

Abstract
Purpose
Many infants develop a postsurgical chylothorax after diaphragmatic hernia repair. The pathogenesis remains elusive but may be due to dysfunctional lymphatic development. This study characterizes pulmonary lymphatic development in the nitrofen mouse model of CDH.
Methods
CD1 pregnant mice were fed nitrofen/bisdiamine (N/B) or olive oil at E8.5. At E14.5 and E15.5, lung buds were categorized by phenotype: normal, N/B without CDH (N/B−CDH), or N/B with CDH (N/B+CDH). Anti-CD31 was used to localize all endothelial cells, while anti-LYVE-1 was used to identify lymphatic endothelial cells in lung buds using immunofluorescence. Differential protein expression of lymphatic-specific markers was analyzed.
Results
Lymphatic endothelial cells localized to the mesenchyme surrounding the airway epithelium at E15.5. CD31 and LYVE-1 colocalization identified lymphatic endothelial cells. LYVE-1 expression was upregulated in N/B+CDH lung buds in comparison to N/B−CDH and normal lung buds by immunofluorescence. Western blotting shows that VEGF-D, LYVE-1, Prox-1, and VEGFR-3 expression was upregulated in N/B+CDH lung buds in comparison to N/B−CDH or control lung buds at E14.5.
Conclusions
Lung lymphatics are hyperplastic in N/B+CDH. Upregulation of lymphatic-specific genes suggest that lymphatic hyperplasia plays an important role in dysfunctional lung lymphatic development in the nitrofen mouse model of CDH.
Keywords: lung lymphatic development, Prox-1, VEGFR-3, VEGF-D, LYVE-1, nitrofen, Congenital Diaphragmatic Hernia, CDH, chylothorax
Introduction
Congenital diaphragmatic hernia (CDH) is a common birth anomaly that affects 1 in 2000 newborns and carries a 30% mortality.1 The mainstay of treatment includes respiratory support and stabilization prior to repair of the diaphragmatic defect. Postoperative chylothorax was first described in 1973,2 and since then, centers report between 5% – 28% of infants develop chylothorax after diaphragmatic hernia repair.3–8 Chylothorax prolongs duration of ventilation, increases rates of sepsis, cost of care, length of stay, and also prolongs the need for parenteral nutrition and central lines.3, 6, 8, 9 Postoperative chylothorax contributes significant morbidity to infants with CDH, yet the pathogenesis remains elusive.
Surgical disruption of lymphatics remains the most widely accepted hypothesis to explain postoperative chylothorax after CDH repair.4, 10 Disruption of lymphatics has been associated with resection of the hernia sac7 or patch repair of the diaphragmatic defect.3 However, most diaphragmatic hernias and chylothoraces are left-sided, whereas the cysterna chyli and thoracic duct normally traverse the diaphragm into the thorax on the right side. In addition, two patients developed chylothorax on extracorporeal membrane oxygenation (ECMO) prior to repair of the diaphragmatic hernia,3 which argues against surgical disruption of lymphatics as the cause of postoperative chylothoraces in CDH. Klin et al11 proposed that visceral herniation obstructs the thoracic duct above the fifth thoracic vertebrae, leading to a back-pressure phenomenon that causes chylothorax through small disruptions in the thoracic duct. Postoperative chylothoraces have also been attributed to increased central venous pressure (secondarily obstructing thoracic duct outflow into the superior vena cava),9, 12 and aberrant lymphatic development as part of the constellation of defects in CDH.
The rodent model of CDH effectively replicates the constellation of defects seen in human infants with CDH: pulmonary hypoplasia, pulmonary hypertension, and a Bochdalek-type, predominantly left-sided diaphragmatic defect.13–15 However, pulmonary lymphatic development has not been well-characterized in the nitrofen rodent model of CDH. Normal lung lymphatics are important for reabsorption of alveolar fluid after the onset of breathing at birth and for reabsorption of interstitial fluid in mature lungs.16 In the embryonic mouse, vascular endothelial growth factor (VEGF) D is first expressed in the lung mesenchyme at E13.5.17 The mature, processed forms of VEGF-C18 and VEGF-D19 activate vascular endothelial growth factor receptor 3 (VEGFR-3) signaling to promote lymphatic development.10, 20 Mice deficient in VEGF-D have a small but significant reduction in the number of lung lymphatics,21 but lymphatic development is greatly impaired in mice deficient in VEGFR-3.22, 23 Impaired VEGFR-3 signaling is associated with lymphedema in humans24 and mice.25 In addition, transgenic overexpression of a soluble form of VEGFR-3 sequesters VEGF-C and VEGF-D in the embryonic lung to prevent endogenous VEGFR-3 activation, impairing the growth of lung lymphatics.16
VEGFR-3 activation has previously been shown to be important for normal lymphatic development in mice. The purpose of this study is to characterize lung lymphatic development in the nitrofen mouse model of CDH. We hypothesize that expression of VEGF-D and VEGFR-3 is abnormal in the nitrofen mouse model of CDH, resulting in aberrant lymphatic development. These studies will give insight into the pathophysiology behind postoperative chylothoraces in CDH, which will help prevent and treat chylothoraces in the future.
Methods
Pregnant CD1 mice were gavage fed 15mg of nitrofen and 10mg of bisdiamine (N/B) or olive oil (control) at E8.5 under IACUC protocol AN081689-03C. At E14.5 and E15.5, pregnant dams underwent laparotomy and hysterotomy. Lung buds were dissected from mouse embryos, snap frozen for protein analysis, or placed in OCT for histological analysis. Mouse embryos were categorized by phenotype: normal (control), N/B without CDH (N/B−CDH), or N/B with CDH (N/B+CDH).
Immunofluorescence Antibodies
Rabbit anti-LYVE-1 (1:1000 for immunofluorescence, 1:500 for whole mount staining) was purchased from Abcam (Cambridge, MA). Rat anti-CD31 (1:500 for immunofluorescence and whole mount staining) was purchased from BD Biosciences (San Jose, CA). Secondary antibodies donkey anti-rabbit-Cy3 (1:1000 for immunofluorescence, 1:200 for whole mount staining), and donkey anti-rat-FITC (1:500) were purchased from Jackson ImmunoResearch (West Grove, PA).
Western Blot Antibodies
Rabbit anti-Prox-1 (1:800), rabbit anti-LYVE-1 (1:3000) and rabbit anti-β-actin (1:5000) primary antibodies were purchased from Abcam (Cambridge, MA). Goat anti-VEGF-D (0.1 micrograms/mL) and goat anti-VEGFR-3 (0.1 micrograms/mL) antibodies were purchased from R&D Systems (Minneapolis, MN). Secondary goat anti-rabbit antibody conjugated with HRP (1:3000 for LYVE-1 and Prox-1, 1:10,000 for β-actin), and horse anti-goat antibody conjugated with HRP (1:3000) were purchased from Cell Signaling (Danvers, MA) and Vector Laboratory (Burlingame, CA), respectively.
Immunofluorescence Protocol
After lung buds were isolated and frozen in OCT, tissue was cut in 8 micron sections and fixed with acetone. Slides were blocked in 3% normal donkey serum/0.2% Triton X-100/PBS for 30 minutes at room temperature. Slides were incubated with primary antibodies in 3% normal donkey serum/0.2% TritonX-100/PBS at 4° Celsius overnight. Slides were washed 3 times for two minutes each time with PBST at room temperature before incubation with secondary antibodies in 2% BSA/3% normal donkey serum/0.2% Triton X-100/PBS at 4° Celsius overnight. They were washed with PBST three times for two minutes each time, equilibrated in PBS, and then counterstained with blue-fluorescent DAPI (Vector Laboratory (Burlingame, CA).
Whole mount lung bud staining protocol
Lung buds were dehydrated in 5% H2O2/100% methanol at room temperature (RT) for 5 hours, and then washed twice for 10 minutes in 100% methanol at RT with gentle agitation. Then they were sequentially washed for 10 minutes each with 75%, 50% and 25% methanol/PBST at RT, and then 100% PBST two times for 10 minutes each time. Lung buds were blocked with 5% normal donkey serum/PBS/0.5% Triton-X at RT for 1 hour and incubated with primary antibody overnight at 4° Celsius. The lung buds were then washed in 5% normal donkey serum/PBS/0.5% Triton-X with gentle agitation for 1 hour, and then incubated in secondary antibody overnight at 4° Celsius. The lung buds were washed three times for 20 minutes each in PBST, and then mounted in mount medium.
Western Blotting Protocol
Two embryonic lung buds were combined for each mouse phenotype: normal, N/B−CDH and N/B+CDH protein was isolated and concentration was determined using the Micro BCA Protein Assay Kit (Thermo Scientific, Rockford, IL). Two micrograms of lung bud protein from each mouse embryonic phenotype at E14.5 and E15.5 were loaded into a 5–20% Tris-HCl gel (Biorad, Hercules, CA) and run at 150V over 45 minutes. Protein was transferred onto a PVDF membrane (Biorad, Hercules, CA) at 100V over 1 hour. PVDF membranes were blocked in 5% milk/TBST for 1 hour, before overnight incubation with primary antibodies in 1% milk/TBST at 4° Celsius. Blots were washed three times with TBST for ten minutes each time and then incubated with secondary antibody overnight at 4° Celsius in 1% milk/TBST. Membranes were washed three times for ten minutes each time with TBST before detection of HRP activity with Pierce ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL). Protein densitometry was determined using Image J (Bethesda, MD).
Results
Mouse embryos were harvested at E14.5 and E15.5, and characterized by CDH phenotype. Nitrofen-bisdiamine (N/B) treated mice were edematous, but control mouse embryos were normal in appearance (Figure 1). Forty-two percent of embryos at E14.5 and 57% of embryos at E15.5 had a left-sided CDH after pregnant dams were treated with N/B.
Figure 1.
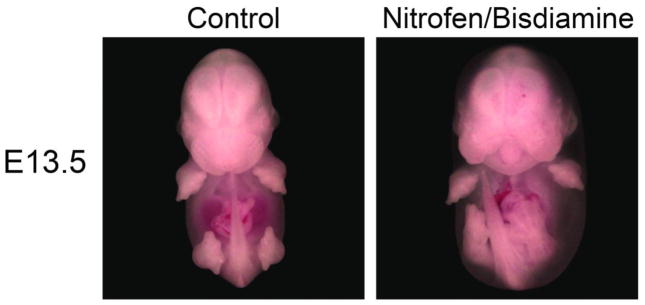
E13.5 mouse embryos. N/B embryos (right) are edematous in comparison to normal (left).
Immunofluorescence
Embryonic lung bud sections are visualized at 20X. Lymphatic endothelial cells identified by LYVE-1-Cy3 (red) were located in the mesenchyme surrounding the airway epithelium in sections of E15.5 lung buds (Figure 2). Endothelial cells were localized with CD31-FITC antibodies (green). CD31 and LYVE-1 colocalization identified lymphatic endothelial cells (yellow). There was increased LYVE-1 expression in N/B+CDH lung buds in comparison to N/B−CDH and normal lung buds by immunofluorescence. In addition, lymphatic cells appeared more linear and organized in the normal lung buds in comparison to N/B+CDH.
Figure 2.

Immunofluorescence of E15.5 lung bud sections at 20X magnification. Endothelial cells were stained with anti-CD31-FITC (green), and lymphatic endothelial cells were localized with anti-LYVE-1-Cy3 (red). Lymphatic endothelial cells can be identified by colocalization of LYVE-1 and CD31 (yellow). Lung lymphatics are identified in the mesenchyme surrounding the airway epithelium. Lymphatics are hyperplastic in N/B+CDH lung buds in comparison to normal and N/B-CDH lung buds.
Whole mount staining
Whole mount lung buds were stained with immunofluorescent antibodies at E14.5 (Figure 3). Normal lung buds are larger than N/B+CDH or N/B-CDH lung buds. N/B+CDH lung bud images demonstrate severe left lung hypoplasia. Lymphatic endothelial cells are identified by LYVE-1-Cy3 (red), and endothelial cells are identified with CD31-FITC (green). Normal lung buds show linear, organized, homogenous distribution of LYVE-1 expression. In contrast, LYVE-1 expression in N/B+CDH lung buds is disorganized and distribution is heterogeneous throughout the surface of the lung bud, even in comparison to the N/B-CDH lung buds.
Figure 3.
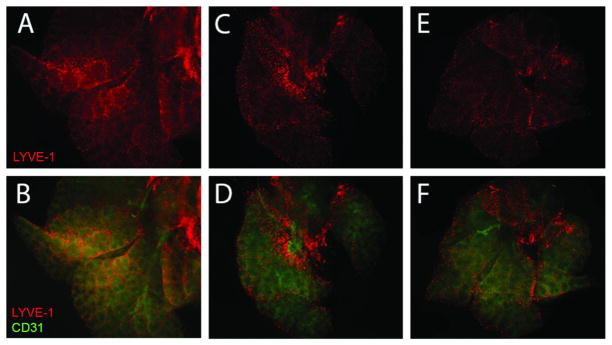
Whole mount lung bud staining at E14.5 at 5X magnification. Lymphatic endothelial cells were localized with anti-LYVE-1-Cy3 (red). Endothelial cells were localized with anti-CD31-FITC (green). Normal lung buds (A and B) have linear, organized lymphatic endothelial cells with homogeneous distribution throughout the lung bud. N/B+CDH lung buds (C and D) are hypoplastic, and lymphatic endothelial cells are disorganized, nonlinear, with heterogeneous distribution throughout the lung bud. N/B-CDH lung buds (E and F) have left lung hypoplasia, but less severe than that seen in N/B+CDH. Lymphatics in N/B-CDH lung buds are more organized and linear than in N/B+CDH lung buds.
Western Blotting
To quantify expression of lymphatic-specific proteins, we performed Western blotting on protein isolated from mouse embryonic lung buds. Lymphatic specific genes, such as VEGF-D, LYVE-1, VEGFR-3 and Prox-1 were tested. Expression of all genes was upregulated in N/B+CDH lung buds by Western blotting at E14.5 in comparison to N/B−CDH or control lung buds at E14.5 (Figures 4 and 5). While VEGF-D and LYVE-1 expression remained elevated at E15.5, VEGFR-3 and Prox-1 expression normalized in the N/B+CDH lung buds. At E14.5, relative expression of LYVE-1, VEGFR-3, and Prox-1 in N/B+CDH lung buds was increased 2-fold compared to normal and N/B−CDH lung buds. Relative expression of VEGF-D in N/B+CDH lung buds was increased 2.5-fold compared to normal and 3-fold compared to N/B−CDH lung buds. At E15.5, N/B+CDH lung buds had a 4-fold increase in relative protein expression of LYVE-1 in comparison to normal, and 1.5-fold increase in comparison to N/B−CDH. At E15.5, relative expression of VEGF-D in N/B+CDH was increased 2.5-fold compared to normal and N/B−CDH. However, expression of VEGFR-3 and Prox-1 normalized in N/B+CDH lung buds at E15.5.
Figure 4.

Western blot of whole lung buds at E14.5 and E15.5. Expression of lymphatic-specific genes LYVE-1, VEGF-D, VEGFR-3 and Prox-1 are upregulated in N/B+CDH lung buds in comparison to normal and N/B-CDH.
Figure 5.

Protein densitometry. Relative expression of LYVE-1, VEGF-D, VEGFR-3 and Prox-1 are increased in N/B+CDH lung buds in comparison to normal and N/B-CDH lung buds at E14.5. At E15.5, relative expression of LYVE-1 and VEGF-D remains high but VEGFR-3 and Prox-1 expression normalizes.
Discussion
Pulmonary hypoplasia and pulmonary hypertension are well-described, but lung lymphatic development has not yet been characterized in the nitrofen rodent model of CDH. Mouse lymphatic development begins around E9.5, when blood endothelial cells from the cardinal vein begin to express Prox-1, a lymphatic-specific nuclear transcription factor that determines lymphatic endothelial cell fate by regulating VEGFR-3 expression.23, 26–28 These cells leave the cardinal vein to begin forming lymphatic sacs. Both VEGFR-3 and Prox-1 knockout mice have defective lymphatic development and generalized edema during development.23, 29 Similarly, we found that mouse embryos exposed to N/B had generalized edema in comparison to control mouse embryos, providing gross evidence of aberrant lymphatic development in this model.
At E15.5, lymphatic endothelial cells appear linear and organized, whereas lymphatics surrounding the airway epithelium appear to be hyperplastic and disorganized in the N/B+CDH lung buds. Though this effect could be attributed solely to the teratogenic effects of nitrofen and bisdiamine, the lymphatic endothelial cells appeared to be more disorganized and hyperplastic in the N/B+CDH lung buds in comparison to the N/B−CDH lung buds, suggesting that disorganized lymphatic hyperplasia characterizes aberrant lung lymphatic development in the nitrofen mouse model of CDH.
We performed Western blotting and quantified expression using protein densitometry. At E14.5, relative expression of lymphatic-specific proteins LYVE-1, VEGF-D, VEGFR-3 and Prox-1 were much higher in the N/B+CDH mouse embryonic lung buds in comparison to the control and N/B−CDH. Relative expression of VEGF-D and LYVE-1 remained high in lung buds at E15.5. However, relative expression of both VEGFR-3 and Prox-1 decreased to levels comparable to control and N/B−CDH lung buds at this time point. This corresponds with previously published data that reports that Prox-1 negative endothelial cells in heterozygous embryos from transgenic mice had low levels of VEGFR-3.26 Our results indicate that expression of lymphatic-specific proteins is upregulated in the N/B+CDH mouse lung buds. In addition, upregulation of VEGFR-3 and Prox-1 expression is greatest at E14.5, and normalizes by E15.5.
Upregulation of lymphatic-specific markers at E14.5 may be a response to generalized lymphedema to restore normal fluid homeostasis. In an injury model of lymphedema, disruption of lymphatics in a mouse tail leads to upregulation of VEGF-C and hyperplastic lymphatics.30 However, hyperplastic lymphatics are not necessarily functional lymphatic vessels. Injection of fluorescent tracer into the injured mouse tail outlined lymphatic vessels, but also leaked into the interstitial space.30 It is difficult to delineate whether upregulation of lymphatic-specific proteins represents a cause or an effect, but aberrant lung lymphatic development in the nitrofen mouse model of CDH is characterized by lymphatic hyperplasia, not lymphatic hypoplasia.
In summary, many studies attribute postoperative chylothorax to traumatic disruption during surgery, but we have demonstrated aberrant lung lymphatic development in the nitrofen mouse model of CDH. Lung lymphatic development is characterized by lymphatic hyperplasia in a heterogeneous distribution in the nitrofen mouse model of CDH. Lymphatic hyperplasia is associated with upregulation of lymphatic-specific markers, which play an important part in dysfunctional lung lymphatic development in the nitrofen mouse model of CDH.
Acknowledgments
This work was supported by the National Institutes of Health K08 HL092062 (DM), T32 2T32HD049303-06A1 (ES), and T32 GM008258 (SCC), as well as the UCSF Academic Senate Individual Investigator Grant (DM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cortes RA, Keller RL, Townsend T, et al. Survival of severe congenital diaphragmatic hernia has morbid consequences. J Pediatr Surg. 2005;40(1):36–45. doi: 10.1016/j.jpedsurg.2004.09.037. discussion 45–6. [DOI] [PubMed] [Google Scholar]
- 2.Wiener ES, Owens L, Salzberg AM. Chylothorax after Bochadalek herniorrhaphy in a neonate. Treatment with intravenous hyperalimentation. J Thorac Cardiovasc Surg. 1973;65(2):200–6. [PubMed] [Google Scholar]
- 3.Gonzalez R, Bryner BS, Teitelbaum DH, et al. Chylothorax after congenital diaphragmatic hernia repair. J Pediatr Surg. 2009;44(6):1181–5. doi: 10.1016/j.jpedsurg.2009.02.021. discussion 1185. [DOI] [PubMed] [Google Scholar]
- 4.Casaccia G, Crescenzi F, Palamides S, et al. Pleural effusion requiring drainage in congenital diaphragmatic hernia: incidence, aetiology and treatment. Pediatr Surg Int. 2006;22(7):585–8. doi: 10.1007/s00383-006-1706-8. [DOI] [PubMed] [Google Scholar]
- 5.Kamiyama M, Usui N, Tani G, et al. Postoperative chylothorax in congenital diaphragmatic hernia. Eur J Pediatr Surg. 2010;20(6):391–4. doi: 10.1055/s-0030-1261956. [DOI] [PubMed] [Google Scholar]
- 6.Kavvadia V, Greenough A, Davenport M, et al. Chylothorax after repair of congenital diaphragmatic hernia--risk factors and morbidity. J Pediatr Surg. 1998;33(3):500–2. doi: 10.1016/s0022-3468(98)90097-5. [DOI] [PubMed] [Google Scholar]
- 7.Mercer S. Factors involved in chylothorax following repair of congenital posterolateral diaphragmatic hernia. J Pediatr Surg. 1986;21(9):809–11. doi: 10.1016/s0022-3468(86)80374-8. [DOI] [PubMed] [Google Scholar]
- 8.Zavala A, Campos JM, Riutort C, et al. Chylothorax in congenital diaphragmatic hernia. Pediatr Surg Int. 2010;26(9):919–22. doi: 10.1007/s00383-010-2677-3. [DOI] [PubMed] [Google Scholar]
- 9.Mills J, Safavi A, Skarsgard ED. Chylothorax after congenital diaphragmatic hernia repair: a population-based study. J Pediatr Surg. 2012;47(5):842–6. doi: 10.1016/j.jpedsurg.2012.01.031. [DOI] [PubMed] [Google Scholar]
- 10.Oliver G, Detmar M. The rediscovery of the lymphatic system: old and new insights into the development and biological function of the lymphatic vasculature. Genes Dev. 2002;16(7):773–83. doi: 10.1101/gad.975002. [DOI] [PubMed] [Google Scholar]
- 11.Klin B, Kohelet D, Bar-Nathan N, et al. Chylothorax complicating repair of congenital diaphragmatic hernia. Isr J Med Sci. 1992;28(12):891–2. [PubMed] [Google Scholar]
- 12.Trevisanuto D, Chiandetti L, Biban P, et al. Chylothorax complicating ECMO and surgical repair of congenital diaphragmatic hernia. Isr J Med Sci. 1995;31(6):388–9. [PubMed] [Google Scholar]
- 13.Luong C, Rey-Perra J, Vadivel A, et al. Antenatal sildenafil treatment attenuates pulmonary hypertension in experimental congenital diaphragmatic hernia. Circulation. 2011;123(19):2120–31. doi: 10.1161/CIRCULATIONAHA.108.845909. [DOI] [PubMed] [Google Scholar]
- 14.Chinoy MR, Nielsen HC, Volpe MV. Mesenchymal nuclear transcription factors in nitrofen-induced hypoplastic lung. J Surg Res. 2002;108(2):203–11. doi: 10.1006/jsre.2002.6550. [DOI] [PubMed] [Google Scholar]
- 15.Mey J, Babiuk RP, Clugston R, et al. Retinal dehydrogenase-2 is inhibited by compounds that induce congenital diaphragmatic hernias in rodents. Am J Pathol. 2003;162(2):673–9. doi: 10.1016/s0002-9440(10)63861-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kulkarni RM, Herman A, Ikegami M, et al. Lymphatic ontogeny and effect of hypoplasia in developing lung. Mech Dev. 2011;128(1–2):29–40. doi: 10.1016/j.mod.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Greenberg JM, Thompson FY, Brooks SK, et al. Mesenchymal expression of vascular endothelial growth factors D and A defines vascular patterning in developing lung. Dev Dyn. 2002;224(2):144–53. doi: 10.1002/dvdy.10095. [DOI] [PubMed] [Google Scholar]
- 18.Joukov V, Sorsa T, Kumar V, et al. Proteolytic processing regulates receptor specificity and activity of VEGF-C. EMBO J. 1997;16(13):3898–911. doi: 10.1093/emboj/16.13.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stacker SA, Stenvers K, Caesar C, et al. Biosynthesis of vascular endothelial growth factor-D involves proteolytic processing which generates non-covalent homodimers. J Biol Chem. 1999;274(45):32127–36. doi: 10.1074/jbc.274.45.32127. [DOI] [PubMed] [Google Scholar]
- 20.Makinen T, Alitalo K. Lymphangiogenesis in development and disease. Novartis Found Symp. 2007;283:87–98. doi: 10.1002/9780470319413.ch8. discussion 98–105, 238–41. [DOI] [PubMed] [Google Scholar]
- 21.Baldwin ME, Halford MM, Roufail S, et al. Vascular endothelial growth factor D is dispensable for development of the lymphatic system. Mol Cell Biol. 2005;25(6):2441–9. doi: 10.1128/MCB.25.6.2441-2449.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karkkainen MJ, Haiko P, Sainio K, et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. 2004;5(1):74–80. doi: 10.1038/ni1013. [DOI] [PubMed] [Google Scholar]
- 23.Haiko P, Makinen T, Keskitalo S, et al. Deletion of vascular endothelial growth factor C (VEGF-C) and VEGF-D is not equivalent to VEGF receptor 3 deletion in mouse embryos. Mol Cell Biol. 2008;28(15):4843–50. doi: 10.1128/MCB.02214-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karkkainen MJ, Saaristo A, Jussila L, et al. A model for gene therapy of human hereditary lymphedema. Proc Natl Acad Sci U S A. 2001;98(22):12677–82. doi: 10.1073/pnas.221449198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irrthum A, Karkkainen MJ, Devriendt K, et al. Congenital hereditary lymphedema caused by a mutation that inactivates VEGFR3 tyrosine kinase. Am J Hum Genet. 2000;67(2):295–301. doi: 10.1086/303019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wigle JT, Harvey N, Detmar M, et al. An essential role for Prox1 in the induction of the lymphatic endothelial cell phenotype. Embo Journal. 2002;21(7):1505–1513. doi: 10.1093/emboj/21.7.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flister MJ, Wilber A, Hall KL, et al. Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-kappaB and Prox1. Blood. 2010;115(2):418–29. doi: 10.1182/blood-2008-12-196840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lohela M, Saaristo A, Veikkola T, et al. Lymphangiogenic growth factors, receptors and therapies. Thromb Haemost. 2003;90(2):167–84. doi: 10.1160/TH03-04-0200. [DOI] [PubMed] [Google Scholar]
- 29.Hong YK, Detmar M. Prox1, master regulator of the lymphatic vasculature phenotype. Cell Tissue Res. 2003;314(1):85–92. doi: 10.1007/s00441-003-0747-8. [DOI] [PubMed] [Google Scholar]
- 30.Rutkowski JM, Moya M, Johannes J, et al. Secondary lymphedema in the mouse tail: Lymphatic hyperplasia, VEGF-C upregulation, and the protective role of MMP-9. Microvasc Res. 2006;72(3):161–71. doi: 10.1016/j.mvr.2006.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]