcript length in Peregrine libraries, as compared with ScriptSeq libraries.
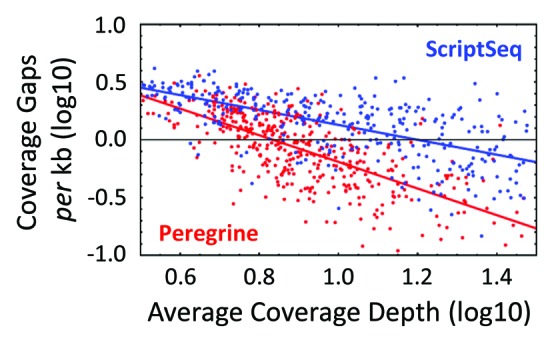
Figure 5. Continuity of transcript coverage in Peregrine and ScriptSeq libraries prepared from E. coli K-12 RNA. Plot of the Log10 number of coverage gaps per kilobase of operon as function of the Log10 coverage depth of the operon. A gap is defined as ≥ five contiguous bases with zero coverage. Linear regression of the gap counts corresponding to coverage values from three to 30 yielded a slope ± standard deviation of -1.15 ± 0.058 and -0.64 ± 0.043 for Peregrine and ScriptSeq, respectively, showing empirically that number of gaps fell twice as fast with increasing coverage for Peregrine libraries. Read statistics for the library sub-samples analyzed are included in Tables S3 and S4.
To directly measure the uniformity of sequence coverage provided by Peregrine and ScriptSeq, for each method we calculated the coefficient of variation (CV) for distribution of coverage depth along operon length, limiting analysis to the top 50% of operons with respect to average coverage depth. The distribution of CV values for each method is shown in Figure 6A. The average CV for the Peregrine and ScriptSeq distributions were 0.82 and 1.46, respectively. These values are comparable to those reported for other methods (ref. 5, see their Fig. 4A). However, one cannot properly estimate the statistical significance of differences in average CV, because CV are not bounded and are nonlinearly sensitive to outliers, and because the distributions of CV values themselves are highly tailed even when low-coverage operons are excluded from analysis. It is also possible that differences in the average CV overemphasize rare spikes in coverage. For these reasons, we also computed for each operon the Gini coefficient (GC), a bounded and robust nonparametric statistical measure of inequality among values of a frequency distribution. The distribution of GC values for each method is shown in Figure 6B. We found that the mean ± standard deviation of GC for all non-zero-coverage operons was 0.41 ± 0.09 for Peregrine, and 0.63 ± 0.13 for ScriptSeq, where smaller GC values indicate greater uniformity of coverage within the operon. Student's t-test for the null hypothesis that the means of the distributions are the same yielded a pvalue of 1.4 × 10−238. Moreover, the mean ± standard deviation GC values are robust to coverage level: Limiting analysis to the top 50% operons ranked by coverage (as for calculation of CV values) produces mean ± standard deviation GC values of 0.40 ± 0.10 for Peregrine, and 0.59 ± 0.12 for ScriptSeq. Together, these results indicate that distribution of coverage depth along the length of the operon was significantly more uniform in Peregrine libraries than in ScriptSeq libraries, at all levels of operon coverage, consistent with our interpretation of the results shown in Figure 5.
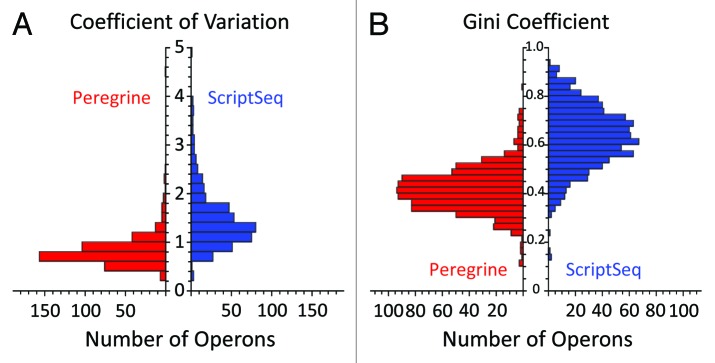
Figure 6. Statistical analysis of transcript coverage uniformity in Peregrine and ScriptSeq libraries prepared from E. coli K-12 RNA. Read statistics for the library sub-samples analyzed are included in Tables S3 and S4. (A) Distribution of coefficient of variation (CV) values describing uniformity of coverage depth within each operon. Analysis was limited to the top 50% of operons ranked by average coverage. Mean CV values are 0.82 and 1.46 for Peregrine and ScriptSeq, respectively. (B) Distribution of Gini coefficient (GC) values describing uniformity of coverage depth within each operon. For reference, lower Gini coefficients indicate greater uniformity. Mean ± standard deviation GC values for all non-zero-coverage operons are 0.41 ± 0.09 and 0.63 ± 0.13 for Peregrine and ScriptSeq, respectively. Student's t-test for the null hypothesis that the means of the distributions are the same yielded a pvalue of 1.4 × 10−238.
As a further means of assessing the uniformity of sequence coverage along transcript length, we measured the depth of coverage for each percentile of length for each operon represented in the library, then calculated the mean coverage depth at each length percentile for the library as a whole. We found that in Peregrine libraries, mean coverage depth varied within a narrow range of values (0.50% to 1.46% at each length percentile) (Fig. 7A) comparable to those reported for other methods (ref. 5, see their Fig. 4B). Relative to Peregrine libraries, ScriptSeq libraries showed greater variation in coverage depth along transcript length (range of 0.25‒ 2.18% at each length percentile) (Fig. 7A). Both sets of libraries showed reduced coverage depth at the length percentiles corresponding to the 3′ ends of the operons. This result was expected, given that both methods depend upon random priming for reverse transcription (synthesis of the first cDNA strand), which typically leads to reduced coverage at the 3′ end.5 To more precisely measure coverage bias at the ends of transcripts, we analyzed the coverage of 23S and 16S rRNA, which have well-defined 5′ and 3′ ends40 and are highly abundant in untreated libraries. As anticipated, we found that in both Peregrine and ScriptSeq libraries, coverage depth dropped at the 3′ end of each transcript, over the final 90–160 nt (Fig. 7B). In summary, our results indicate that Peregrine, with or without an rRNA depletion step, provides sequence coverage of high continuity and uniformity along the full length of the transcript, with a coverage drop at the 3′ end that is comparable to that of ScriptSeq and other library preparation methods that rely upon random priming of reverse transcription.5

Figure 7. Uniformity of coverage along length of transcripts in Peregrine and ScriptSeq libraries prepared from E. coli K-12 RNA. Read statistics for the library sub-samples analyzed are included in Tables S3 and S4. (A) Average coverage at each percentile of length for all operons. Coverage depth was tabulated at each nucleotide position within each operon of > 1,000 bp. Each operon was length-normalized by percentile, and coverage was calculated as the total number of sequenced bases in each percentile divided by the total number of sequenced bases. The first percentile represents the 5′ end and the 100th percentile represents the 3′ end of the operon. (B) Average coverage at each nucleotide within the 23S (left panel) and 16S (right panel) rRNA, which have well-defined 5′ and 3′ ends.
Use of Peregrine for RNA-Seq analysis of human PBMC transcriptome
Peripheral blood mononuclear cells (PBMC) are comprised of diverse cell types (lymphocytes, monocytes, macrophages, dendritic cells) that play vital roles in both innate and adaptive immunity. Due to their importance to immunity, PBMC are commonly studied primary cells and often the focus of transcriptional profiling studies, including those using RNA-Seq.41-47 We used the Peregrine method, alone or in combination with DSN-mediated normalization, to prepare libraries from human PBMC for RNA-Seq analysis. As with our E. coli K-12 transcriptome analysis, we found that the majority of reads from Peregrine-prepared PBMC libraries mapped with high confidence to the reference human (hg19) genome (Table S5). In absence of rRNA depletion, ~81% of the mapped reads were assigned to rRNA (Fig. 9; Table S5). DSN treatment led to markedly reduced numbers of rRNA reads (to ~8% of mapped reads), and concomitant gains in numbers of reads mapping to other constituents of the transcriptome (Fig. 8; Fig. S5 and Table S4). Technical replicates of Peregrine library preparation, with or without DSN treatment, showed strong similarity, as seen in small standard deviation values (typically < 2%) in read mapping statistics (Table S5) and high R2 values (≥ 0.95) in scatter plots (Fig. S5). Strand specificity values were comparable to those seen in Peregrine and ScriptSeq libraries generated from E. coli RNA (Tables 1 and 2). The uniformity of coverage depth along transcript length was comparable as well, though negative bias at the 3′ end was less pronounced (Fig. S6). These results indicate that Peregrine consistently generated high-quality libraries from human primary cells, and that its use in combination with an rRNA depletion method (DSN-mediated normalization) favored sequencing of non-rRNA constituents of the transcriptome.

Figure 9. Categorization of reads mapping to different RNA species comprising the human carcinoma cell line (A549) transcriptome, as a function of starting material quantity. Peregrine libraries were prepared from 100 ng, 50 ng, 10 ng or 1 ng of A549 total RNA, normalized via DSN treatment, and sequenced. Reads were aligned to the reference human genome, and categorized according to the type of RNA species to which they mapped. Reads failing to pass the quality filter criteria were rejected (dark gray). Reads that passed the quality filter but failed to align to the genome were categorized as unmapped (light gray). Aligned reads were categorized according to the type of RNA species to which they mapped. Additional read statistics for these libraries are included in Table S6.
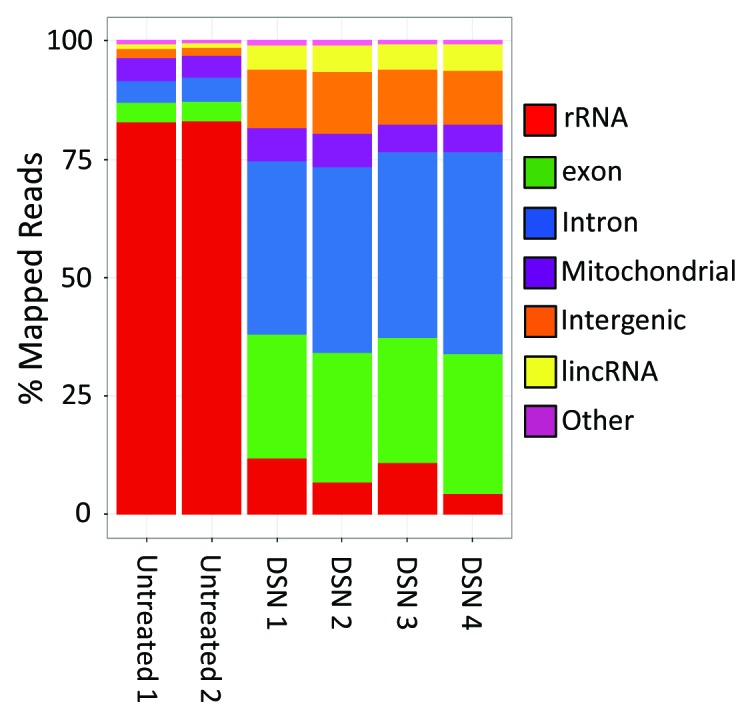
Figure 8. Categorization of reads mapping to different RNA species comprising the human PBMC transcriptome. Peregrine libraries were prepared from human PBMC total RNA; two control libraries were untreated, and four were normalized via DSN. Each library was sequenced, and reads aligning to the reference human genome were categorized according to the type of RNA species to which they mapped. Additional read statistics for these libraries are included in Table 2; Table S5.
Table 2. Strand specificity of Peregrine libraries prepared from human PBMC RNA.
| Treatment | rRNA | CDS |
|---|---|---|
| Untreated |
99.50 ± 0.28 |
96.37 ± 0.00 |
| DSN | 99.71 ± 0.15 | 97.03 ± 0.00 |
Peregrine libraries were prepared from human PBMC total RNA, sequenced, and aligned to the reference human genome. Read statistics for the libraries analyzed are included in Table 2; Table S5. Reads mapping to rRNA genes or CDS were assessed for strand specificity. Values indicate the proportion of reads mapping to the sense strand (mean ± standard deviation).
RNA-Seq analysis of the human carcinoma cell line (A549) transcriptome, using Peregrine libraries prepared from reduced amounts of starting material
An important consideration for any SGS library preparation method is its starting material requirements. Methods that generate representative libraries from little starting material are particularly useful, because they allow analysis of small, precious samples, such as those collected in animal and clinical studies. To determine the starting material requirements of Peregrine, we created a dilution series of fragmented total RNA derived from the well-characterized human lung carcinoma cell line A549,44 and prepared libraries from each dilution using Peregrine in combination with DSN-mediated normalization. We found that libraries prepared from 100 ng, 50 ng or 10 ng of total RNA were essentially indistinguishable from one another, with respect to the results generated from their analysis by SGS (Fig. 9; Table S6). In each case, ~80% of the reads passed the quality filter criteria, ~88% of those reads mapped to the reference human genome, and ≤ 15% of those were assigned to rRNA. Indeed, the only noticeable trend in the results from these libraries was that those generated from smaller amounts of starting material yielded fewer reads assigned to rRNA, suggesting that DSN was more effective in depleting rRNA when smaller amounts of starting material were used. In contrast, libraries prepared from 1 ng of fragmented total RNA were of poor quality: Only ~16% of the reads passed the quality filter, fewer than half of those (~45%) mapped to the reference human genome, and > 25% of the mapped reads were assigned to rRNA. This difference in library quality was reflected in the genome coverage provided by the libraries: Those generated from ≥ 10 ng of starting material yielded reads mapping to ~66% of human exons, at an average coverage depth of ≥ 34 reads/exon; whereas those generated from 1 ng of starting material yielded reads mapping to fewer than half as many exons (29%), at an average coverage depth of only two reads/exon. These results, together with those from libraries generated from a variety of other starting materials (data not shown), indicate that as little as 10 ng of fragmented total RNA is sufficient starting material for reproducible preparation of high-quality RNA-Seq libraries using the Peregrine method.
Discussion
In this study, we evaluated Peregrine, a new technique for fast, simple and cost-effective preparation of RNA-Seq libraries from small amounts of starting material. In this method, reverse transcription is initiated by a random priming event that incorporates a short tag sequence at the 5′ end of the first strand of cDNA. Through use of the MMLV reverse transcriptase in combination with an oligo that promotes template switching, a different tag is incorporated at the 3′ end of the cDNA strand. This produces cDNA fragments flanked by short, non-identical tags that preserve strand specificity information and are compatible with DSN-mediated normalization. The tags serve as primer binding sites for incorporation of full-length Illumina adaptors and barcodes during second strand synthesis. Adaptor incorporation via MMLV reverse transcriptase and SMART technology17 was first implemented for the SOLiD platform by Cloonan et al.,48 and modified for Illumina sequencing by Levin et al.5 Peregrine improves upon these predecessor methods by: (1) Using shorter tag sequences, which prevents interference with annealing reactions, such as those required for normalization; (2) Introducing the template-switching oligo at initiation of second strand synthesis (rather than late in the process), which greatly improves reaction efficiency and (3) Using a custom read primer that directs the sequencer to begin reads at the 5′ end of the cDNA (rather than within the adaptor) ensures that sequence diversity is high during the cluster-calling stage, thereby preventing the problem of “monotemplate sequencing.”5 Another key innovation is use of a novel qPCR assay to precisely determine the number of cycles required for optimal amplification in the final library enrichment step. We have found this assay to be especially useful in enabling comparison of samples that have yielded dramatically different amounts of first strand cDNA synthesis product. In these cases, it is particularly important keep the balance between cycling enough (to generate sufficient product for sequencing) but not too much (to prevent over-cycling effects such as primer concatemers), to ensure comparison of high-quality libraries. Our qPCR assay enables this balance to be kept with precision, with little time invested (~35 min).
To demonstrate the versatility of the Peregrine method, we used it to prepare RNA-Seq libraries from total RNA extracts from a bacterial strain (E. coli K-12), human primary cells (PBMC), and a human cell line (A549). In each case, we found that the Peregrine method consistently generated representative, strand-specific libraries yielding high-quality sequence data. For direct comparison with Peregrine, we used ScriptSeq to prepare libraries from the same E. coli RNA starting material. We found that Peregrine sampled the diversity of transcriptome constituents at least as uniformly as did ScriptSeq. Both methods provided less sequence coverage, to comparable degrees, at the 3′ end (90–160 nt) of transcripts, which is typical for library preparation methods that rely upon random priming of reverse transcription.5 Analysis of overall coverage within transcripts revealed that Peregrine provided more continuous and uniform coverage than did ScriptSeq, and in these respects performed as well as the top ranked methods tested by Levin et al.5
Inclusion of an rRNA depletion step (DSN-mediated normalization, or Ribo-Zero treatment) facilitated sampling of the other, less abundant constituents of the transcriptome. However, in-depth analysis of the E. coli sequencing results revealed that use of Ribo-Zero with Peregine led to increased numbers of reads mapping to antisense rRNA (> 99% sense in untreated and DSN-normalized libraries vs. ~2% sense in Ribo-Zero treated libraries). These antisense rRNA reads presumably derived from inadvertant sequencing of the Ribo-Zero probe. We also detected an increase in reads mapping to antisense CDS in these libraries. ScriptSeq libraries prepared from the same starting material showed the former, but not the latter, effect. These results indicate that use of Ribo-Zero can affect measurement of strand specificity in RNA-Seq libraries, in ways that may differ depending upon the library preparation method.
It should be noted that while short RNA species such as tRNA and miRNA were represented in the Peregrine libraries analyzed, their levels were lower than expected (data not shown). This is likely due to the fact that the method we used for size selection during library preparation (Agencourt AMPure XP beads) is not recommended for recovery of fragments < 100 bp. It is possible that use of alternative size selection methods favoring recovery of shorter fragments would be sufficient to enable analysis of short RNA species via Peregrine. For the library preparation pipeline described herein, however, we recommend a lower size limit for RNA species of ≥ 200 nt to ensure robust and consistent recovery for analysis.
In summary, we have found that Peregrine libraries can be reliably prepared from as little as 10 ng of total RNA, in as few as 5 h, at a per-sample cost (~$5) significantly lower than that of ScriptSeq (≥ $100/sample) and other commercially available products.5,6 The Peregrine method offers several important advantages over other cDNA library preparation methods, and holds great promise for adding value to a wide variety of RNA-Seq studies.
Materials and Methods
Samples and RNA extraction
Escherichia coli strain K-12 was obtained in lyophilized form from ATCC. Bacteria were revived with the addition of 300 μl of LB broth (Difco) followed by plating on LB agar (Difco) and incubation at 37°C overnight. Four individual colonies were used to inoculate separate tubes of 5 ml fresh LB broth, and grown at 37°C with shaking for 3 h. The four cultures were then combined, and 1 ml of the mixture was centrifuged at 5,000 rpm at 4°C for 5 min. RNA was extracted from the cells using the RNeasy Protect Bacterial Mini Kit with on-column DNase treatment (Qiagen) following the manufacturer’s protocol.
Human peripheral blood mononuclear cells (PBMC) were obtained from Astarte Biologics (lot #515SE10), and human A549 cells were purchased from ATCC. Five to 10 million cells were centrifuged at 1,000 rpm at 4°C for 5 min, and re-suspended in 1 ml of RNAzol (Molecular Research Center), followed by addition of 400 μl of sterile nuclease-free water. Following incubation at room temperature (RT) for 15 min, the tubes were centrifuged at 16,000 rpm at 4°C for 15 min, and ~800 μl of the aqueous phase from each tube transferred to a new 2-ml tube and mixed 1:1 with 100% nuclease-free ethanol (Sigma). RNA was extracted using the Direct-zol kit (Zymo Research), following the manufacturer’s instructions; total RNA was eluted in sterile nuclease-free water, and stored at -80°C.
The concentration and purity of each RNA sample was measured using a NanoDrop 2000 (Thermo-Fisher). In all cases, the A260/A280 and A260/A230 ratios were > 2.0, indicating a pure RNA product. To assess the RNA integrity and population size, samples were run on a BioAnalyzer (Agilent) using the RNA Nano 6000 chip; the human samples were analyzed using the total eukaryotic RNA program, while the bacterial samples were analyzed using the total prokaryotic RNA program. In all cases, the RNA integrity numbers (RIN) were ≥ nine, indicating negligible degradation during RNA extraction and purification.
Preparing RNA for cDNA synthesis
E. coli RNA samples were separated into two aliquots, and one was depleted of rRNA via Ribo-Zero treatment using the “Gram-Negative Bacteria” kit (Epicentre); RNA samples from human PBMC or human cell line A549 were not treated with Ribo-Zero. In all cases, 200 ng aliquots of RNA (total or rRNA-depleted via Ribo-Zero) were subjected to random fragmentation in 20 μl reactions, through addition of 2 μl of 10X NEBNext RNA fragmentation buffer (New England BioLabs) and incubation at 94°C for 3 min, followed by immediate cooling on ice and addition of 2 μl of NEBNext RNA fragmentation stop solution (New England Biolabs). Fragmented RNA was purified using the Zymo RNA Clean and Concentrator-5 system (Zymo Research), following the manufacturer's general procedure (≥ 17 bp) and eluting in 6 μl of nuclease-free water. RNA concentrations were determined by NanoDrop 2000 (Thermo-Fisher). RNA integrity and fragment size distribution were assessed by BioAnalyzer (Agilent) using the RNA Nano 6000 chip, as described in the previous section; in all cases, RIN were ≥ nine, and fragment sizes averaged 200–500 nt. For experiments evaluating the starting material requirements of the Peregrine library preparation method, aliquots of total RNA from the human cell line A549 were diluted to 100 ng, 50 ng, 10 ng and 1 ng, in duplicate.
First strand cDNA synthesis
Three and a half μl (10–200 ng) aliquots of fragmented RNA (total or Ribo-Zero treated) were mixed with 1 μl of 25 mM primer PP_RT (5′-CAGACGTGTGCTCTTCCGATCTNNNNNN-3′), incubated at 65°C for 2 min, and then immediately cooled on ice. While on ice, 4.5 μl of a master mix containing 2 μl of SMARTScribe 5X First-Strand Buffer, 0.25 μl of 20 mM DTT, 1 μl of 10 mM dNTP mix, 0.25 μl of RiboGuard RNase inhibitor and 1 μl of SMARTScribe Reverse Transcriptase (all products from Takara) were added, and the mixture incubated at 25°C for 3 min followed by 42°C for 1 min. At this point, 1 μl of 12 mM template-switching oligo PP_TS (5′-CAGGACGCTGTTCCGTTCTauggg-3′) (lower-case letters indicating ribonucleotides) were added while the reaction mixture remained in the thermocycler, and incubation continued at 42°C for 1 h. The reaction was then terminated through incubation at 70°C for 10 min. The reaction products (first strand of cDNA) were purified using 18 μl (1.8X volumes) of AMPure XP beads (Beckman Coulter Genomics) and eluting in 25–50 μl of nuclease-free water, following the manufacturer’s protocol.
Quantification of cDNA libraries for second strand synthesis
A new qPCR-based assay was used to determine the number of PCR cycles needed for production and optimal amplification of high-quality double-stranded (ds) cDNA libraries from first strand cDNA synthesis reaction products. After diluting the first strand cDNA 1:10 in nuclease-free water, 1 μl of the dilution was combined with 5 μl of SsoFast EvaGreen SuperMix (Bio-Rad), 3 μl of nuclease-free water, 0.5 μl of 10 mM primer PP_P1 (5′-CAGGACGCTGTTCCGTTCTATGGG-3′) and 0.5 μl of 10 mM primer PP_P2 (5′-CAGACGTGTGCTCTTCCGATCT-3′). The assays were run in quadruplicate on a CFX96 qPCR machine (Bio-Rad), using the following cycle parameters: 95°C for 45 sec, followed by 25 cycles of 95°C for 5 sec and 60°C for 30 sec. The cycle number at which fluorescence intensity exceeded the detection threshold [i.e., the cycle threshold (Ct)] was identified as optimal (maximum yield of SGS-ready cDNA with minimal over-amplification bias) for production of ds cDNA libraries from the undiluted first strand cDNA synthesis reaction products.
Second strand synthesis and PCR amplification to prepare ds cDNA for DSN-mediated normalization
To generate the second strand of cDNA for normalization treatment, 10 μl of the first strand cDNA synthesis reaction products were mixed with 1 μl of 10 mM primer PP_P1, 1 μl of 10 mM primer PP_P2, 12.5 μl of water, 25 μl of Premix E from the Failsafe PCR system and 0.5 μl of FailSafe Enzyme mix (all products from Epicentre) and subjected to the following PCR conditions: 94°C for 1 min, followed by 10–14 cycles (determined by qPCR result) of 94°C for 30 sec, 55°C for 30 sec and 68°C for 3 min. After a final extension at 68°C for 7 min, the reaction products (ds cDNA libraries) were purified using the Zymo DNA Clean and Concentrator-5 kit and eluting in 10 μl of nuclease-free water. The concentration of each cDNA library was measured by Nanodrop 2000.
DSN-mediated normalization was performed using the Trimmer kit (Evrogen), following the manufacturer's protocol. Briefly, the cDNA library concentration was adjusted to 20 ng/μl using nuclease-free water, its ds cDNA species denatured at 98°C for 3 min in the presence of hybridization buffer [50 mM HEPES (pH 7.5), 0.5M NaCl], and the cDNA strands allowed to re-anneal at 68°C for 5 h. At this point, a master buffer was added to each hybridization reaction, followed by 1.5 μl of DSN enzyme, and incubation at 68°C continued for 25 min. After adding 2 μl of stop solution (100 mM EDTA) to each reaction, the products (predominantly single-stranded cDNA) were purified using 1.6X volumes of AMpure XP beads and eluted in 25 μl of nuclease-free water. An aliquot of each purified product was diluted 1:10 in nuclease-free water for qPCR-based quantitation, as described above.
Second strand synthesis, PCR amplification and size selection to prepare SGS-ready ds cDNA
To generate the second strand of cDNA and add adaptors that support SGS analysis, 10 μl of the first strand cDNA synthesis reaction products were mixed with 1 μl of 10 mM PP_A (5′-AATGATACGGCGACCACCGAGATCTACACTTCGCTACAGGACGCTGTTCCGTTCTATGGG- 3′), 1 μl of 10 mM ScriptSeq Index PCR Primer (PP_I) (5′-CAAGCAGAAGACGGCATACGAGAT-BARCODE-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT -3′, where BARCODE signifies one of several specific 6-mer sequences) (Epicentre), 12.5 μl of water, 25 μl of Premix E from the Failsafe PCR system and 0.5 μl of FailSafe Enzyme mix, and subjected to the following PCR conditions: 94°C for 1 min, followed by 10–14 cycles (determined by qPCR result) of 94°C for 30 sec, 55°C for 30 sec and 68°C for 3 min, and a final extension at 68°C for 7 min. The reaction products (ds cDNA libraries) were purified using 0.8X volumes of Agencourt AMPure XP beads, which enriched for PCR products of 200–500 bp as previously described;1 each size-selected cDNA library was eluted in 20 μl of nuclease-free water. As an exception, the Ribo-Zero treated E. coli libraries were size selected using a DNA 300 chip on a LabChip XT machine (Caliper Life Sciences), collecting a 250 bp ± 15 bp fraction. To assess size distribution and concentration, a 1:3 dilution of each size-selected library was run on a DNA High Sensitivity chip on the Bioanalyzer (Agilent Technologies).
Peregrine library multiplexing and sequencing
Four to eight cDNA libraries bearing different barcodes were mixed in equal molar ratios, based on the concentration measurements made using the DNA High Sensitivity chip on the Bioanalyzer; to maximize the sequence diversity of the mixtures, each included both human and E. coli cDNA libraries. Each library mixture was then concentrated using the Zymo DNA Clean and Concentrator-5 system, eluting in 20 μl of nuclease-free water, and its final concentration measured using the Kapa qPCR assay (Kapa Biosystems).
Each library mixture was loaded into a lane of a HiSeq 2000 (Illumina) at 10 pM concentration, for a 100 bp single-end run. Note that although all of the SGS data generated in this study were from single-end runs, there is no impediment to using our methods for paired-end SGS, and indeed we have done so successfully (data not shown). The custom read 1 primer PP_R1 (5′-ACACTTCGCTACAGGACGCTGTTCCGTTCTATGGG-3′) was used instead of the standard Illumina read primer HP1. Note that the underlined nucleotides drive annealing to the adaptor/cDNA junction site, directing the sequencer to begin reads at the 5′ end of the cDNA, rather than within the adaptor; this ensures sufficient sequence diversity in the initial stages of sequencing to enable accurate cluster calling, thereby circumventing the problem of “monotemplate sequencing.”5 The standard Illumina index read primer was used.
ScriptSeq library preparation and sequencing
One hundred ng of E. coli K-12 total RNA, or RNA that had been depleted of rRNA using Ribo-Zero, were used as starting material for ScriptSeq mRNA-Seq library preparation (Epicentre/Illumina). The two sets of libraries were prepared in triplicate, following the manufacturer's instructions. Following purification and size selection via AMPure XP beads (as described above), each set of three indexed libraries was pooled to generate two multiplexed libraries. The final concentrations of the multiplexed libraries were measured using Kapa qPCR, and 8 pM of each were loaded onto a MiSeq (Illumina) for a 300 bp single-end run using V2 chemistry.
Sequence data pre-processing
Raw FASTQ sequence files from HiSeq and MiSeq runs were demultiplexed using CASAVA v1.8 and MiSeq Reporter, respectively. The sequence files were further processed with our custom qfilter.pl perl script, which trims low-quality bases, detects and trims internal barcodes and primer fragments, masks low-complexity sequence, and removes any sequence with an overall quality or length below acceptable thresholds. First, internal barcodes and 3′ and 5′ tails with minimal quality scores were trimmed off. At this and subsequent trimming steps, a length test was applied; sequences below a minimum length (default 30 bp) were rejected. Each remaining unique sequence was passed through three filters that do not query quality. In the first filter, sequences of primers used in library construction were identified and trimmed in the following way. Primer “parts” of length 14 nt were collected, first taking the 14-mer DNA oligo sequence from both the 3′ and 5′ end of each primer, unless this sequence was homopolymeric, in which case it was substituted with the 14-mer taken from an internal position such that only 6 nt of the homopolymer were included. The reverse complement of each 14-mer was then added to the primer part list. In the second filter, sequences with any remaining positions uncalled (i.e., called as “N”) were rejected. Then, in the final filter, dustmasker (from the NCBI C++ Toolkit) was used to identify low-complexity sequences; sequences were rejected when masking left less than 30 bp, otherwise, low-complexity sequences were allowed to remain. Returning to individual reads and their quality strings, the quality markings were converted to a 2–40 score scale, the average score for all remaining positions was calculated, and the read was rejected if the average was below a default threshold of 30. The qfilter.pl script is available from the authors by request. Tables S1, S2, S5 and S6 summarize the total number of reads, and the proportion of reads passing qfilter, for each set of libraries analyzed.
Although the Peregrine and ScriptSeq data sets were analyzed in full for some analyses, we also sub-sampled both data sets to permit meaningful direct comparisons. To do this, we combined the quality-filtered reads for all replicates within each preparation/treatment, discarded all reads shorter than 100 bp, trimmed all reads down to a maximum length of 100 bp and randomly sampled 2 million reads from each preparation/treatment type. These comparable data sets are referred to as the “sampled data.” Table S3 summarizes the total number of reads, and the proportion of reads passing qfilter, for each sub-sampled library analyzed.
Bioinformatic analysis of RNA-Seq data
E. coli K-12 reads were aligned to the MG1655 genome (accession number NC_000913.2) using Bowtie249 (v 2.0.6). For human reads, alignment was first performed against a Bowtie2 index composed of just the four ribosomal subunit sequences (5S, 5.8S, 18S and 28S) to provide accurate quantification of rRNA. Next, TopHat50 (v 2.0.4) was used to map remaining non-rRNA reads to the human genome, using the Ensembl GRCh37.68 assembly and annotation. For both Bowtie2 and TopHat, reads were mapped using default parameters; in particular, the default setting of zero mismatches for the seed of length 22 bp was used. Mapped reads were assigned to genome compartments by using the “intersect” command from BEDTools51 to count intersections between alignment BAM files and a set of non-overlapping BED files; in this way, each read was assigned to one, and only one, of the following categories: For E. coli, rRNA, CDS, intergenic and tRNA; and for human, rRNA, mtRNA, lincRNA, exon, intron and intergenic. Cufflinks52 (v.2.0.2) was used to estimate transcript abundance [i.e., fragments per Kilobase of exon model per million mapped reads (FPKM)], and to assess sample-to-sample variability. Tables S1, S2, S3, S5 and S6 summarize the percent of reads mapping to the reference genome, the percent of aligned reads found in CDS or rRNA, and the percent of CDS to which at least one read mapped (a measure of breadth of genome coverage) for each of the libraries analyzed. To visualize potential 5′/3′ mRNA coverage bias, the “coverage” command from BEDTools was used to tabulate the depth of coverage at each nucleotide of each transcript. For the E. coli data, the set of transcriptional units analyzed was composed of Arkin polycistronic operons53 (downloaded from the Lowe Lab website: microbes.ucsf.edu) plus annotated genes located outside of these operons. For the human data, we selected ~12,000 genes from the mean FPKM interquartile and for each gene used the highest expressed Ensembl transcript. Both analyses were limited to transcriptional units of 1–5 kb in length. For each transcriptional unit, we divided the length into percentiles (spanning 10–50 bases each) and calculated the coverage percentage as the number of sequenced bases in each percentile divided by the total number of sequenced bases for the entire transcript.
Supplementary Material
Acknowledgments
This research was fully supported by the Laboratory Directed Research and Development program at Sandia National Laboratories. Sandia is a multi-program laboratory managed and operated by Sandia Corporation, a wholly owned subsidiary of Lockheed Martin Corporation, for the US Department of Energy's National Nuclear Security Administration under contract DE-AC04-94AL85000. The authors would like to thank the staff of the Vincent J. Coates Sequencing Laboratory for their assistance and insight.
Glossary
Abbreviations:
- RNA
ribonucleic acid
- mRNA
messenger RNA
- rRNA
ribosomal RNA
- miRNA
micro RNA
- DNA
deoxyribonucleic acid
- cDNA
complementary DNA
- SGS
second generation sequencing
- NGS
next generation sequencing
- RNA-Seq
RNA (cDNA) sequencing
- PCR
polymerase chain reaction
- qPCR
quantitative PCR
- DSN
duplex-specific nuclease
- MMLV
Moloney murine leukemia virus
- ds
double-stranded
- C(t)
threshold cycle
- PBMC
peripheral blood mononuclear cells
- CDS
coding DNA sequence
- SMART
switching mechanism at 5′ end of ribonucleic acid template
- SOLiD
sequencing by oligonucleotide ligation and detection
- ATCC
American type culture collection
- LB
Luria broth
- A260
absorbance at 260 nm
- RIN
RNA integrity number
- CASAVA
consensus assessment of sequence and variation
- FPKM
fragments per kilobase of exon model per million mapped reads
- FU
fluorescence units
- RFU
relative fluorescence units
- RT
room temperature
Disclosure of Potential Conflicts of Interest
The authors declare that they have no competing financial interests.
Author Contributions
S.A.L., Z.W.B., K.P.W., J.S.S, T.W.L. and S.S.B conceived the research. S.A.L., Z.W.B., S.S.B. and T.W.L. designed experiments. S.A.L., Z.W.B, D.J.C., P.D.L. and A.S. performed experiments. S.A.L., Z.W.B., O.D.S., K.P.W. and J.S.S. analyzed sequencing data. S.A.L., Z.W.B., O.D.S., D.J.C., K.P.W., J.S.S. and S.S.B. created figures and tables for the manuscript. All authors participated in writing and editing the manuscript, and agreed with the final submitted draft.
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/24284
References
- 1.Malone JH, Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011;9:34. doi: 10.1186/1741-7007-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–17. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, et al. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods. 2010;7:709–15. doi: 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Z, Theurkauf WE, Weng Z, Zamore PD. Strand-specific libraries for high throughput RNA sequencing (RNA-Seq) prepared without poly(A) selection. Silence. 2012;3:9. doi: 10.1186/1758-907X-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozsolak F, Platt AR, Jones DR, Reifenberger JG, Sass LE, McInerney P, et al. Direct RNA sequencing. Nature. 2009;461:814–8. doi: 10.1038/nature08390. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z, Duan X. Ribosomal RNA depletion for massively parallel bacterial RNA-sequencing applications. Methods Mol Biol. 2011;733:93–103. doi: 10.1007/978-1-61779-089-8_7. [DOI] [PubMed] [Google Scholar]
- 9.Vandernoot VA, Langevin SA, Solberg OD, Lane PD, Curtis DJ, Bent ZW, et al. cDNA normalization by hydroxyapatite chromatography to enrich transcriptome diversity in RNA-seq applications. Biotechniques. 2012;53:373–80. doi: 10.2144/000113937. [DOI] [PubMed] [Google Scholar]
- 10.Morlan JD, Qu K, Sinicropi DV. Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS One. 2012;7:e42882. doi: 10.1371/journal.pone.0042882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bogdanova EA, Barsova EV, Shagina IA, Scheglov A, Anisimova V, Vagner LL, et al. Normalization of full-length-enriched cDNA. Methods Mol Biol. 2011;729:85–98. doi: 10.1007/978-1-61779-065-2_6. [DOI] [PubMed] [Google Scholar]
- 12.Kumar N, Creasy T, Sun Y, Flowers M, Tallon LJ, Dunning Hotopp JC. Efficient subtraction of insect rRNA prior to transcriptome analysis of Wolbachia-Drosophila lateral gene transfer. BMC Res Notes. 2012;5:230. doi: 10.1186/1756-0500-5-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reuter M, Berninger P, Chuma S, Shah H, Hosokawa M, Funaya C, et al. Miwi catalysis is required for piRNA amplification-independent LINE1 transposon silencing. Nature. 2011;480:264–7. doi: 10.1038/nature10672. [DOI] [PubMed] [Google Scholar]
- 14.Sinicropi DV, Qu K, Collin F, Crager M, Liu ML, Pelham RJ, et al. Whole transcriptome RNA-Seq analysis of breast cancer recurrence risk using formalin-fixed paraffin-embedded tumor tissue. PLoS One. 2012;7:e40092. doi: 10.1371/journal.pone.0040092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie W, Lei Y, Fu W, Yang Z, Zhu X, Guo Z, et al. Tissue-specific transcriptome profiling of Plutella xylostella third instar larval midgut. Int J Biol Sci. 2012;8:1142–55. doi: 10.7150/ijbs.4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marks H, Kalkan T, Menafra R, Denissov S, Jones K, Hofemeister H, et al. The transcriptional and epigenomic foundations of ground state pluripotency. Cell. 2012;149:590–604. doi: 10.1016/j.cell.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu YY, Machleder EM, Chenchik A, Li R, Siebert PD. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Biotechniques. 2001;30:892–7. doi: 10.2144/01304pf02. [DOI] [PubMed] [Google Scholar]
- 18.Li JW, Robison K, Martin M, Sjödin A, Usadel B, Young M, et al. The SEQanswers wiki: a wiki database of tools for high-throughput sequencing analysis. Nucleic Acids Res. 2012;40(Database issue):D1313–7. doi: 10.1093/nar/gkr1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mao F, Leung WY, Xin X. Characterization of EvaGreen and the implication of its physicochemical properties for qPCR applications. BMC Biotechnol. 2007;7:76. doi: 10.1186/1472-6750-7-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riley M, Abe T, Arnaud MB, Berlyn MK, Blattner FR, Chaudhuri RR, et al. Escherichia coli K-12: a cooperatively developed annotation snapshot--2005. Nucleic Acids Res. 2006;34:1–9. doi: 10.1093/nar/gkj405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, Riley M, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–62. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 22.Yi H, Cho YJ, Won S, Lee JE, Jin Yu H, Kim S, et al. Duplex-specific nuclease efficiently removes rRNA for prokaryotic RNA-seq. Nucleic Acids Res. 2011;39:e140. doi: 10.1093/nar/gkr617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dornenburg JE, Devita AM, Palumbo MJ, Wade JT. Widespread antisense transcription in Escherichia coli. MBio. 2010;1:e00024–10. doi: 10.1128/mBio.00024-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raghavan R, Sloan DB, Ochman H. Antisense transcription is pervasive but rarely conserved in enteric bacteria. MBio. 2012;3:e00156–12. doi: 10.1128/mBio.00156-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt WM, Spiel AO, Jilma B, Wolzt M, Müller M. In vivo profile of the human leukocyte microRNA response to endotoxemia. Biochem Biophys Res Commun. 2009;380:437–41. doi: 10.1016/j.bbrc.2008.12.190. [DOI] [PubMed] [Google Scholar]
- 26.Gaarz A, Debey-Pascher S, Classen S, Eggle D, Gathof B, Chen J, et al. Bead array-based microrna expression profiling of peripheral blood and the impact of different RNA isolation approaches. J Mol Diagn. 2010;12:335–44. doi: 10.2353/jmoldx.2010.090116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaz C, Ahmad HM, Sharma P, Gupta R, Kumar L, Kulshreshtha R, et al. Analysis of microRNA transcriptome by deep sequencing of small RNA libraries of peripheral blood. BMC Genomics. 2010;11:288. doi: 10.1186/1471-2164-11-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voellenkle C, van Rooij J, Cappuzzello C, Greco S, Arcelli D, Di Vito L, et al. MicroRNA signatures in peripheral blood mononuclear cells of chronic heart failure patients. Physiol Genomics. 2010;42:420–6. doi: 10.1152/physiolgenomics.00211.2009. [DOI] [PubMed] [Google Scholar]
- 29.Keller A, Backes C, Leidinger P, Kefer N, Boisguerin V, Barbacioru C, et al. Next-generation sequencing identifies novel microRNAs in peripheral blood of lung cancer patients. Mol Biosyst. 2011;7:3187–99. doi: 10.1039/c1mb05353a. [DOI] [PubMed] [Google Scholar]
- 30.Allantaz F, Cheng DT, Bergauer T, Ravindran P, Rossier MF, Ebeling M, et al. Expression profiling of human immune cell subsets identifies miRNA-mRNA regulatory relationships correlated with cell type specific expression. PLoS One. 2012;7:e29979. doi: 10.1371/journal.pone.0029979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oshikawa M, Tsutsui C, Ikegami T, Fuchida Y, Matsubara M, Toyama S, et al. Full-length transcriptome analysis of human retina-derived cell lines ARPE-19 and Y79 using the vector-capping method. Invest Ophthalmol Vis Sci. 2011;52:6662–70. doi: 10.1167/iovs.11-7479. [DOI] [PubMed] [Google Scholar]
- 32.Rosenberg BR, Hamilton CE, Mwangi MM, Dewell S, Papavasiliou FN. Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA-editing targets in transcript 3′ UTRs. Nat Struct Mol Biol. 2011;18:230–6. doi: 10.1038/nsmb.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng Z, Cheng Y, Tan BC, Kang L, Tian Z, Zhu Y, et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol. 2012;30:253–60. doi: 10.1038/nbt.2122. [DOI] [PubMed] [Google Scholar]
- 34.Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–60. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 35.Hassan MA, Melo MB, Haas B, Jensen KD, Saeij JP. De novo reconstruction of the Toxoplasma gondii transcriptome improves on the current genome annotation and reveals alternatively spliced transcripts and putative long non-coding RNAs. BMC Genomics. 2012;13:696. doi: 10.1186/1471-2164-13-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin J, Zhu W, Passalacqua KD, Bergman N, Borodovsky M. Bacillus anthracis genome organization in light of whole transcriptome sequencing. BMC Bioinformatics. 2010;11(Suppl 3):S10. doi: 10.1186/1471-2105-11-S3-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. MetaHIT Consortium A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Runckel C, Flenniken ML, Engel JC, Ruby JG, Ganem D, Andino R, et al. Temporal analysis of the honey bee microbiome reveals four novel viruses and seasonal prevalence of known viruses, Nosema, and Crithidia. PLoS One. 2011;6:e20656. doi: 10.1371/journal.pone.0020656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Z, Bruno DP, Martens CA, Porcella SF, Moss B. Genome-wide analysis of the 5′ and 3′ ends of vaccinia virus early mRNAs delineates regulatory sequences of annotated and anomalous transcripts. J Virol. 2011;85:5897–909. doi: 10.1128/JVI.00428-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deutscher MP. Maturation and degradation of ribosomal RNA in bacteria. Prog Mol Biol Transl Sci. 2009;85:369–91. doi: 10.1016/S0079-6603(08)00809-X. [DOI] [PubMed] [Google Scholar]
- 41.Ramilo O, Allman W, Chung W, Mejias A, Ardura M, Glaser C, et al. Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood. 2007;109:2066–77. doi: 10.1182/blood-2006-02-002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang BMP, McLean AS, Dawes IW, Huang SJ, Lin RCY. Gene-expression profiling of peripheral blood mononuclear cells in sepsis. Crit Care Med. 2009;37:882–8. doi: 10.1097/CCM.0b013e31819b52fd. [DOI] [PubMed] [Google Scholar]
- 43.McHale CM, Zhang L, Lan Q, Li G, Hubbard AE, Forrest MS, et al. Changes in the peripheral blood transcriptome associated with occupational benzene exposure identified by cross-comparison on two microarray platforms. Genomics. 2009;93:343–9. doi: 10.1016/j.ygeno.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chaussabel D, Pascual V, Banchereau J. Assessing the human immune system through blood transcriptomics. BMC Biol. 2010;8:84. doi: 10.1186/1741-7007-8-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glatt SJ, Stone WS, Nossova N, Liew CC, Seidman LJ, Tsuang MT. Similarities and differences in peripheral blood gene-expression signatures of individuals with schizophrenia and their first-degree biological relatives. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:869–87. doi: 10.1002/ajmg.b.31239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bolen CR, Uduman M, Kleinstein SH. Cell subset prediction for blood genomic studies. BMC Bioinformatics. 2011;12:258. doi: 10.1186/1471-2105-12-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen KD, Chang PT, Ping YH, Lee HC, Yeh CW, Wang PN. Gene expression profiling of peripheral blood leukocytes identifies and validates ABCB1 as a novel biomarker for Alzheimer’s disease. Neurobiol Dis. 2011;43:698–705. doi: 10.1016/j.nbd.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 48.Cloonan N, Forrest AR, Kolle G, Gardiner BB, Faulkner GJ, Brown MK, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods. 2008;5:613–9. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- 49.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodrigue S, Materna AC, Timberlake SC, Blackburn MC, Malmstrom RR, Alm EJ, et al. Unlocking short read sequencing for metagenomics. PLoS One. 2010;5:e11840. doi: 10.1371/journal.pone.0011840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Price MN, Huang KH, Alm EJ, Arkin AP. A novel method for accurate operon predictions in all sequenced prokaryotes. Nucleic Acids Res. 2005;33:880–92. doi: 10.1093/nar/gki232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.