

Summary
Hemochromatosis is a genetic disorder of iron overload resulting from loss-of-function mutations in genes coding for the iron-regulatory proteins HFE, transferrin receptor 2, ferroportin, hepcidin, and hemojuvelin (HJV). Recent studies have established the expression of all the five genes in retina, indicating their importance in retinal iron homeostasis. We previously demonstrated that Hjv is expressed in retinal pigment epithelium (RPE), outer and inner nuclear layers, and ganglion cell layer. Here we report on the consequences of Hjv deletion on the retina in mice. Hjv-null mice at ≥ 18 months of age had increased iron accumulation in retina with marked morphological damage compared to age-matched controls; these changes were not found in younger mice. The retinal phenotype in Hjv-null mice included hyperplasia of RPE. We isolated RPE cells from wild type and Hjv-null mice and examined their growth patterns. Hjv-null RPE cells were less senescent and exhibited a hyperproliferative phenotype. Hjv-null RPE cells also showed upregulation of Slc7a11 that codes for the ‘transporter proper’ subunit xCT in the heterodimeric amino acid transporter cystine/glutamate exchanger (xCT/4F2hc). Bone morphogenic protein (BMP) 6 could not induce hepcidin expression in Hjv-null RPE cells, confirming that retinal cells require Hjv for induction of hepcidin via BMP6 signaling. Hjv is a glycosylphosphatidylinositol-anchored protein, and the membrane-associated Hjv is necessary for BMP6-mediated activation of hepcidin promoter in RPE cells. Taken together, these studies confirm the biological importance of Hjv in the regulation of iron homeostasis in the retina and in RPE.
Keywords: hemojuvelin, hemochromatosis, retinal pigment epithelium, hepcidin, bone morphogenic protein, knockout mouse
INTRODUCTION
Iron is an essential nutrient obligatory for vital cellular functions. However it can provoke oxidative stress and cellular dysfunction when it accumulates excessively in tissues. In recent years, we have been investigating the retinal expression of various genes involved in the maintenance of iron homeostasis to understand the role of these genes in retinal health and disease [1–5]. The rationale for these investigations was the growing evidence that retina also is susceptible to iron-induced oxidative damage similar to other organs. The presence of excess iron in retina has been demonstrated in patients with age-related macular degeneration (AMD) [6, 7] and aceruloplasminemia with associated macular degeneration [8]. Similarly, knockout mice with disruption of two iron-oxidizing enzymes, ceruloplasmin and hephaestin, have excessive iron accumulation in retinal pigment epithelium (RPE) [9, 10]. Hereditary hemochromatosis, an autosomal recessive disorder of iron metabolism, is the most common genetic disease in Caucasians with homozygosity in the range of approximately 1 in 300. Hemochromatosis patients accumulate excess iron in various organs, including the liver, pancreas, kidney, heart, and brain [11–14]. This results in hepatocarcinoma/cirrhosis, diabetes, nephropathy, cardiomyopathy, and pituitary dysfunction. Almost 85% of the cases with hemochromatosis represent the classical form, which results in iron overload mainly in older patients due to mutations in HFE [Histocompatability leukocyte antigen class I-like protein involved in iron (FE) homeostasis]. The remaining ~15% of mutations occur in hepcidin, hemojuvelin (HJV), ferroportin, and transferrin receptor 2 (TfR2), all of which are also important determinants of iron homeostasis. Mutations in HJV and hepcidin lead to iron overload at a much younger age resulting in juvenile hemochromatosis [14].
While there is unequivocal evidence for the accumulation of iron in various systemic organs in hemochromatosis in humans with consequent dysfunction of these organs, little information is available on retinal involvement in this disease. Indeed, there have been only two reports of retinal iron accumulation in hemochromatosis patients [15, 16]. The apparent lack of focus on the retina as a potential target organ for iron overload in hemochromatosis is likely due to the widespread notion that the blood-retinal barrier dissociates retinal iron status from systemic iron levels, thereby protecting the retina from the consequences of systemic iron overload. It is important to note that for a number of years, a similar notion persisted with respect to brain. However, recent studies have provided evidence that though the brain is separated from systemic circulation by the blood-brain barrier, iron accumulation in certain areas of the brain such as the basal ganglia occurs in patients with hemochromatosis [17–20]. Similarly, in the last few years there has been overwhelming evidence from animal models for the involvement of retina in hemochromatosis and other disorders of iron overload. We now know that retina expresses all the five genes involved in hemochromatosis [1–3, 21]. TfR2 and ferroportin are expressed throughout the retina [1, 21]. HFE, the most commonly mutated gene in hemochromatosis, is expressed predominantly in the basolateral membrane of RPE [1]. In our recent studies, we demonstrated that Hfe-null mice have retinal iron accumulation as they get older and have hyperproliferative RPE [4]. Hepcidin is expressed in RPE, Müller cells, and photoreceptor cells [2]. Also, hepcidin knockout mice have age-dependent iron accumulation in the retina with consequent retinal degeneration [22]. The expression of Hjv in retina is widespread; the protein is found in RPE, Muller cells, photoreceptor cells, and retinal ganglion cells [3]. In RPE, the expression of Hjv is polarized with exclusive localization in the apical membrane facing the subretinal space. Although mutations in HJV in humans result in a severe form of iron overload (juvenile hemochromatosis) and induce oxidative stress and cellular dysfunction in several systemic organs, there have been no reports of the consequences of these mutations on retinal health. Here we used Hjv-null mice to investigate the potential involvement of retina as a target organ for iron overload in juvenile hemochromatosis and assess the consequences in terms of retinal phenotype.
MATERIALS AND METHODS
Materials
Reagents were obtained from the following sources: RNA extraction reagent (TRIzol; Invitrogen-Gibco Corp., Grand Island, NY, U. S. A.), GeneAmp RT PCR kit (Applied Biosystems, Inc., Foster City, CA, U. S. A.), Taq polymerase kit (TaKaRa, Tokyo, Japan), Power Block (Biogenex, San Ramon, CA, U. S. A.), mouse monoclonal RPE65 antibody (Abcam, MA, U. S. A.), rabbit polyclonal anti-HJV antibody (GenScript Corporatioin, Piscataway Township, NJ, U. S. A.), goat anti-rabbit IgG coupled to Alexa Fluor 568 and goat anti-mouse IgG coupled to Alexa Fluor 488 (Molecular Probes, Eugene, OR, U. S. A.), and JB-4 solutions (Polysciences Inc., Warrington, PA, U. S. A.). DMEM/F12 medium (Invitrogen-Gibco) with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin was used for growing RPE cells. PI-PLC was obtained from Sigma-Aldrich (St. Louis, MO, U. S. A.). [3H]-Glutamate (specific radioactivity, 43 Ci/mmol) was from American Radiolabeled Chemicals, Inc. (St. Louis, MO, U. S. A.) and [3H]-thymidine (specific radioactivity, 74.8 Ci/mmole) was from PerkinElmer (Waltham, MA, U. S. A.).
Animals
Breeding pairs of Hjv+/− mice on 129/SvEvTac (129/S) background were kindly provided by Dr. Nancy Andrews, Duke University (Durham, NC, U. S. A.). Genotyping was done to identify wild type (Hjv+/+), heterozygous (Hjv+/−), and homozygous (Hjv−/−) mice in the litters. Age-matched wild type (Hjv+/+) and knockout (Hjv−/−) male mice were selected from the same litters for comparison studies. All experimental procedures involving these animals adhered to the “Principles of Laboratory Animal Care” (National Institutes of Health publication #85-23, revised in 1985) and were approved by the Institutional Committee for Animal Use in Research and Education.
Preparation of eye sections and morphometric analysis
Mice were killed by CO2 asphyxiation followed by cervical dislocation. For studies using frozen sections, eyes were enucleated and oriented in Tissue-Tech O.C.T. compound so that the 10-μm-thick sections included the full length of retina approximately along the horizontal meridian, passing through the ora serrata and the optic nerve in both the temporal and nasal hemispheres. After slow freezing, the cryosections were prepared and mounted on slides (Superfrost; Fisher Scientific, Pittsburgh, PA, U. S. A.). They were stained with hematoxylin and eosin and used for morphometric studies. Additional cryosections were used for immunohistochemical studies.
Hematoxylin-eosin-stained sections of retina from different ages of Hjv−/− mice along with age-matched Hjv+/+ mice were used for systematic morphometric analysis with a fluorescence microscope (Axioplan2 equipped with HRM camera and the Axiovision 4.6.3 program; Carl Zeiss Meditec, Dublin, CA, U. S. A.). Measurements included the thickness of the total retina, RPE, inner plexiform layer (IPL), inner nuclear layer (INL), outer plexiform layer (OPL), and the photoreceptor inner/outer segments. The number of cell bodies in the ganglion cell layer (GCL) was quantified by counting cells from the temporal ora serrata to the nasal ora serrata and expressing the data as number of cells per 100-μm length of retina. Three measurements were made on each side (temporal and nasal) of the optic nerve at 200- to 300-μm intervals resulting in six measurements obtained per eye. Two measurements on either side of the optic nerve were taken as central retina and the remaining 4 measurements from that eye were considered as peripheral retina. Both eyes were analyzed in each mouse.
JB-4 plastic sections and histological analysis
Eyes were enucleated and fixed for 1 h at room temperature (22 °C) in 2% (w/v) paraformaldehyde/0.2% glutaraldehyde in 0.1 M cacodylate buffer in sucrose and post-fixed for 1 h with 1% tannic acid in 0.1 M sodium cacodylate buffer. Tissues were dehydrated using a graded series of ethanol and infiltrated overnight in 1.25% benzoyl peroxide using the JB-4 embedding kit. The next day, the eyecups were oriented and embedded in plastic as recommended in the embedding kit. Sections, 3-μm thick, were cut and used for histological analysis after hematoxylin and eosin staining.
Immunofluorescence analysis
Cryosections of mouse eyes were fixed in 4% paraformaldehyde for 10 min, washed with 0.01 M phosphate-buffered saline (pH 7.4), and blocked with 1x blocking agent for 60 min. Sections were then incubated overnight at 4 °C with rabbit polyclonal anti-HJV antibody. Negative control sections were treated identically but in the absence of the primary antibody. Sections were rinsed and incubated for 1 h with goat anti-rabbit IgG coupled to Alexa Fluor 568 at a dilution of 1:1000. Coverslips were mounted after staining with Hoechst nuclear stain, and sections were examined with a wide-field epifluorescence microscope (Carl Zeiss Meditec, Oberkochen, Germany). The RPE cell layer in retinal sections was identified using an antibody specific for RPE65, a marker for RPE cells.
Establishment of primary RPE cell cultures from mouse eyes
Primary cultures of RPE were prepared as described previously [2–4]. Age-matched Hjv+/+ and Hjv−/− mice were obtained from the same litter originating from the mating of heterozygous mice. 3-week-old mice were then used to establish primary cultures of RPE. Purity of the cultures was verified as described previously by immunodetection of RPE65, a known marker for RPE cells [23].
ELISA for determination of ferritin levels
A mouse ferritin ELISA kit (Kamiya Biomedical Company, Seattle, WA, U. S. A.) was used for quantification of ferritin levels in Hjv+/+ and Hjv−/− mouse retinas and RPE cells. The absorbance of the final reaction mixture was measured at 450 nm. A calibration curve was used as instructed in the kit to determine the ferritin levels in these samples.
[3H]-Thymidine incorporation assay
Hjv+/+ and Hjv−/− primary RPE cells were plated at a density of 10,000 cells per well in 24-well plates, and grown for 24 h in serum-containing medium. Cells were then serum-starved for 48 h and then [3H]-thymidine (1 μCi per well) was added to the wells and incubated for 12 h. The cells were then washed twice with 5% trichloroacetic acid, the resultant precipitate was solubilized in 0.1M NaOH, and the radioactivity was measured using a scintillation counter.
Senescence assay
Senescence assay was performed in Hjv+/+ and Hjv−/− primary RPE cells using a commercially available senescence β-galactosidase staining kit (Cell Signaling Technology, Danvers, MA, U. S. A.). Development of blue color after incubation with the reagents provided in the kit identifies the senescent cells.
Reverse transcriptase-PCR
RNA was isolated from wild type and Hjv-null primary RPE cells and used for RT-PCR using GeneAmp RT PCR kit. PCR primers were designed based on the sequence information available in GenBank™ for mouse cDNAs. The following primers were used: mouse Hfe forward: 5′-GGCTTCTGGAGATATGGTTAT-3′; reverse: 5′-GACTCCACTGATGATTCCGATA-3′; mouse hepcidin forward: 5′-GCACCACCTATCTCCATCAACAGA-3′; reverse: 5′-GGTCAGGATGTGGCTCTAGGCTAT-3′; mouse Hjv forward: 5′-GGCTGAGGTGGACAATCTTC-3′; reverse: 5′-GAACAAAGAGGGCCGAAAG-3′; mouse TfR1 forward: 5′-GCCCAAGTATTCTCAGATATGAT-3′; reverse: 5′-TAGAAGTAGCACGGAAGTAGTCTC-3′; mouse TfR2 forward: 5′-GAGGATCCGGAAGTCTACTGTC-3′; reverse: 5′-TCGATGCACGCAAAGATGTTACTG-3′; mouse DMT1 forward: 5′-CACTATCATGGCCCTCACGTTT-3′; reverse: 5′-GCTGCAGGCCCGAAGTAACA-3′; mouse β2M forward: 5′-CCGAACATACTGAACTGCTAC-3′ reverse: 5′-CATACTGGCATGCTTAACTCT-3′; mouse xCT forward 5′-AAGTGGTTCAGACGATTATCAG-3′; reverse 5′-AAGAAACGTGGTAGAGGAATG-3′; mouse 4F2hc forward: 5′-CTCCCAGGAAGATTTTAAAGACCTTCT-3′; reverse 5′-TTCATTTTGGTGGCTACAATGTCAG-3. Mouse 18S primers were used as an internal control. PCR was performed using a commercially available Taq polymerase kit.
Assay of system xc− transport activity
The heterodimeric amino acid transporter xCT/4F2hc is responsible for the activity of the amino acid transport system known as xc−. It is a Na+-independent system that mediates the cellular entry of cystine in exchange for intracellular glutamate under physiological conditions. However, we routinely measure the activity of this transporter by cellular uptake of [3H]-glutamate under Na+-free conditions [24, 25]. Under these conditions, xc− mediates the cellular entry of [3H]-glutamate in exchange for intracellular unlabeled glutamate. Transport activity was measured using a Na+-free uptake buffer (25 mM Hepes/Tris, 140 mM N-methyl-D-glucamine chloride, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4, and 5 mM glucose, pH 7.5). L-[3H]-glutamic acid was used as the substrate for uptake experiments. Uptake was initiated by the addition of 250 μl uptake buffer containing 2.5 μM glutamate spiked with 2 μCi/ml [3H]-glutamate. Cells were incubated for 15 min at 37 °C, after which time, the buffer was removed, and the cells were washed twice with ice-cold uptake buffer. The cells were then solubilized with 0.5 ml 1% SDS/0.2 N NaOH, and radioactivity was determined by liquid scintillation spectrometry. Protein was measured using the Bio-Rad protein assay reagent. Non-carrier-mediated uptake (i.e, diffusional component) was determined by measuring the uptake of [3H]-glutamate under identical conditions but in the presence of excess unlabeled glutamate (5 mM). The diffusional component was less than 10% of total uptake. The transport activity of xc− was calculated by subtracting the diffusional component from total uptake.
Western blot
Plasma membrane proteins from Hjv+/+ and Hjv−/− RPE cells were prepared using Pierce Cell Surface Protein Isolation Kit (Thermo Fisher Scientific, Waltham, MA, U. S. A.). Protein lysates were subjected to 10% SDS-PAGE. The proteins were then transferred on to a polyvinylidene fluoride membrane and probed with specific antibodies. The positive bands were detected with appropriate secondary antibodies coupled to horseradish peroxidase. The signals were developed using an enhanced-chemiluminescence detection kit (Thermo Fisher Scientific, Waltham, MA, U. S. A.).
Cleavage of cell-surface HJV by phosphatidylinositol-specific phospholipase C (PI-PLC)
ARPE19 cells, a human RPE cell line, were cultured in Dulbecco’s modified Eagle’s medium-F12 medium, supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. For PI-PLC treatment, the cells were seeded in 24-well plates with coverslips in Dulbecco’s modified Eagle’s medium-F12 and incubated in the presence or absence of PI-PLC. (Sigma, St. Louis, MO, U. S. A.) at a concentration of 1 unit/ml for 2 h at 37 °C in 5% CO2 incubator. Non-permeabilised cells were then stained for immunodetection of HJV using rabbit anti-HJV antibody (GenScript Corporation, Piscataway, NJ, USA) at 1:100 dilution.
Generation of hepcidin-specific promoter-reporter constructs
The human hepcidin-specific promoter-EGFP construct was generated by first subcloning the 2-kb hepcidin promoter (obtained by PCR using human genomic DNA as the template) into the pGEM-T Easy vector. The promoter insert was then released by digestion with HindIII/XhoI, and the insert was then subcloned into pUIIR3-EGFP vector. The primers used for PCR were 5′-ATACTCGAGACTCTCACCCAGGCTGGG-3′ (sense) and 5′-AAGCTTCATCGTGCCGTCTGTCTGGCT-3′ (antisense).
Treatment of retinal cells with exogenous BMP6
Cells were serum-starved for 6 h by culturing in the presence of 1% fetal bovine serum and then treated with 100 ng/ml of recombinant human BMP6 (R&D Systems, Minneapolis, MN, U. S. A.) in normal medium for 12 h.
RESULTS
Age-dependent morphological changes in the retina in Hjv−/− mice
To analyze the function of Hjv in retina, Hjv−/− mouse retinas of different ages were used to compare with age-matched Hjv+/+ retinas stained with hematoxylin and eosin. We did not find any morphological differences in the retina between the two genotypes in younger mice. In JB-4 plastic sections, we found drastic morphological changes in the retina of Hjv−/− mice at ≥ 18 months of age compared to the age-matched Hjv+/+ controls, both in the central and peripheral retina (Fig. 1A). We performed a systematic morphometric analysis of the retinas from frozen sections. The morphological changes evident in Hjv−/− mice include significant loss of ganglion cells and increase in RPE cell thickness, specifically in the central retina (Fig. 1B). The loss of ganglion cells in 18-month-old Hjv−/− mouse retinas was evident also in JB-4 plastic sections (Fig. 1A).
Figure 1. Deletion of Hjv in mice causes significant morphological changes in retina.
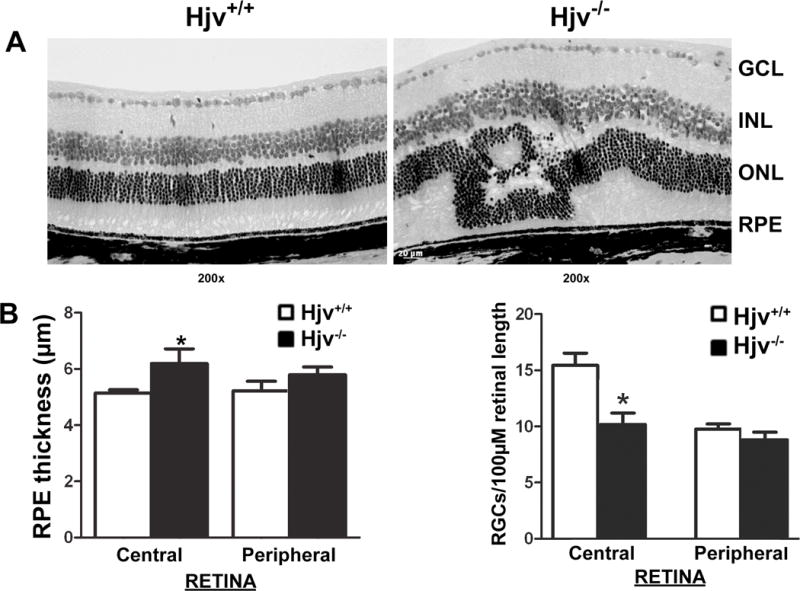
(A) Representative hematoxylin and eosin-stained JB-4 plastic sections of Hjv+/+ and Hjv−/− mouse retinas of 18-month-old mice. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium. (B) Hematoxylin and eosin-stained retinal frozen sections from 18-month-old Hjv+/+ and Hjv−/− mice were used for morphometric analysis. Bar diagram represents RPE thickness and number of cell bodies in GCL per 100-μm length of retina calculated using measurements from central retina and peripheral retina of Hjv+/+ and Hjv−/− mice. Data are the means ± SE of measurements from retinas of six Hjv+/+ and six Hjv−/− mice (12 eyes each). *p < 0.05.
Our previous studies and reports from other labs have shown that iron accumulation in the retina is associated with RPE hypertrophy and hyperplasia [4, 9, 22]. These studies used the ceruloplasmin/hephaestin double-knockout mice as well as Hfe- and hepcidin-knockout mice as models of retinal iron overload. Since loss-of-function mutations in HJV also lead to excessive iron accumulation in various tissues in humans, we examined the RPE cell layer in the retinas of Hjv−/− mice (18-month-old). Age-matched retinas from Hjv+/+ mice were used for comparison. Hjv knockout was confirmed by the presence and absence of Hjv immunostaining in wild type and Hjv−/− retinas respectively (data not shown). There were focal areas of RPE hyperplasia in Hjv−/− mouse retina as evidenced by the presence of multinucleated regions in the RPE cell layer when examined after immunostaining with RPE65 (marker for RPE) and the nuclear stain Hoechst (Fig. 2A). There were many regions with hyperplasia in RPE of 18-month-old Hjv−/− mouse retinas as shown in Fig. 2B.
Figure 2. Hjv-null mice display focal areas of RPE hyperplasia.
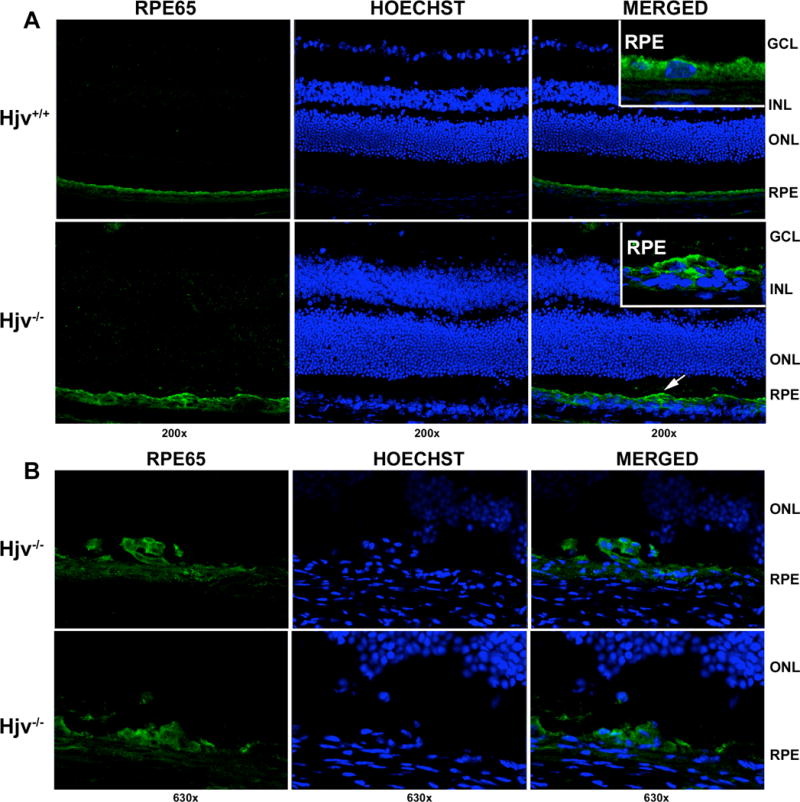
(A) Immunostaining of 18-month-old Hjv+/+ and Hjv−/− mouse retinas with RPE65 antibody showing focal areas of RPE hyperplasia in the knockout mice. Left most panel shows staining for RPE65 (green), middle panel represents Hoechst nuclear stain, and right most panel is a merged image. Retinal sections treated similarly but in the absence of the primary antibody were used as a negative control (data not shown). Inset in the right most panel represents a higher magnification of the RPE cell layer. (B) Additional representative regions of RPE hyperplasia in Hjv−/− mice retinas.
Iron accumulation in Hjv-null mouse retina
HJV is an important iron-regulatory protein; loss-of-function mutations in this protein result in iron overload at much younger age and consequently lead to juvenile hemochromatosis. The levels of cytosolic iron-storage protein ferritin are regulated by intracellular iron through the iron-regulatory proteins IRP1 and IRP2 (iron-regulatory proteins 1 and 2). Increase in intracellular iron leads to increased ferritin mRNA translation, resulting in increased ferritin protein levels [26, 27]. Ferritin is a 24-subunit protein complex composed of heavy (H) and light (L) chains. To determine whether the retinal morphological changes observed in Hjv−/− mice are accompanied by increased iron accumulation, ferritin levels in retinas and primary RPE cells from Hjv−/− mice and age-matched Hjv+/+ mice were measured with using ELISA (Fig. 3A). The ferritin levels were significantly higher in Hjv−/− mice compared to the controls. We also examined the changes in other proteins involved in iron homeostasis in 3-month and 8-month-old Hjv−/− mouse retinas. Hfe and hepcidin expression were lower in Hjv−/− mouse retinas (Fig. 3B), indicating additional mechanisms of iron accumulation in Hjv−/− mouse retinas. Also, the expression of transferrin receptors TfR1 and TfR2, as well as that of the iron-importing transporter DMT1 decreased in Hjv−/− mouse retina (Fig. 3B). Increase in intracellular iron is known to decrease TfRs and DMT1 expression. Thus, changes in the expression levels of TfR1, TfR2, and DMT1 also provide evidence for increased iron accumulation in Hjv−/− mouse retinas.
Figure 3. Iron levels are increased in Hjv−/− mouse retina.
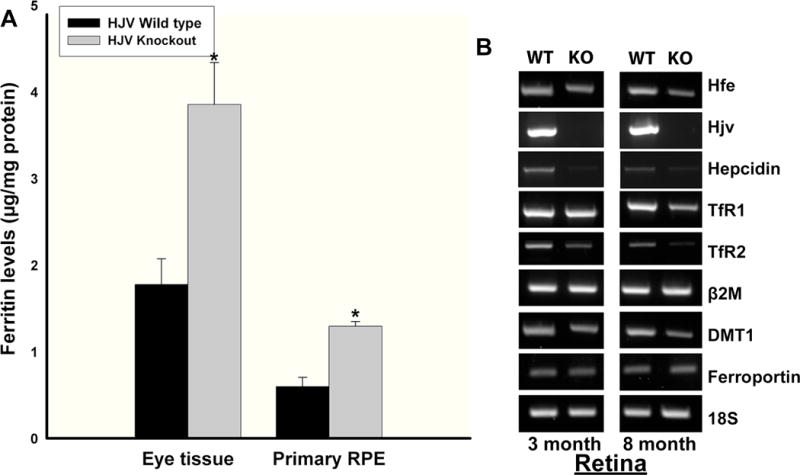
(A) Ferritin levels were determined by ELISA in tissue lysates from 18-month-old Hjv+/+ and age-matched Hjv−/− mouse retinal tissues (n = 5) and primary RPE cell preparations (n = 4). *p < 0.001. (B) RT-PCR analysis of iron-regulatory genes in 3-month-old and 8-month-old Hjv+/+ (WT) and Hjv−/− (KO) mouse retinas.
Hyperproliferation of Hjv−/− primary RPE cells
Hjv-null retinas in situ showed evidence of RPE hyperplasia. Therefore, we isolated primary RPE cells from Hjv+/+ and Hjv−/− mouse retinas and examined their growth and proliferation pattern. Using β -galactosidase senescence assay, we found that Hjv−/− RPE cells senesce at a much slower rate than Hjv+/+ RPE cells grown under the identical conditions and passage number (Fig. 4A). We then used thymidine incorporation assay to compare the proliferation of Hjv+/+ and Hjv−/− RPE cells. Thymidine incorporation is a measure of DNA synthesis and hence cell proliferation. The proliferation rate of Hjv−/− RPE cells was significantly greater (~3-fold) than that of wild type RPE cells (Fig. 4B).
Figure 4. RPE cells from Hjv−/− mice exhibit a decreased senescence and hyperproliferative phenotype.
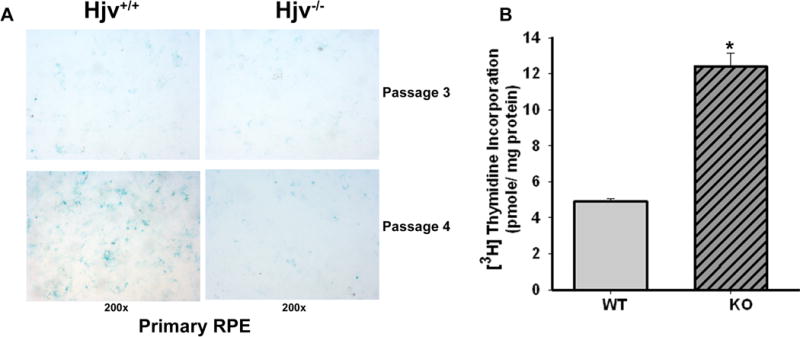
(A) Senescence assay in Hjv−/− primary RPE cells compared with Hjv+/+ primary RPE cells. Blue color indicates senescent cells. (B) [3H]-Thymidine incorporation assay with Hjv+/+ and Hjv−/− RPE cells. *p < 0.001 (n = 12).
Up-regulation of xCT mRNA and protein in Hjv−/− primary RPE cells
Several recent studies have implicated the cystine/glutamate exchanger in cell cycle progression including our previous article on Hfe-null mouse retina [4, 28–31]. Slc7a11 codes for the ‘transporter proper’ subunit xCT in the heterodimeric amino acid transport system xc− that mediates the cellular uptake of cystine coupled with the efflux of glutamate. The other subunit is 4F2hc that functions as a chaperone for xCT for optimal recruitment into the plasma membrane. Since cellular cysteine is the rate-limiting factor for glutathione synthesis, xc− is one of the key determinants of cellular glutathione status. RT-PCR was done for xCT and 4F2hc with RNA from Hjv+/+ and Hjv−/− RPE cells. RT-PCR showed that xCT mRNA expression was higher in Hjv−/− RPE cells whereas the 4F2hc mRNA levels remained unchanged (Fig. 5A). We also monitored the levels of xCT protein in the plasma membrane of Hjv+/+ and Hjv−/− RPE cells by western blot. The protein levels in these cells changed in parallel with their mRNA levels (Fig. 5B).
Figure 5. The cystine/glutamate exchanger xCT is upregulated in Hjv−/− RPE cells.
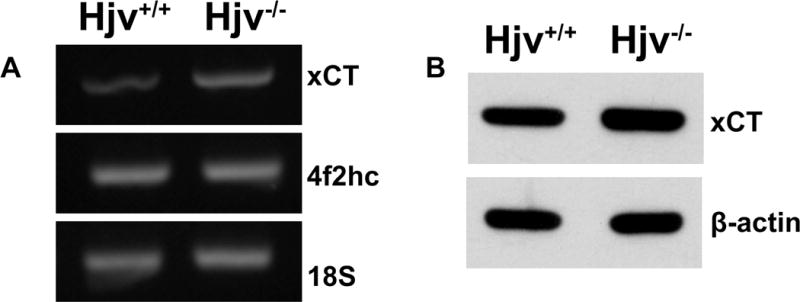
(A) RT–PCR analysis of mRNA transcripts specific for xCT and 4F2hc in Hjv+/+ and Hjv−/− RPE cells. 18S was used as an internal control. (B) Western blot for xCT protein in Hjv+/+ and Hjv−/− RPE cells. β-actin was used as a loading control.
Functional activity of system xc− in wild type and Hjv−/− RPE cells
To obtain functional evidence for the increase in the expression of Slc7a11 in Hjv−/− RPE cells, we compared the activity of system xc− between wild type and Hjv−/− RPE cells. Under the experimental conditions employed during the measurement, xc− mediates the cellular uptake of [3H]-glutamate in exchange for intracellular unlabeled glutamate. The Na+-independent glutamate uptake, which is a measure of xc− functional activity, was ~3-fold higher in Hjv−/− RPE cells than in Hjv+/+ RPE cells (Fig. 6A). To confirm that the observed glutamate uptake occurred via system xc−, we performed substrate selectivity studies (Fig. 6B). Uptake of [3H]-glutamate in Hjv+/+ and Hjv−/− RPE cells was inhibited by unlabeled glutamate and cystine, but not by valine and aspartate. This mirrors the substrate specificity of system xc−, strongly suggesting that the observed uptake of glutamate in RPE cells occurred principally via system xc−.
Figure 6. The activity of the cystine/glutamate exchanger xc− is increased in Hjv−/− RPE cells.
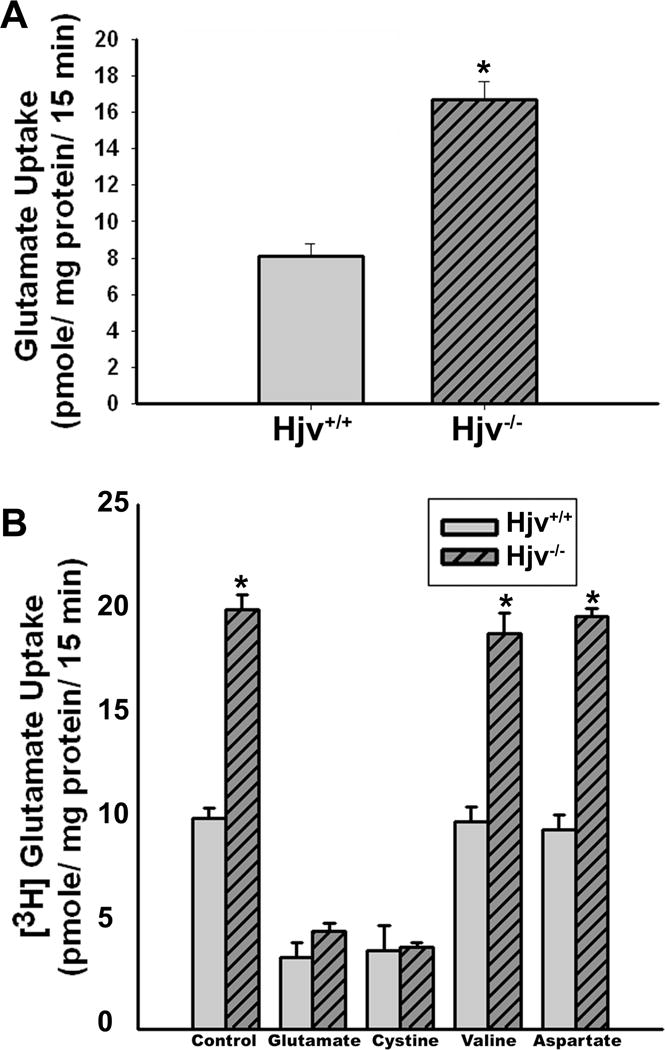
(A) Uptake of glutamate in Hjv+/+ and Hjv−/− RPE cells. Uptake of [3H]-glutamate (2.5 μM) was measured for 15 min at 37 °C in the absence of Na+ as a readout of xc− transport activity. Data (means ± S. E.) represent transport activity specific for system xc−. *p < 0.001 (n = 8). (B) Substrate selectivity of glutamate uptake in Hjv+/+ and Hjv−/− RPE cells. Uptake of [3H]-glutamate (2.5 μM) was measured in Hjv+/+ and Hjv−/− RPE cells in the absence of Na+ for 15 min at 37 °C in the absence or presence of unlabelled amino acids glutamate, cystine, valine and aspartate, each at a concentration of 1 mM. *p < 0.001 (n = 6). Again, the data (means ± S. E.) represent only the transport activity specific for xc−.
BMP signaling in Hjv-null retinal cells
BMP6 has been shown to be a co-ligand along with HJV for BMP receptor-mediated signaling involved in the induction of hepcidin expression and consequently in the modulation of iron homeostasis [32–35]. We treated Hjv+/+ and Hjv−/− primary RPE cells with BMP6 and monitored the expression of hepcidin by RT-PCR. We found that Hjv+/+ RPE cells had increased hepcidin mRNA levels after BMP6 treatment; in contrast, Hjv−/− primary RPE cells did not have any change in hepcidin expression (Fig. 7A), confirming the need for Hjv in BMP signaling. HJV is a GPI-anchored protein and hence treatment with PI-PLC, an enzyme that specifically hydrolyzes the anchoring lipid, releases the GPI-anchored HJV from the membrane [36]. We confirmed the anchoring of HJV to GPI in ARPE19 cells. Treatment of non-permeabilized ARPE19 cells with PI-PLC led to a significant reduction in membrane-associated HJV (Fig. 7B). We then transfected ARPE19 cells with a hepcidin-specific promoter-EGFP construct to assess the role of BMP6 on the promoter activity. Addition of BMP6 to these transfected cells induced the expression of GFP (Fig. 7C). However, when these cells were treated with PI-PLC to remove the membrane-anchored HJV, BMP6 failed to activate the hepcidin promoter as evidenced from the lack of expression of the reporter GFP (Fig. 7C). The experiments were repeated with three independent transfections, and four different regions were imaged for each experiment. The number of GFP-positive cells per field of view was counted and the data as a quantitative representation of the reporter activity in control ARPE19 cells (i.e., in the absence of PI-PLC) with and without treatment with BMP6 are given in Fig. 7D. The results from hepcidin-specific promoter activity were corroborated by RT-PCR for hepcidin mRNA levels (data not shown).
Figure 7. Regulation of hepcidin expression by BMP6 in RPE cells requires Hjv.
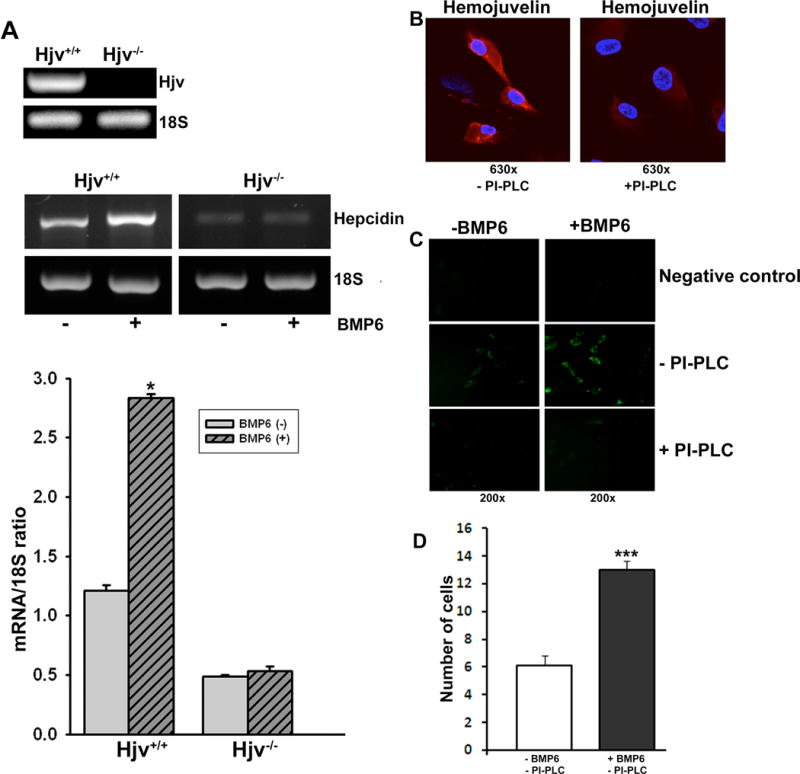
(A) RT-PCR analysis of hepcidin expression in the absence or presence of BMP6 in Hjv+/+ and Hjv−/− RPE cells. Bottom bar diagram is a quantitative representation of hepcidin expression after normalizing with 18S internal control. *p < 0.001 (B) ARPE19 cells, a human RPE cell line, were treated with (right panel) or without (left panel) phosphatidylinositol-specific phospholipase C (PI-PLC) and then immunostained using an anti-HJV antibody without permeabilization. Experiments were repeated three times with consistent results. No staining was detected when HJV antibody was not added in the negative control (data not shown). (C) Effects of BMP6 in the presence or absence of membrane-associated HJV on hepcidin promoter activity using EGFP reporter assay in ARPE19 cells. HJV was removed from the cell surface of ARPE19 cells by treatment with PI-PLC. (D) Quantitative representation of the number of GFP-positive cells per field of view in control cells and in BMP6-treated cells, both in the absence of PI-PLC (three independent transfections with the reporter construct and examination of four different sections per experiment). There were no GFP-positive cells, with or without BMP6 treatment, when PI-PLC was used.
DISCUSSION
Retinas in patients with aceruloplasminemia [8] as well as in ceruloplasmin/hephaestin double-knockout mice [9, 10] have marked morphological changes demonstrating that excessive iron accumulation is detrimental to the retina. Recent studies with mouse models have also brought to light the retinal involvement in hemochromatosis, a highly prevalent autosomal recessive genetic disorder in humans, which is associated with iron overload in circulation as well as in a variety of organs [4, 22]. To date, there have been only two studies, reported almost four decades ago, showing evidence of excessive iron accumulation in the retina in patients with hemochromatosis [15, 16]. We reported recently that Hfe-null mice, a widely used animal model of hemochromatosis, have increased iron accumulation with characteristic hypertrophic and hyperproliferative RPE [4]. Another group of investigators have also reported on the age-dependent iron accumulation and retinal degeneration in hepcidin-null mice, another model of hemochromatosis [22]. Since all five genes associated with hemochromatosis are expressed in retina, and Hfe- and hepcidin-null mice have retinal damage, it seems reasonable that the disease involves retina as a target organ for excessive iron accumulation as has been demonstrated in patients with hemochromatosis many years ago [15, 16]. Here we used a different mouse model of hemochromatosis, namely the Hjv-null mouse, to investigate the relevance of the disease to retinal iron homeostasis. To date, this represents the third mouse model to be used to study the retinal involvement in hemochromatosis.
In the present study, we show for the first time that there are marked morphological changes in the retina of 18-month and older Hjv−/− mice. It is important to note that the retinal changes seen in 18-month-old Hjv−/− mice are not evident in younger mice. Interestingly, Hfe−/− mice also had iron accumulation and retinal disruption at ≥ 18 months of age. Based on these data, we speculated that human patients with hemochromatosis resulting from loss-of-function mutations in HFE would develop iron overload in the retina at later stages of their life. Patients with mutations in HJV or hepcidin develop severe iron overload at much younger ages (juvenile hemochromatosis). Though the systemic iron overload in Hjv−/− mice occurs by 10-weeks of age, we find retinal iron accumulation and morphological changes only in older mice. Interestingly, hepcidin knockout mice have also been recently reported to develop iron accumulation and retinal degeneration when they are 18 months or older [22]. It is known that HJV regulates iron homeostasis through hepcidin. Hence it is not surprising that just like hepcidin-null mice, Hjv−/− mice also have retinal degeneration only when they get older. Though juvenile hemochromatosis patients with mutations in HJV or hepcidin develop other systemic disorders at a much younger age, they might be susceptible to retinal problems only at later stages.
The morphological changes in Hjv−/− mice include increase in RPE thickness and decreased number of cells in the ganglion cell layer, both of which are predominant especially in the central retina. There were marked disruptions of the inner and outer nuclear layers. This morphological damage in the old Hjv−/− mouse retina was due to increased iron accumulation as evident from ferritin levels. Also, we found that Hjv−/− mouse retina had decreased expression of the iron-regulatory proteins TfR1 and divalent metal transporter (DMT1), which are known to be down-regulated under conditions of increased intracellular iron levels. Whether these morphological changes accompanied by increased iron in the retina are associated with detectable changes in retinal function needs to be investigated. We also provide evidence of hyperplasia in the RPE cell layer. Iron accumulation in the retina observed in the ceruloplasmin/hephaestin double-knockout mouse and hepcidin knockout mice are associated with hypertrophy and hyperplasia of RPE [9, 22]. We reported a similar phenomenon in the Hfe-null hemochromatosis mouse [4]. In the present study, we find the same phenotype in Hjv-null mice. These findings demonstrate that excessive iron accumulation underlies the cellular and growth pattern changes seen in RPE. Similar to Hfe-null mice, the hyperplasia phenotype of RPE observed in vivo in intact retina of Hjv−/− mouse is also seen with primary RPE cell cultures established from Hjv−/− mouse retinas. Many studies including ours have previously shown that Slc7a11 plays a critical role in cell proliferation [4, 37, 38]. The expression of this transporter is upregulated in Hjv−/− RPE cells, suggesting that cystine uptake might play an important role in the hyperproliferative phenotype of Hjv−/− cells. The increased expression of Slc7a11 that codes for the ‘transporter proper’ subunit xCT in the cystine/glutamate exchange system xc− is demonstrable at the mRNA level, protein level, and functional level in Hjv−/− RPE cells.
The present studies also show that loss-of-function of Hjv affects not only the RPE cell layer but also other cellular layers of the retina. The changes observed in the retina in the mouse model of juvenile hemochromatosis may have relevance to age-related macular degeneration (AMD). AMD is a major cause of gradual bilateral loss of central vision in elderly people [29]. There is overwhelming evidence in support of a genetic component in the etiology of AMD [30, 31]. Several genes have been identified either as the cause or contributors to AMD-associated retinal pathology, but still the etiology of the disease in most cases remains unknown. Irrespective of the genetic factors, patients with AMD show evidence of excessive iron accumulation in retina [6, 7]. Iron is a pro-oxidant and increased concentration of free iron in cells can cause oxidative stress. Fenton reaction is the primary mechanism underlying the iron-dependent oxidative stress in which Fe2+ mediates the conversion of hydrogen peroxide into highly reactive hydroxyl radical. Since AMD occurs mostly in older patients and also since oxidation-induced cellular damage increases with age, the possible contribution of oxidative stress to the etiology of AMD has been speculated previously [31]. Published reports on patients with hemochromatosis [15, 16], and recent findings from studies with hemochromatosis mouse models (Hfe and hepcidin knockout mice) [4, 22] along with our present study on Hjv−/− mouse model clearly show that iron does accumulate in the retina in this disease. This raises the possibility that the genetic disease hemochromatosis may promote the progression and pathological features of AMD in humans. The increasing evidence for the presence of excessive iron in AMD, emerging reports on the retinal involvement in hemochromatosis, and the RPE hyperplasia as a common feature found in hemochromatosis mouse models and AMD patients strongly suggest that further studies to investigate the potential association between hemochromatosis and AMD are highly warranted.
Hepcidin is an important regulator of systemic iron homeostasis, and hepcidin deficiency induces severe iron overload [22]. HJV is a member of the repulsive guidance molecule (RGM) family, which also includes the bone morphogenetic protein (BMP) co-receptors RGMa and DRAGON (RGMb). Mutations in HJV (HFE2 or RGMc) cause severe iron overload and correlate with low hepcidin levels, suggesting that HJV positively regulates hepcidin expression [32]. Membrane-anchored HJV has been shown to up-regulate the expression of hepcidin through the bone morphogenic protein (BMP) signaling pathway by acting as a BMP co-receptor [32]. HJV is a GPI-anchored protein that can be cleaved by the furin family of proprotein convertases, which releases a soluble form of HJV that suppresses BMP signaling and hepcidin expression by acting as a bait that competes with membrane-bound HJV for BMP ligands [33]. In the present study, we found that HJV expressed in RPE cells is indeed a GPI-anchored protein as evident from the findings that treatment of the cells with PI-PLC resulted in the loss of membrane-bound HJV. Among the different BMP ligands, BMP6 has been shown to play a key role as a ligand for hemojuvelin and as an endogenous regulator of hepcidin expression and iron homeostasis in vivo[34, 35]. In the present study, we have shown for the first time that in RPE cells, addition of BMP6 activates hepcidin expression and hepcidin-promoter activity only in the presence of Hjv, and more specifically, only in the presence of membrane-bound Hjv. This confirms that retina requires membrane-anchored HJV to act as a co-receptor of BMP6 for the regulation of hepcidin expression within the retina and consequently for the regulation of retinal iron homeostasis.
In summary, deletion of the gene coding for the iron-regulatory protein Hjv in mice leads to excessive iron accumulation with consequent age-related morphological disruption in the retina. Deletion of Hjv in RPE cells leads to a hyperproliferative phenotype both in vivo and in vitro. Upregulation of the cystine/glutamate exchanger is a likely contributor to the enhanced proliferation of Hjv−/− RPE cells. Similar to the published reports on Hfe-null and hepcidin-null mouse models of hemochromatosis, the present studies suggest that mutations, dysregulation or dysfunction of HJV in humans may also have a role in the etiology and/or progression of AMD. In addition, the present studies also show that HJV is expressed as a GPI-anchored protein in retinal cells and that the membrane-bound form of this protein is obligatory for BMP6-mediated hepcidin expression in the retina.
Acknowledgments
This work was supported by the National Institutes Health grant EY019672.
Abbreviations used
- HJV
human hemojuvelin
- Hjv
mouse hemojuvelin
- HFE
human HLA-like protein involved in iron (FE) homeostasis
- Hfe
mouse HLA-like protein involved in iron (FE) homeostasis
- DMT1
divalent metal transporter 1
- β2M
β2-microglobulin
- RPE
retinal pigment epithelium
- ONL
outer nuclear layer
- INL
inner nuclear layer
- GCL
ganglion cell layer
- TfR
transferrin receptor
- IRP
iron-regulatory protein
- BMP
bone morphogenic protein
- AMD
age-related macular degeneration
- PI-PLC
phosphatidylinositol-specific phospholipase C
- xc−
cystine/glutamate exchanger
- Slc
solute-linked carrier
- RGM
repulsive guidance molecule
- GPI
glycosylphosphatidylinositol
References
- 1.Martin PM, Gnana-Prakasam JP, Roon P, Smith RG, Smith SB, Ganapathy V. Expression and polarized localization of the hemochromatosis gene product HFE in the retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2006;47:4238–4244. doi: 10.1167/iovs.06-0026. [DOI] [PubMed] [Google Scholar]
- 2.Gnana-Prakasam JP, Martin PM, Mysona BA, Roon P, Smith SB, Ganapathy V. Hepcidin expression in mouse retina and its regulation via lipopolysaccharide/Toll-like receptor-4 pathway independent of Hfe. Biochem J. 2008;411:79–88. doi: 10.1042/BJ20071377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gnana-Prakasam JP, Martin PM, Zhang M, Atherton SS, Smith SB, Ganapathy V. Expression of the iron-regulatory protein hemojuvelin in retina and its regulation during cytomegalovirus infection. Biochem J. 2009;419:533–543. doi: 10.1042/BJ20082240. [DOI] [PubMed] [Google Scholar]
- 4.Gnana-Prakasam JP, Thangaraju M, Liu K, Martin PM, Smith SB, Ganapathy V. Absence of iron-regulatory protein HFE results in hyper-proliferation of retinal pigment epithelium mediated by induction of cystine/glutamate transporter. Biochem J. 2009;424:243–52. doi: 10.1042/BJ20090424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gnana-Prakasam JP, Ananth S, Prasad PD, Zhang M, Atherton SS, Martin PM, Smith SB, Ganapathy V. Expression and iron-dependent regulation of succinate receptor GPR91 in retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2011;52:3751–3758. doi: 10.1167/iovs.10-6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hahn P, Milam AH, Dunaief JL. Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch’s membrane. Arch Ophthalmol. 2003;121:1099–1105. doi: 10.1001/archopht.121.8.1099. [DOI] [PubMed] [Google Scholar]
- 7.Dentchev T, Hahn P, Dunaief JL. Strong labeling for iron and the iron-handling proteins ferritin and ferroportin in the photoreceptor layer in age-related macular degeneration. Arch Ophthalmol. 2005;123:1745–1746. doi: 10.1001/archopht.123.12.1745. [DOI] [PubMed] [Google Scholar]
- 8.Dunaief JL, Richa C, Franks EP, Schultze RL, Aleman TS, Schenck JF, Zimmerman EA, Brooks DG. Macular degeneration in a patient with aceruloplasminemia, a disease associated with retinal iron overload. Ophthalmology. 2005;112:1062–1065. doi: 10.1016/j.ophtha.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 9.Hahn P, Qian Y, Dentchev T, Chen L, Beard J, Harris ZL, Dunaief JL. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc Natl Acad Sci USA. 2004;101:13850–13855. doi: 10.1073/pnas.0405146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hadziahmetovic M, Denntchev T, Song Y, Haddad N, He X, Hahn P, Pratico D, Wen R, Harris ZL, Lambris JD, et al. Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD. Invest Ophthalmol Vis Sci. 2008;49:2728–2736. doi: 10.1167/iovs.07-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wrighting DM, Andrews NC. Iron homeostasis and erythropoiesis. Curr Top Dev Biol. 2008;82:141–167. doi: 10.1016/S0070-2153(07)00006-3. [DOI] [PubMed] [Google Scholar]
- 12.Fleming RE, Britton RS, Waheed A, Sly WS, Bacon BR. Pathophysiology of hereditary hemochromatosis. Semin Liver Dis. 2005;25:411–419. doi: 10.1055/s-2005-923313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beutler E. Hemochromatosis: genetics and pathophysiology. Annu Rev Med. 2006;57:331–347. doi: 10.1146/annurev.med.57.121304.131310. [DOI] [PubMed] [Google Scholar]
- 14.Wallace DF, Subramaniam VN. Non-HFE hemochromatosis. World J Gastroenterol. 2007;13:4690–4698. doi: 10.3748/wjg.v13.i35.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies G, Dymock I, Harry J, Williams R. Deposition of melanin and iron in ocular structures in haemochromatosis. Br J Ophthalmol. 1972;56:338–342. doi: 10.1136/bjo.56.4.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roth AM, Foos RY. Ocular pathologic changes in primary hemochromatosis. Arch Ophthalmol. 1972;87:507–514. doi: 10.1001/archopht.1972.01000020509003. [DOI] [PubMed] [Google Scholar]
- 17.Miyasaki K, Murao S, Koizumi N. Hemochromatosis associated with brain lesions: a disorder of trace-metal binding proteins and/or polymers. J Neuropathol Exp Neurol. 1977;36:964–976. doi: 10.1097/00005072-197711000-00008. [DOI] [PubMed] [Google Scholar]
- 18.Nielsen JE, Jensen LN, Krabbe K. Hereditary haemochromatosis: a case of iron accumulation in the basal ganglia associated with a Parkinsonian syndrome. J Neurol Neurosurg Psychiatry. 1995;59:318–321. doi: 10.1136/jnnp.59.3.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berg D, Hoggenmuller U, Hofmann E, Fischer R, Kraus M, Scheurlen M, Becker G. The basal ganglia in haemochromatosis. Neuroradiology. 2000;42:9–13. doi: 10.1007/s002340050002. [DOI] [PubMed] [Google Scholar]
- 20.Costello DJ, Walsh SL, Harrington HJ, Walsh CH. Concurrent hereditary haemochromatosis and idiopathic Parkinson’s disease: a case report series. J Neurol Neurosurg Psychiatry. 2004;75:631–633. doi: 10.1136/jnnp.2003.027441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hahn P, Dentchev T, Qian Y, Rouault T, Harris ZL, Dunaief JL. Immunolocalization and regulation of iron handling proteins ferritin and ferroportin in the retina. Mol Vis. 2004;10:598–607. [PubMed] [Google Scholar]
- 22.Hadziahmetovic M, Song Y, Ponnuru P, Iacovelli J, Hunter A, Haddad N, Beard J, Connor JR, Vaulont S, Dunaief JL. Age-dependent retinal iron accumulation and degeneration in hepcidin knockout mice. Invest Ophthalmol Vis Sci. 2011;52:109–118. doi: 10.1167/iovs.10-6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin PM, Ananth S, Cresci GA, Roon P, Smith SB, Ganapathy V. Expression and localization of GPR109A (PUMA-G/HM74A) mRNA and protein in mammalian retinal pigment epithelium. Mol Vis. 2009;15:362–372. [PMC free article] [PubMed] [Google Scholar]
- 24.Bridges CD, Kekuda R, Wang H, Prasad PD, Mehta P, Huang W, Smith SB, Ganapathy V. Structure, function, and regulation of human cystine/glutamate transporter in retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2001;42:47–54. [PubMed] [Google Scholar]
- 25.Bridges CC, Hu H, Miyauchi S, Siddaramappa UN, Ganapathy ME, Iganatowicz L, Maddox DM, Smith SB, Ganapathy V. Induction of cystine/glutamate transporter xc− by human immunodeficiency virus type 1 transactivator protein Tat in retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2004;45:2906–2914. doi: 10.1167/iovs.03-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomson AM, Rogers JT, Leedman PJ. Iron-regulatory proteins, iron-responsive elements and ferritin mRNA translation. Int J Biochem Cell Biol. 1999;31:1139–1152. doi: 10.1016/s1357-2725(99)00080-1. [DOI] [PubMed] [Google Scholar]
- 27.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 28.Reddy NM, Kleeberger SR, Bream JH, Fallon PG, Kensler TW, Yamamoto M, Reddy SP. Genetic disruption of the Nrf2 compromises cell-cycle progression by impairing GSH-induced redox signaling. Oncogene. 2008;27:5821–5832. doi: 10.1038/onc.2008.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bird AC. Towards an understanding of age-related macular disease. Eye. 2003;17:457–466. doi: 10.1038/sj.eye.6700562. [DOI] [PubMed] [Google Scholar]
- 30.Fine SL, Berger JW, Maguire MG, Ho AC. Age-related macular degeneration. N Engl J Med. 2000;342:483–492. doi: 10.1056/NEJM200002173420707. [DOI] [PubMed] [Google Scholar]
- 31.Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003;48:257–293. doi: 10.1016/s0039-6257(03)00030-4. [DOI] [PubMed] [Google Scholar]
- 32.Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, Campagna JA, Chung RT, Schneyer AL, Woolf CJ, Andrews NC, Lin HY. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38:531–539. doi: 10.1038/ng1777. [DOI] [PubMed] [Google Scholar]
- 33.Nili M, Shinde U, Rotwein P. Soluble repulsive guidance molecule c/hemojuvelin is a broad spectrum bone morphogenetic protein (BMP) antagonist and inhibits both BMP2- and BMP6-mediated signaling and gene expression. J Biol Chem. 2010;285:24783–24792. doi: 10.1074/jbc.M110.130286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andriopoulos B, Jr, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, Knutson MD, Pietrangelo A, Vukicevic S, Lin HY, Babitt JL. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009;41:482–487. doi: 10.1038/ng.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hadziahmetovic M, Song Y, Wolkow N, Iacovelli J, Kautz L, Roth MP, Dunaief JL. Bmp6 regulates retinal iron homeostasis and has altered expression in age-related macular degeneration. Am J Pathol. 2011;179:335–348. doi: 10.1016/j.ajpath.2011.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang AS, West AP, Jr, Wyman AE, Bjorkman PJ, Enns CA. Interaction of hemojuvelin with neogenin results in iron accumulation in human embryonic kidney 293 cells. J Biol Chem. 2005;280:33885–33894. doi: 10.1074/jbc.M506207200. [DOI] [PubMed] [Google Scholar]
- 37.Lo M, Wang YZ, Gout PW. The xc− cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol. 2008;215:593–602. doi: 10.1002/jcp.21366. [DOI] [PubMed] [Google Scholar]
- 38.Lastro M, Kourtidis A, Farley K, Conklin DS. xCT expression reduces the early cell cycle requirement for calcium signaling. Cell Signaling. 2008;20:390–399. doi: 10.1016/j.cellsig.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]