

Abstract
AIM: To determine whether the human giant larvae homolog 1 gene (Hugl-1/Llg1/Lgl1) exerts tumor suppressor effects in esophageal cancer.
METHODS: We constructed a Hugl-1 expression plasmid, pEZ-M29-Hugl1, for gene transfection. We transfected the pEZ-M29-Hugl1 plasmid into Eca109 esophageal cancer cell lines with Lipofectamine 2000 to overexpress Hugl-1. Real-time reverse transcription-polymerase chain reaction (RT-PCR) and Western blotting were performed to determine the effects of the plasmid on Hugl-1 expression. In vitro cell proliferation and apoptosis were examined separately by cell counting Kit-8 (CCK-8) assay, flow cytometry, and Western blotting before and after the transfection of the plasmid into Eca109 cells. Cell cycle distribution was assessed with flow cytometry. The effect of Hugl-1 overexpressing on tumor growth in vivo was performed with a xenograft tumor model in nude mice. Expression of Hugl-1 in xenograft tumor was analyzed by immunohistochemistry. The transferase-mediated dUTP nick end-labeling (TUNEL) technique was performed to detect and quantitate apoptotic cell.
RESULTS: The transfection efficiency was confirmed with real-time RT-PCR and Western blotting. Our results show that compared with control groups the mRNA levels and protein levels of Hugl-1 in pEZ-M29-Hugl1-treated group were remarkably increased (P < 0.05). The CCK-8 assay demonstrated that the growth of cells overexpressing Hugl-1 was significantly lower than control cells. Cell cycle distribution showed there was a G0/G1 cell cycle arrest in cells overexpressing Hugl-1 (64.09% ± 3.14% vs 50.32% ± 4.60%, 64.09% ± 3.14% vs 49.13% ± 2.24%). Annexin V-fluorescein isothiocyanate revealed that apoptosis was significantly increased in cells overexpressing Hugl-1 compared with control group (17.33% ± 4.76% vs 6.90% ± 1.61%, 17.33% ± 4.76% vs 6.27% ± 0.38%). Moreover, we found that Hugl-1 changes the level of the anti-apoptotic protein Bcl-2 and the pro-apoptotic protein Bax and the activation of both caspase-3 and caspase-9. With a TUNEL assay, we found that Hugl-1 markedly increased the apoptosis rate of Eca109 cells in vivo (60.50% ± 9.11% vs 25.00% ± 12.25%). It was shown that Hugl-1 represents a significantly more effective tumor suppressor gene alone in a xenograft tumor mouse model. This data suggest that Hugl-1 inhibited tumor growth and induced cell apoptosis in vivo.
CONCLUSION: These results suggest that Hugl-1 induces growth suppression and apoptosis in a human esophageal squamous cell carcinoma cell line both in vitro and in vivo.
Keywords: Esophageal squamous cell carcinoma, Human giant larvae homolog 1, Proliferation, Apoptosis
Core tip: In this paper, we constructed a plasmid to express Hugl-1 which has significant homology to the Drosophila tumor suppressor gene lethal giant larvae. The human esophageal squamous cell carcinoma cell line Eca109 was used as the object of study. We found a positive correlation between Hugl-1 expression and cell apoptosis in Eca109 cells both in vitro and in vivo. These data suggest that Hugl-1 is a tumor suppressor gene in esophageal cancer and may provide a novel target for the treatment of esophageal cancer patients.
INTRODUCTION
Esophageal cancer has two major histological types: squamous cell carcinoma (ESCC) and adenocarcinoma (EAC)[1,2]. ESCC is one of the most frequently diagnosed cancers in China[3]. It has been well established that surgical treatment can prolong the survival time of cancer patients, yet the 5-year survival rate for ESCC after surgery is still low (ranging 14%-22%)[4]. Most esophageal cancers are diagnosed in the advanced stages[5]. Thus, detecting gene alternations that promote the carcinogenesis process leading to esophageal cancer will have a profound impact on the diagnosis and treatment of the disease.
Lethal giant larvae (lgl), an evolutionarily conserved and widely expressed cytoskeletal protein, is indispensable for the establishment and maintenance of cell polarity and is a regulator of cell proliferation[6,7]. In Drosophila, mutations in three neoplastic tumor suppressor genes, discs large (dlg), scribble (scrib) and lgl, have revealed a link between the regulation of cell polarity and cell proliferation[8-13]. The human homologs of lgl are Hugl-1 (Llgl1) and Llgl2. The Hugl-1 protein shares 62.5% similarity with Lgl[14]. Several studies have shown that Hugl-1 transcripts are reduced or absent in a high proportion of breast cancers, lung cancers, prostate cancers, ovarian cancers, colorectal cancers, melanomas, endometrial cancers and hepatocellular carcinomas[15-19]. These studies have also shown that Hugl-1 may function as a tumor suppressor gene in various cancer types. In ESCC tissue samples, Hugl-1 is notably lower than in normal tissues[20]; however, the effect of Hugl-1 on tumor progression and prognosis in ESCC is not clear.
In the present study, we analyzed Hugl-1 expression in the esophageal carcinoma cell line Eca109 as well as in tissue samples. By using a forced overexpression technique, we explored the biological activity of Hugl-1 and the underlying mechanism in vitro and in vivo. We demonstrated that Hugl-1 inhibits proliferation in the esophageal carcinoma cell line as well as in ESCC tissue samples and that it promotes apoptosis in esophageal carcinoma cells and xenograft tumors through a mitochondria-related pathway.
MATERIALS AND METHODS
Cells, cell culture
The human ESCC cell line, Eca109, purchased from the China Center for Type Culture Collection (Wuhan Province, China) and cultured in RPMI-1640 medium (Gibco, United States) containing 10% fetal bovine serum (Gibco, United States), in a humidified atmosphere of 5% CO2 at 37 °C.
Plasmid construction and purification of cultured Eca109 cells
Hugl-1 expression plasmids were constructed with pEZ-M29 as the vector, Hugl-1 as the expression gene and ampicillin resistance for antibiotic selection (GeneCopoeia, United States). An empty expression plasmid of the same type was used as a control. Eca109 cells were seeded into a 6 cm dish at a density of 5 × 105 cells per well and incubated overnight with 5% CO2 at 37 °C. For each transfection, 9 μL of lipofectamine 2000 (Invitrogen, United States) and 3 μg of the Hugl-1 expression plasmid were added to 1 mL of Opti-MEM (Invitrogen, United States) and incubated for 5 min at room temperature. The diluted plasmid and lipofectamine were mixed together and incubated for 30 min before adding them directly to the cells. Eca109 cells overexpressing Hugl-1 were grown in RPMI-1640 medium with 200 μg/mL of G418 for stable clone selection.
Real-time RT-PCR
Total RNA was prepared from the Eca109 cells with TRIzol reagent (Invitrogen, United States) according to the manufacturer’s protocol. First-strand cDNA was synthesized using the PrimeScriptTM RT reagent kit (Takara, Japan). The isolated RNA (1 μg) was used as template to perform one-step RT-PCR according to the protocol, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. All reactions were conducted in a 25 μL volume.
Real-time RT-PCR was conducted on the resulting cDNA with the SYBR Green method and the AB7500 Real-time RT-PCR system. The sequences of the primer sets used were as follows: forward 5’-AGAAGGCTGGGGCTCATTTG-3’ and reverse 5’-AGGGGCCATCCACAGTCTTC-3’ for GAPDH (258 bp); forward 5’-GCTGCTTCGATCCCTACAGTGAC-3’ and reverse 5’-CGGCACATCCTAAGCTCCAG-3’ for Hugl-1 (131 bp). PCR was performed by initial denaturation at 95 °C for 30 s followed by 40 cycles of 5 s at 95 °C, 30 s at 60 °C and 1 min at 72 °C. The threshold cycle (Ct) values of each sample were used in the 2-ΔΔCT data analysis method.
Western blotting
Cells were harvested from 6-well culture plates, and aliquots of cell extracts were separated on an 8%-12% SDS-polyacrylamide gel. The proteins were then transferred to a polyvinylidene difluoride membrane (Millipore, United States) and incubated overnight at 4 °C with the following rabbit polyclonal antibodies: anti-Hugl1 (ab39292, Abcam), anti-Bcl2 (SC-492, Santa Cruzc), anti-Bax (5023, Cell Signaling), anti-p21 (2947, Cell Signaling), anti-cyclin D1 (2978, Cell Signaling), anti-survivin (2808, Cell Signaling), anti-caspase9 (9502, Cell Signaling), anti-caspase3 (9662, Cell Signaling), anti-p65 (3037, Cell Signaling), anti-p-p65 (3033, Cell Signaling) or anti-GAPDH (2118, Cell Signaling).
The blots were rinsed three times in TBST and incubated with a 1:10000 diluted goat-anti-rabbit secondary antibody (LICOR, United States) conjugated to horseradish peroxidase for 1 h at room temperature before they were washed extensively with TBST. Finally, the membranes were scanned with a two-color infrared imaging system (Odyssey, LICOR, United States). Membranes were also probed for GAPDH as an additional loading control.
Cell proliferation analysis
Cells were seeded into 96-well plates at a density of 3000 cells per well 48 h after transfection. The effects of let-7a on cell proliferation were examined with CCK-8 (Dojindo, Japan) according to the manufacturer’s instruction 0, 24, 48, 72 and 96 h after seeding.
Cell cycle analysis
Cell cycle analysis was performed with flow cytometry (BD FACS Aria III, United States). Cultured cells were harvested 48 h after transfection with pEZ-M29-eGFP and pEZ-M29-Hugl1, respectively, washed with ice-cold phosphate buffered solution (PBS), and fixed in 70% ethanol overnight at 4 °C. After centrifugation at 500 × g for 5 min at 4 °C, the cell pellets were stained with 10 μg/mL propidium iodide (PI) and 10 μg/mL RNase A in phosphate buffered saline (PBS) buffer for 20 min at room temperature in the dark. Cell cycle analysis was performed with three independent experiments.
Flow cytometric analysis of apoptotic cells using Annexin V-fluorescein isothiocyanate kit
The cultured cells were harvested after treatment with pEZ-M29-eGFP and pEZ-M29-Hugl1, respectively, washed with ice-cold PBS and centrifuged for 5 min at 500 × g at 4 °C. The supernatants were discarded, and the cell pellets were resuspended in ice-cold binding buffer. Double staining with Annexin V-fluorescein isothiocyanate (FITC) and PI was performed using the Annexin V-FITC kit (Beyotime, China) according to the manufacturer’s recommendations, and the cells were then analyzed by FACS (BD FACS Aria III, United States).
Nude mice xenograft experiments
BALB/c nude mice (5-6 week old) were obtained from the Beijing HFK Experimental Animal Center and were quarantined for one week before tumor implantation. Animal welfare and experimental procedures were performed in strict accordance with guidelines. Mice were randomly divided into two groups (six mice per group). A xenograft tumor model was established by subcutaneously injecting either Hugl1-overexpressing cells or PBS-treated cells (2 × 106) suspended in 0.1 mL of PBS into the right flank of mice, and the tumor volume was measured every week until the mice were sacrificed. At the end of the experiment (day 21), tumors were harvested for additional analyses. Differences in tumor growth were tested for statistical significance.
Immunohistochemistry analysis
The xenograft tumors were embedded in paraffin, cut into 4 μm sections, and either stained with hematoxylin and eosin or treated with Hugl-1 antibody for immunohistochemical evaluation. The results were captured by microscopy (Olympus, Japan).
Transferase-mediated dUTP nick end-labeling assay
The transferase-mediated dUTP nick end-labeling (TUNEL) technique was performed to detect and quantitate apoptotic cell death using the in situ Cell Death Detection Kit (Roche, United States) according to the manufacturer’s instructions. Chamber slides were fixed with 4% paraformaldehyde and permeabilized in 0.1% Triton X-100. The slides were then incubated with the TUNEL reaction mixture for 1 h at 37 °C. After the slides were washed with PBS, they were incubated with peroxidase-conjugated antibody for 30 min at 37 °C and were developed with the DAB system. A minimum of 3 fields were randomly selected, and the total cells were counted in each field to achieve a minimum number of 100 total cells. Apoptotic rates (the number of apoptotic cells/total cells) were expressed as mean ± SD from different fields.
Statistical analysis
The statistical analysis was performed using SPSS software (version 17.0 for Windows). Data were presented as means ± SD and comparisons were made using Student’s t test. A probability of 0.05 or less was considered statistically significant.
RESULTS
Overexpression of Hugl-1 in vitro
The eGFP was used as an marker to detect whether the pEZ-M29-Hugl1 plasmid vectors were successfully transduced in vitro. The transfection efficiency is shown in Figure 1A, in vitro approach to 60%.
Figure 1.
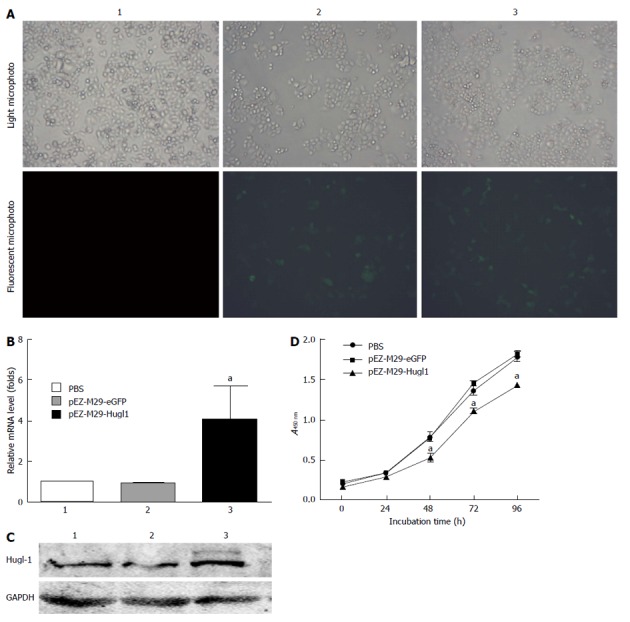
Transfection with pEZ-M29-Hugl1 increased Hugl-1 expression and inhibited the proliferation in Eca109 cells. A: Fluorescent expression in Eca109 cells (× 200); B: Real-time reverse transcription-polymerase chain reaction data of Hugl-1 mRNA levels following transfection with pEZ-M29-Hugl1 plasmids (group 3), pEZ-M29-eGFP (group 2), or treatment with phosphate buffered saline (PBS) (group 1); C: Western blotting data showing Hugl-1 protein expression levels following pEZ-M29-Hugl1 transfection compared with control groups; D: The effect of Hugl-1 on cell proliferation was assessed by cell counting Kit-8. Results represent mean values of three experiments and are indicated as mean ± SD. aP < 0.05 vs the pEZ-M29-eGFP-treated and PBS-treated groups. GAPDH: Glyceraldehyde 3-phosphate dehydrogenase.
To analyze the effect of pEZ-M29-Hugl1 on the expression of cancer genes, we assessed the mRNA levels of Hugl-1 in the treated cells by Real-time RT-PCR. Our results demonstrated that, compared with group 1 (PBS-treated), the mRNA levels of Hugl-1 in group 3 (pEZ-M29-Hugl1-treated) were remarkably increased (P < 0.05), but the mRNA levels in group 2 (pEZ-M29-eGFP-treated) were not noticeably different (Figure 1B).
We next assessed the expression of Hugl-1 protein by Western blotting. The expression of Hugl-1 was consistent with results from real-time RT-PCR, and compared with group 1, the protein level of group 3 was increased (Figure 1C).
Effect of Hugl-1expression on the proliferation of Eca109 cells
Cell proliferation assays were performed with the cell counting Kit 8 assay 48 h after transfection. The proliferation of Eca109 cells 48 h after being transfected with pEZ-M29-Hugl1 was slower than that of the other two control groups (P < 0.05). Therefore, Hugl-1 inhibited the proliferation of Eca109 cells (Figure 1D).
Effect of Hugl-1 protein on the cell cycle of Eca109 cells
The mechanism underlying the inhibition of cell proliferation in Eca109 cells was investigated by analyzing the cell cycle with FACS following pEZ-M29-Hugl1 transfection. It was observed that Hugl-1 overexpression arrested the cell cycle in the G1 phase (Figure 2A). The pEZ-M29-Hugl1 transfected cells were found to contain 64.09% ± 3.14% of cells in the G1 phase and 31.47% ± 4.90% of cells in the S phase, whereas in the PBS-treated group, 50.32% ± 4.60% of cells were in the G1 phase and 49.30% ± 4.98% of cells were in the S phase (Figure 2B). There was no difference between the group 3 and the group 2 (49.13% ± 2.24% in the G1 phase and 43.47% ± 2.09% in the S phase).
Figure 2.
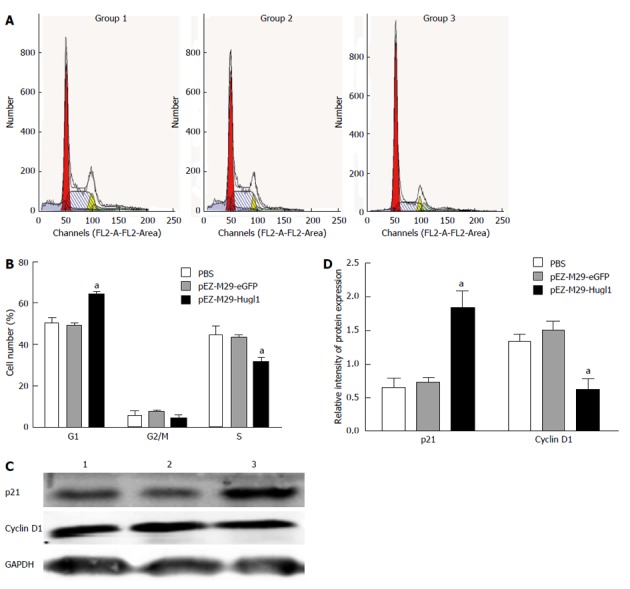
Effect of Hugl-1 on cell cycle distribution of Eca109 cells in vitro. A: Cells were treated with pEZ-M29-Hugl1, pEZ-M29-eGFP or phosphate buffered saline (PBS) for 48 h and were then prepared for fluorescence-activated cell sorting analysis; B: Data are presented as mean ± SD of three independent experiments; C: Western blotting data of p21 and cyclin D1 protein expression levels following transfection with pEZ-M29-Hugl1 or controls; D: Analysis of the expression of proteins. aP < 0.05 vs the pEZ-M29-eGFP-treated and PBS-treated groups. GAPDH: Glyceraldehyde 3-phosphate dehydrogenase.
Figure 2C shows that cells overexpressing Hugl-1 exhibited down-regulation of cyclin D1 and up-regulation of p21; these results suggest that the G0/G1 cell cycle arrest induced by Hugl-1 involved a reduced level of cyclin D1 and an increased level of p21. The protein levels of p21 was upregulated (1.83 ± 0.25 vs 0.64 ± 0.14, 1.83 ± 0.25 vs 0.72 ± 0.08, P < 0.05) and cyclin D1 was downregulated (0.61 ± 0.18 vs 1.33 ± 0.12, 0.61 ± 0.18 vs 1.48 ± 0.15, P < 0.05) in the pEZ-M29-Hugl1 transfected cells (Figure 2D).
Effect of Hugl-1 protein on Eca109 cell apoptosis
After transfection, cells were incubated with Annexin V-FITC in a buffer containing PI and were then analyzed by flow cytometry (Figure 3A). The results show that group 3 had a higher apoptosis rate (17.33% ± 4.76%) compared with group 1 (6.90% ± 1.61%) (P < 0.05), and as we expected, no difference was observed between the apoptosis rates of group 1 and group 2 (6.27% ± 0.38%) (P > 0.05) (Figure 3B).
Figure 3.
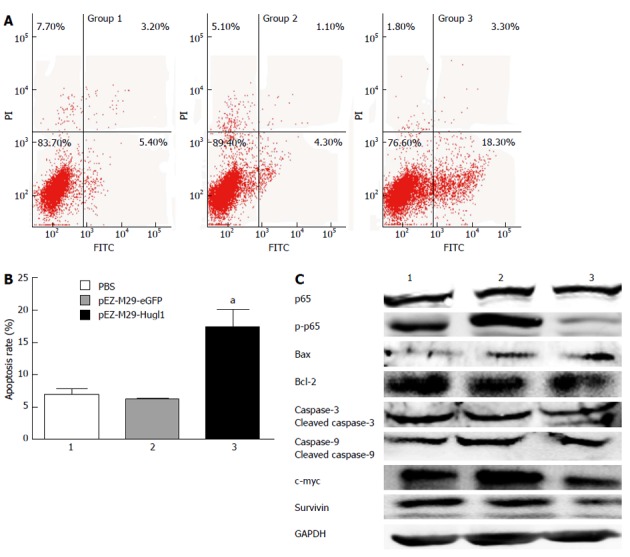
Effect of Hugl-1 on apoptosis of Eca109 cells in vitro. A: Cells were treated for 48 h and were then processed for FACS by staining with Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI); B: After transfection with pEZ-M29-Hugl1, a significant number of cells were in an early state of apoptosis, and a population of cells had progressed to a later stage of apoptosis; C: Up-regulation of Hugl-1 led to a change of the protein levels of p65, p-p65, Bax, Bcl-2, caspase-3 and -9, survivin and c-myc among the three cell lines. All experiments were performed three times independently. aP < 0.05 vs the pEZ-M29-eGFP-treated and phosphate buffered solution (PBS)-treated groups. GAPDH: Glyceraldehyde 3-phosphate dehydrogenase.
To identify the mechanisms that were affected by Hugl-1 expression, Eca109 cells expressing Hugl-1 were analyzed by Western blotting for changes in the levels of various cell-signaling proteins. Figure 3C shows that phospho-p65 was essentially absent in Hugl-1-overexpressing cells, but the total p65 level decreased only slightly. These results suggest that Hugl-1 down-regulated the nuclear factor kappa B (NF-κB) signaling pathway through inhibition of IKKα/β and p65 phosphorylation.
To establish that Hugl-1 induced apoptosis, we examined the activation of the classical caspases and the Bcl-2 family of proteins by Western blotting. Figure 3C shows that Hugl-1 up-regulated the expression of Bax, of cleaved caspase-3, and of cleaved caspase-9, and it down-regulated Bcl-2, survivin, and c-myc expression. These results suggest that Hugl-1 induced apoptosis in Eca109 cells through activation of the mitochondrial apoptotic pathway.
Effect of Hugl-1 expression on xenograft tumor growth
To study the effect of Hugl-1 on tumor growth, nude mice were inoculated with Hugl-1-overexpressing cells, and the resulting tumor growth was compared to that in a control group of mice injected with PBS-treated cells. The difference in tumor growth between the two groups of mice was statistically significant at P < 0.05. In the control group, tumors displayed rapid and continued outgrowth during the course of the experiment, and the mean tumor volume was 1126.56 ± 141.70 mm3. In contrast, the mean tumor size for the experimental group was 606.03 ± 22.49 mm3 (Figure 4A).
Figure 4.
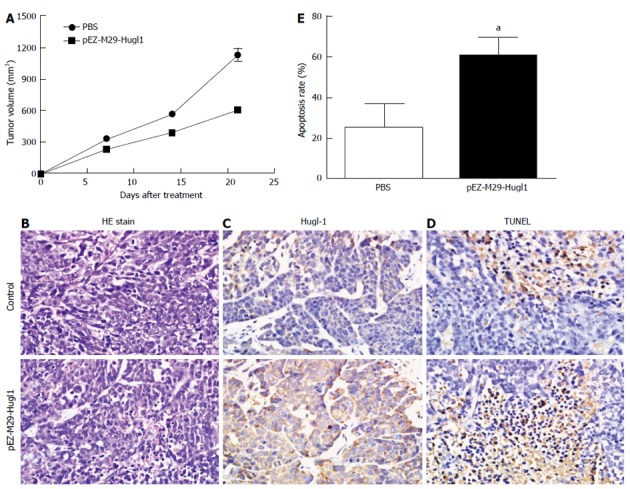
Effect of Hugl-1 on xenograft tumor in vivo. A: Cells were injected subcutaneously into nude mice, and with one group of mice receiving pEZ-M29-Hugl1-treated cells and another receiving phosphate buffered saline (PBS). Tumor volume was measured at 7-d intervals for 21 d; B: Tumor sections were observed by hematoxylin and eosin (HE) staining (× 400); C: Expression of Hugl-1 in tumor tissues was analyzed by immunohistochemistry. Many cells were strongly positive for Hugl-1 in the pEZ-M29-Hugl1-treated tumor sections (× 400); D: Representative photomicrographs showing transferase-mediated dUTP nick end-labeling (TUNEL) staining for evidence of apoptosis in transplantation tumors undergoing various treatments (× 400); E: Quantitative analysis of apoptotic cells in tumors treated with PBS or pEZ-M29-Hugl1. Apoptotic cells, shown by TUNEL, were significantly increased in tumors treated with pEZ-M29-Hugl1. Data are presented as mean ± SD (× 400). aP < 0.05 vs the pEZ-M29-eGFP-treated and PBS-treated groups.
Hematoxylin and eosin staining revealed a significant level of cell death in tumor tissues treated with the pEZ-M29-Hugl1 plasmid compared to the control group (Figure 4B). As shown in Figure 4C, many cells were strongly positive for Hugl-1 in the pEZ-M29-Hugl1-treated tumor sections. Apoptotic cells in the tumor sections were analyzed by TUNEL staining (Figure 4D), which showed markedly more positive cells in the pEZ-M29-Hugl1-treated group (60.50% ± 9.11%) than in the PBS-treated group (25.00% ± 12.25%) (Figure 4E).
DISCUSSION
The oncogenesis of esophageal cancer involves accumulated alternations of oncogenes, tumor suppresser genes and other epigenetic regulations[21,22]. Hugl-1 gene is one of the tumor suppresser genes involved in tumor cell proliferation. In this study, we reported that Hugl-1 was a potent anticancer gene for Eca109 cells through inducing G0/G1 cell cycle arrest and apoptosis. We concluded that up-regulation of Hugl-1 suppressed esophageal cancer cell proliferation by CCK-8 assay. Flow cytometry showed that overexpression of Hugl-1 reduced the number of cells in S-phase while increasing the number of cells in G0/G1-phase, indicating a G0/G1 arrest. To understand the mechanism by which Hugl-1 induces esophageal cancer apoptosis, we analyzed the expression of p65, p-p65, Bcl-2, Bax, survivin, c-myc, cyclin D1, p21, and caspase-3 and -9 between cells transfected with the Hugl-1-expressing plasmid and those transfected with the control plasmid. Immunoblotting analysis revealed that Hugl-1 overexpression significantly decreased the expression of p-p65, cyclin D1, Bcl-2, survivin, and c-myc and that it increased the expression of p21 and Bax. In addition, Hugl-1 significantly increased the expression and activity of caspase-3 and caspase-9. More importantly, Hugl-1 potently suppressed the growth of Eca109 cells xenografted in nude mice by inducing cell apoptosis.
Cell cycle arrest and apoptosis are two main ways by which cell growth can be inhibited. In higher eukaryotes, multiple cyclin-dependent kinases associate with multiple cyclins to regulate cell cycle progression[23,24]. Cyclin D1, a member of the G1 cyclins, controls the cell cycle transit from G1 to S phase[25]. The activities of CDKs and CDK/cyclin complexes are known to be regulated by the CIP/KIP family member p21[26]. P21 is, in turn, under transcriptional control of the tumor suppressor p53 and is required for p53-dependent cell cycle arrest[27]. In this study, we found that up-regulation of Hugl-1 in Eca109 cells resulted in a G0/G1 cell cycle arrest that was accompanied by down-regulation of cyclin D1 and up-regulation of p21. These data suggest that the mechanism of Hugl-1-induced cell cycle arrest involves down-regulation of cyclin D1 and up-regulation of the CDK inhibitor p21, causing inhibition of CDK activity.
Furthermore, the increase in Hugl-1 expression induced down-regulation of the anti-apoptotic gene Bcl-2 and up-regulation of the pro-apoptotic gene Bax. The central cast of players in the mitochondrial pathway of programmed cell death is the extended Bcl-2 family of proteins[28,29]. Therefore, the balance between the levels of Bcl-2 and Bax is critical in determining the fate of cells in terms of survival or death[30]. Homo-oligomerization of Bax leads to permeabilization of the mitochondrial membrane and subsequent release of cytochrome C to activate apoptosis[31]. Bcl-2 interacts with Bax, preventing its homo-oligomerization and, ultimately, apoptosis[31]. In this case, we found that Hugl-1 induced apoptosis in Eca109 cells, as evidenced by the increase in Annexin V-positive cells, the reduced cell proliferation, and the changes in caspase activation. Currently, there are two known pathways that activate the apoptotic caspase cascade, the intrinsic (mitochondrial) and extrinsic pathways[32]. Our results disclosed that the caspase-9-regulated intrinsic pathway was involved in Hugl-1-induced cell apoptosis. In Hugl-1-treated cells, we observed an increase in the cleaved caspases-9 and caspases-3. These results suggest that Hugl-1 induces apoptosis in Eca109 cells through activation of the mitochondrial pathway.
Considerable evidence indicates that NF-κB is constitutively active in esophageal cancer and that its activation is correlated with tumor progression[33]. The relationship between the NF-κB signaling pathway and tumor cell apoptosis has been extensively studied[34]. It has been presumed that the NF-κB pathway was involved in suppressing apoptosis[35]. The NF-κB family is composed of homodimers and heterodimers of the Rel family of proteins, including p65 (RelA), c-Rel, RelB, p52 and p50[36]. The most abundant form of NF-κB is a heterodimer with two subunits: p50 and p65. Our study showed that phospho-p65 was essentially absent in Hugl-1-overexpressing cells, but the total level of p65 was only slightly reduced. It has been suggested that Hugl-1 down-regulates the NF-κB signaling pathway through inhibition of the IKKα/β and p65 phosphorylation.
In addition, in the nude mice xenografted with Eca109 cells, we found that up-regulation of Hugl-1 reduced tumor growth (606.03 ± 22.49 mm3 vs 1126.56 ± 141.70 mm3). With a TUNEL assay, we found that Hugl-1 markedly increased the apoptosis rate of Eca109 cells in vivo (60.50% ± 9.11% vs 25.00% ± 12.25%). Thus, our results indicate that Hugl-1 may be a tumor suppressor of esophageal cancer.
In summary, our study has demonstrated that Hugl-1 exerts tumor suppressor effects by inducing growth suppression and apoptosis both in vitro and in vivo. G0/G1 cell cycle arrest induced by Hugl-1 occurs through a pathway that is mediated by p53-dependent p21 and cyclin D1 and that apoptosis induced by Hugl-1 occurs through the mitochondria pathway. The data presented here also indicate that Hugl-1 interferes with cell proliferation by affecting the NF-κB signaling pathway. The observations that Hugl-1 expression led to the loss of activated IKKα/β and p65 suggest that Hugl-1 is a negative regulator of NF-κB signaling. More importantly, Hugl-1 induced growth suppression and apoptosis in a human esophageal carcinoma cell line in vivo. Taken together, we show that Hugl-1 induces growth suppression and apoptosis in a human esophageal squamous cell carcinoma cell line both in vitro and in vivo. These data suggest that Hugl-1 may provide a novel target for treatment of esophageal cancer patients.
ACKNOWLEDGMENTS
We sincerely thank Xia H (Key Laboratory of Hubei Province for Digestive System Disease, Wuhan, China) for their support.
COMMENTS
Background
Esophageal squamous cell carcinoma (ESCC) is one of the most frequently diagnosed cancers in China. Most esophageal cancers are diagnosed in the advanced stages. Thus, detecting gene alternations that promote the carcinogenesis process leading to esophageal cancer will have a profound impact on the diagnosis and treatment of the disease. The human homologs of lethal giant larvae (lgl) are Hugl-1 (Llgl1) and Llgl2. The Hugl-1 protein shares 62.5% similarity with lgl. Several studies have shown that Hugl-1 transcripts are reduced or absent in a high proportion of breast cancers, lung cancers, prostate cancers, ovarian cancers, colorectal cancers and hepatocellular carcinomas. However, the effect of Hugl-1 on tumor progression and prognosis in ESCC is not clear. The authors aimed to determine whether the Hugl-1 exerts tumor suppressor effects in esophageal cancer.
Research frontiers
Targeted molecular therapy is a new effective treatment for cancer including esophageal cancer. Hugl-1 is a potential tumor suppressor in several cancers, but the role of Hugl-1 remains controversial and its exact role in ESCC remains unknown.
Innovations and breakthroughs
Authors constructed a Hugl-1 expression plasmid, pEZ-M29-Hugl1, for gene transfection. Authors transfected the pEZ-M29-Hugl1 plasmid into Eca109 esophageal cancer cell lines to overexpress Hugl-1. The results showed that overexpression of Hugl-1 could inhibit the growth of Eca109 cells and promote cell apoptosis, and modulate the expression of Bcl-2, Bax, caspase-3, caspase-9, etc. Hugl-1 may serve as a potential therapeutic target in ESCC. It suggested that Hugl-1 is a tumor suppressor and interact with the mitochondrial pathway in ESCC.
Applications
The results showed that Hugl-1 is a tumor suppressor and interact with the mitochondrial pathway in ESCC. It may contribute to the future research of ESCC and be a promising target for therapeutic intervention in ESCC.
Terminology
Lethal giant larvae homolog 1 (Human): This gene encodes a protein that is similar to a tumor suppressor in Drosophila.
Peer review
The function of Hugl-1 as a tumor suppressor has been studied in other cancers, but not in ESCC. Thus authors examined the effects of Hugl-1 up-regulation on cell growth and apoptosis in the ESCC cell line Eca109 and determined the conclusion. This conclusion is meaningful in the sense characterized the function of Hugl-1 in ESCC cells.
Footnotes
Supported by The Fundamental Research Funds for the Central Universities, No. 302274546
P- Reviewer Singh PK S- Editor Gou SX L- Editor A E- Editor Li JY
References
- 1.Brown J, Bothma H, Veale R, Willem P. Genomic imbalances in esophageal carcinoma cell lines involve Wnt pathway genes. World J Gastroenterol. 2011;17:2909–2923. doi: 10.3748/wjg.v17.i24.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tran B, Lucas R, Kimlin M, Whiteman D, Neale R. Association between ambient ultraviolet radiation and risk of esophageal cancer. Am J Gastroenterol. 2012;107:1803–1813. doi: 10.1038/ajg.2012.329. [DOI] [PubMed] [Google Scholar]
- 3.Law S, Wong J. Current management of esophageal cancer. J Gastrointest Surg. 2005;9:291–310. doi: 10.1016/j.gassur.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 4.Gamliel Z, Krasna MJ. Multimodality treatment of esophageal cancer. Surg Clin North Am. 2005;85:621–630. doi: 10.1016/j.suc.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Revels SL, Morris AM, Reddy RM, Akateh C, Wong SL. Racial disparities in esophageal cancer outcomes. Ann Surg Oncol. 2013;20:1136–1141. doi: 10.1245/s10434-012-2807-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vasioukhin V. Lethal giant puzzle of Lgl. Dev Neurosci. 2006;28:13–24. doi: 10.1159/000090749. [DOI] [PubMed] [Google Scholar]
- 7.Betschinger J, Mechtler K, Knoblich JA. The Par complex directs asymmetric cell division by phosphorylating the cytoskeletal protein Lgl. Nature. 2003;422:326–330. doi: 10.1038/nature01486. [DOI] [PubMed] [Google Scholar]
- 8.Jacob L, Opper M, Metzroth B, Phannavong B, Mechler BM. Structure of the l(2)gl gene of Drosophila and delimitation of its tumor suppressor domain. Cell. 1987;50:215–225. doi: 10.1016/0092-8674(87)90217-0. [DOI] [PubMed] [Google Scholar]
- 9.Woods DF, Bryant PJ. The discs-large tumor suppressor gene of Drosophila encodes a guanylate kinase homolog localized at septate junctions. Cell. 1991;66:451–464. doi: 10.1016/0092-8674(81)90009-x. [DOI] [PubMed] [Google Scholar]
- 10.Bilder D, Li M, Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. 2000;289:113–116. doi: 10.1126/science.289.5476.113. [DOI] [PubMed] [Google Scholar]
- 11.Bilder D. Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev. 2004;18:1909–1925. doi: 10.1101/gad.1211604. [DOI] [PubMed] [Google Scholar]
- 12.Vieira V, de la Houssaye G, Lacassagne E, Dufier JL, Jaïs JP, Beermann F, Menasche M, Abitbol M. Differential regulation of Dlg1, Scrib, and Lgl1 expression in a transgenic mouse model of ocular cancer. Mol Vis. 2008;14:2390–2403. [PMC free article] [PubMed] [Google Scholar]
- 13.Grifoni D, Garoia F, Bellosta P, Parisi F, De Biase D, Collina G, Strand D, Cavicchi S, Pession A. aPKCzeta cortical loading is associated with Lgl cytoplasmic release and tumor growth in Drosophila and human epithelia. Oncogene. 2007;26:5960–5965. doi: 10.1038/sj.onc.1210389. [DOI] [PubMed] [Google Scholar]
- 14.Strand D, Unger S, Corvi R, Hartenstein K, Schenkel H, Kalmes A, Merdes G, Neumann B, Krieg-Schneider F, Coy JF. A human homologue of the Drosophila tumour suppressor gene l(2)gl maps to 17p11.2-12 and codes for a cytoskeletal protein that associates with nonmuscle myosin II heavy chain. Oncogene. 1995;11:291–301. [PubMed] [Google Scholar]
- 15.Grifoni D, Garoia F, Schimanski CC, Schmitz G, Laurenti E, Galle PR, Pession A, Cavicchi S, Strand D. The human protein Hugl-1 substitutes for Drosophila lethal giant larvae tumour suppressor function in vivo. Oncogene. 2004;23:8688–8694. doi: 10.1038/sj.onc.1208023. [DOI] [PubMed] [Google Scholar]
- 16.Schimanski CC, Schmitz G, Kashyap A, Bosserhoff AK, Bataille F, Schäfer SC, Lehr HA, Berger MR, Galle PR, Strand S, et al. Reduced expression of Hugl-1, the human homologue of Drosophila tumour suppressor gene lgl, contributes to progression of colorectal cancer. Oncogene. 2005;24:3100–3109. doi: 10.1038/sj.onc.1208520. [DOI] [PubMed] [Google Scholar]
- 17.Kuphal S, Wallner S, Schimanski CC, Bataille F, Hofer P, Strand S, Strand D, Bosserhoff AK. Expression of Hugl-1 is strongly reduced in malignant melanoma. Oncogene. 2006;25:103–110. doi: 10.1038/sj.onc.1209008. [DOI] [PubMed] [Google Scholar]
- 18.Tsuruga T, Nakagawa S, Watanabe M, Takizawa S, Matsumoto Y, Nagasaka K, Sone K, Hiraike H, Miyamoto Y, Hiraike O, et al. Loss of Hugl-1 expression associates with lymph node metastasis in endometrial cancer. Oncol Res. 2007;16:431–435. doi: 10.3727/000000007783980855. [DOI] [PubMed] [Google Scholar]
- 19.Lu X, Feng X, Man X, Yang G, Tang L, Du D, Zhang F, Yuan H, Huang Q, Zhang Z, et al. Aberrant splicing of Hugl-1 is associated with hepatocellular carcinoma progression. Clin Cancer Res. 2009;15:3287–3296. doi: 10.1158/1078-0432.CCR-08-2078. [DOI] [PubMed] [Google Scholar]
- 20.Wang HY, Dong WG, Wang Q, Luo HS, Zhang Y. Expression of Human Lethal-giant-lavae 1 and its significance in esophageal carcinoma. Zhonghua Xiaohua Zazhi. 2010;30:271–272. [Google Scholar]
- 21.Qi YJ, Chao WX, Chiu JF. An overview of esophageal squamous cell carcinoma proteomics. J Proteomics. 2012;75:3129–3137. doi: 10.1016/j.jprot.2012.04.025. [DOI] [PubMed] [Google Scholar]
- 22.Tougeron D, Richer JP, Silvain C. Management of esophageal adenocarcinoma. J Visc Surg. 2011;148:e161–e170. doi: 10.1016/j.jviscsurg.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 23.Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Woo RA, Poon RY. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle. 2003;2:316–324. [PubMed] [Google Scholar]
- 25.Zhang LQ, Jiang F, Xu L, Wang J, Bai JL, Yin R, Wu YQ, Meng LJ. The role of cyclin D1 expression and patient’s survival in non-small-cell lung cancer: a systematic review with meta-analysis. Clin Lung Cancer. 2012;13:188–195. doi: 10.1016/j.cllc.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Kaldis P, Aleem E. Cell cycle sibling rivalry: Cdc2 vs. Cdk2. Cell Cycle. 2005;4:1491–1494. doi: 10.4161/cc.4.11.2124. [DOI] [PubMed] [Google Scholar]
- 27.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011;18:1414–1424. doi: 10.1038/cdd.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barillé-Nion S, Bah N, Véquaud E, Juin P. Regulation of cancer cell survival by BCL2 family members upon prolonged mitotic arrest: opportunities for anticancer therapy. Anticancer Res. 2012;32:4225–4233. [PubMed] [Google Scholar]
- 30.Coultas L, Strasser A. The role of the Bcl-2 protein family in cancer. Semin Cancer Biol. 2003;13:115–123. doi: 10.1016/s1044-579x(02)00129-3. [DOI] [PubMed] [Google Scholar]
- 31.Ding J, Zhang Z, Roberts GJ, Falcone M, Miao Y, Shao Y, Zhang XC, Andrews DW, Lin J. Bcl-2 and Bax interact via the BH1-3 groove-BH3 motif interface and a novel interface involving the BH4 motif. J Biol Chem. 2010;285:28749–28763. doi: 10.1074/jbc.M110.148361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87. doi: 10.1186/1756-9966-30-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Changhui M, Tianzhong M, Zhongjing S, Ling C, Ning W, Ningxia Z, Xiancai C, Haibin C. Silencing of tumor necrosis factor receptor 1 by siRNA in EC109 cells affects cell proliferation and apoptosis. J Biomed Biotechnol. 2009;2009:760540. doi: 10.1155/2009/760540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Varfolomeev E, Goncharov T, Maecker H, Zobel K, Kömüves LG, Deshayes K, Vucic D. Cellular inhibitors of apoptosis are global regulators of NF-κB and MAPK activation by members of the TNF family of receptors. Sci Signal. 2012;5:ra22. doi: 10.1126/scisignal.2001878. [DOI] [PubMed] [Google Scholar]
- 35.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 36.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109 Suppl:S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]