

Abstract
Polyglutamine diseases are dominantly inherited neurodegenerative diseases caused by an expansion of a CAG trinucleotide repeat encoding a glutamine tract in the respective disease-causing proteins. Extensive studies have been performed to unravel disease pathogenesis and to develop therapeutics. Here, we report on several lines of evidence demonstrating that Nemo-like kinase (NLK) is a key molecule modulating disease toxicity in spinocerebellar ataxia type 1 (SCA1), a disease caused by a polyglutamine expansion in the protein ATAXIN1 (ATXN1). Specifically, we show that NLK, a serine/threonine kinase that interacts with ATXN1, modulates disease phenotypes of polyglutamine-expanded ATXN1 in a Drosophila model of SCA1. Importantly, the effect of NLK on SCA1 pathology is dependent upon NLK's enzymatic activity. Consistent with this, reduced Nlk expression suppresses the behavioral and neuropathological phenotypes in SCA1 knock-in mice. These data clearly indicate that either reducing NLK enzymatic activity or decreasing NLK expression levels can have beneficial effects against the toxicity induced by polyglutamine-expanded ATXN1.
Introduction
Expansion of an unstable translated CAG repeat causes at least nine dominantly inherited neurodegenerative disorders known as the “polyglutamine diseases.” This family of diseases includes Huntington disease, spinal and bulbar muscular atrophy, dentatorubropallidoluysian atrophy, and six autosomal dominant spinocerebellar ataxias (SCA1, 2, 3, 6, 7, and 17) (Ross, 2002). The main goal of research in the field is to understand the pathogenic mechanisms by which glutamine-expanded mutant proteins mediate neurodegeneration and to identify modulators of disease toxicity, which will lead to the development of therapeutic interventions. We have used SCA1 (MIM #164400) as a prototype to understand some of the fundamental pathogenic mechanisms of polyglutamine disorders. SCA1 is characterized by progressive ataxia, mild cognitive impairments, difficulty with speaking and swallowing, and eventually respiratory failure. The clinical features of SCA1 result from the degeneration of cerebellar Purkinje cells, brainstem cranial nerve nuclei and inferior olive neurons, and spinocerebellar tracts (Orr and Zoghbi, 2007). SCA1 is caused by the expansion of a CAG repeat encoding a glutamine tract in the protein ATAXIN1 (ATXN1) (Orr et al., 1993). Building on our studies of SCA1 and ATXN1, we found that neurodegeneration in SCA1 results from a gain of endogenous functions mediated by the polyglutamine-expanded mutant ATXN1, and from a concurrent partial loss of the beneficial functions of wild-type (WT) ATXN1 (Lim et al., 2008; Crespo-Barreto et al., 2010; Fryer et al., 2011).
To understand the disease pathogenesis better and to identify an effector protein that can be potentially used as a therapeutic target in SCA1, we examined candidate proteins identified from our previous physical interaction or genetic modifier studies (Fernandez-Funez et al., 2000; Lim et al., 2006). We paid special attention to proteins that are drug-targetable and/or capable of differentially affecting WT and polyglutamine-expanded mutant ATXN1, as we think such proteins are more directly relevant to disease pathogenesis and therapy. One of these proteins is Nemo-like kinase (NLK). NLK is an evolutionarily conserved serine/threonine protein kinase (Choi and Benzer, 1994; Banfi et al., 1996; Brott et al., 1998; Meneghini et al., 1999). Across species, NLK proteins function as important regulators of cell growth, patterning, and death by controlling diverse signaling pathways, including Wnt and Notch pathways (Ishitani et al., 1999; Meneghini et al., 1999; Rocheleau et al., 1999; Verheyen et al., 2001; Rottinger et al., 2006). Disruption of the Nlk gene in mice results in diverse neuronal and non-neuronal phenotypes (Kortenjann et al., 2001). Despite its essential role in animal development, however, the cellular and mechanistic basis by which loss of NLK function results in numerous neurological abnormalities has not been established. In this study, we examined the effect of NLK on SCA1 pathogenesis. Using biochemical and cell biological studies, as well as genetic studies in Drosophila and mice, we demonstrated that decreased expression of NLK has beneficial effects against neuronal toxicity in SCA1.
Materials and Methods
Generation and evaluation of Nlk gene trap mice.
Two independent gene trap insertion clones (RRJ297 and XN619) of murine embryonic stem (ES) cells that target the Nlk locus were purchased from BayGenomics (now available from the International Gene Trap Consortium). These two clones were injected independently into 129 blastocysts to generate chimeric mice, which were subsequently implanted into pseudopregnant female mice. Male chimeric mice were mated to C57BL/6J females, and agouti pups born from these matings were further characterized to monitor germline transmission of the trapped clone. After successful germline transmission, the precise site of the gene trap insertion in Nlk was determined by sequence analysis of a PCR product amplified from genomic tail DNA. For the RRJ297 gene trap line, the following primers were used: P1 (5′-GTACTTGTGGCACACTGTTTC-3′), P2 (5′-TGGAGGTTGGAGTATGTCAA-3′), and P3 (5′-CCCACTGACCAGAAGGAAAG-3′). For the XN619 gene trap line, the following primers were used: P1 (5′-TCTGCACATGCACAGAGACAC-3′), P2 (5′-GAGGCAGAAGGAGTGAGTGC-3′), and P3 (5′-CCCACTGACCAGAAGGAAAG-3′).
Mouse husbandry, behavioral analysis, and data analyses.
Mice were maintained on a 12 h light and 12 h dark cycle with standard mouse chow and water ad libitum. All research and animal care procedures were approved by the Yale University and the Baylor College of Medicine Institutional Animal Care and Use Committee. Nlk gene trap (NlkRRJ297/+ or NlkXN619/+ or simply Nlk+/−) and SCA1 knock-in (Atxn1154Q/+) animals were maintained on the pure C57BL/6J background after backcrossing them into the C57BL/6J strain for >10 generations. To perform the genetic interaction study, Atxn1154Q/+ male mice were bred to NlkRRJ297/+ heterozygote females on the pure C57BL/6J background. All four subsequent F1 progeny (WT, NlkRRJ297/+, Atxn1154Q/+, and Atxn1154Q/+; NlkRRJ297/+) were tested for motor function and coordination performance. Dowel rod walking test was performed as described previously (Crespo-Barreto et al., 2010) using 9- to 10-week old animals. Data were analyzed with ANOVA using SPSS 18 software for Mac OSX or Student's t test (two-tailed, not assuming equal variances).
Immunofluorescence and confocal microscopy.
Immunofluorescence staining was performed using frozen mouse tissues as described previously (Lim et al., 2008). Sections from frozen fixed cerebella of four genotypes (WT, Nlk+/−, Atxn1154Q/+, and Atxn1154Q/+; Nlk+/−) were stained with mouse anti-calbindin (1:1000; Sigma-Aldrich). Immunofluorescence staining of cerebellar sections and indirect quantitative analysis of Purkinje cell dendritic arborization were performed as described previously (Watase et al., 2002; Jafar-Nejad et al., 2011). Measurements of cerebellar molecular layer thickness were performed as described previously (Duvick et al., 2010). Fluorescent images were scanned using a Zeiss LSM510 or LSM710 confocal microscope and processed with ImageJ software (National Institutes of Health) and Adobe Photoshop.
X-gal staining.
Whole brains were dissected from 6-week-old WT (negative control) and NlkXN619/+ mice and were fixed with 4% paraformaldehyde in PBS for 4 h at 4°C. Brains were then washed three times with PBS. The 50-μm-thick sections were prepared by vibratome. Sections were rinsed in staining buffer (2 mm MgCl2, 0.01% sodium deoxycholate, and 0.02% NP-40 in PBS) and stained overnight with 3 mg/ml X-gal, 5 mm potassium ferricyanide, and 5 mm potassium ferrocyanide in staining buffer at 37°C. After staining, sections were washed with PBS three times and mounted.
Coaffinity purification and coimmunoprecipitation experiments.
The coaffinity purification or coimmunoprecipitation assays were performed using HEK293T cell or mouse brain extracts as described previously (Lim et al., 2006, 2008). HEK293T cells were plated, and cDNA constructs were transfected into the cells 24 h later using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). Cells were cultured in DMEM with 10% FBS. Approximately 48 h after transfection, cells were harvested and lysed with lysis buffer (0.5% NP-40, 0.5% Triton X-100, 20 mm Tris-HCl, pH 8.0, 150–180 mm NaCl, 1 mm EDTA, and complete protease inhibitor mixture, Roche) for 20 min at 4°C, rotating. After centrifugation for 10 min at 13,000 rpm at 4°C, protein extracts were incubated with glutathione-Sepharose 4B beads (GE Healthcare) or anti-myc or anti-NLK agarose beads, washed with lysis buffer, and analyzed by SDS-PAGE and Western blot.
Western blot analysis.
To examine protein expression levels, tissues were prepared from whole brain or cerebellum from each genotype, collected into 1 ml lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1% SDS, 1% Triton X-100, 1 mm PMSF, and Roche complete protease inhibitor mixture) and briefly homogenized with a Dounce homogenizer. The lysate was rotated at 4°C for 20 min and then centrifuged for 15 min at 13,000 × g at 4°C. The protein concentration of the supernatant was quantified and 40 μg total protein from each sample was boiled for 10 min in SDS sample buffer, loaded onto a SDS-PAGE gel, and transferred to a nitrocellulose or PVDF membrane for Western blot analysis. The following primary antibodies were used for Western blot analysis: mouse anti-β-galactosidase (Developmental Studies Hybridoma Bank), mouse anti-Flag (Sigma-Aldrich), mouse anti-Gapdh (Sigma-Aldrich), mouse anti-MPM2 (Novus Biologicals), mouse anti-myc (Sigma-Aldrich), guinea pig anti-Capicua (CIC) (Lam et al., 2006), rabbit anti-actin (Sigma-Aldrich), rabbit anti-ATXN1 (11750) (Lim et al., 2008), rabbit anti-GFP (Abcam), rabbit anti-GST (Sigma-Aldrich), rabbit anti-NLK (Abcam), and rabbit anti-RBM17 (Lim et al., 2008).
Column fractionation and analysis.
Gel-filtration (sizing) chromatography was performed as described previously with slight modifications (Lam et al., 2006; Lim et al., 2008). An AKTA purifier system from GE Healthcare with a Superose 6 GL300 column was used on soluble extracts from ∼20-week-old mouse cerebellum. The protein extraction buffer used was 0.5% Triton X-100, 50 mm Tris-HCl, pH 8.0, 75 mm NaCl, complete protease inhibitor mixture, phosphatase inhibitors, and PMSF. The column buffer was identical to the protein extraction buffer, except that Triton X-100 was 0.1% and NaCl was 50 mm. Thyroglobulin (669 kDa), ADH (150 kDa), and cytochrome C (12.4 kDa) were used for gel-filtration standards.
Luciferase reporter assay.
HEK293T cells were transfected, using Lipofectamine 2000 according to the manufacturer's instructions, with the pGL3-luciferase reporter construct containing six copies of CIC binding sites (50 ng) (Lam et al., 2006), the pRL-TK vector (1 ng, Promega) as an internal transfection control, and HA-CIC-S (short isoform of Capicua, 100 ng), Flag-ATXN1(82Q) (100 ng) and Flag-NLK (100 ng) expression vectors. Total plasmid amounts were equalized by adding empty plasmid. Transfectants were maintained in DMEM with 10% FBS. After ∼1 d later, cells were lysed and subjected to a dual luciferase reporter assay (Promega). Normalized luciferase activity was obtained by dividing the firefly luciferase activity from the reporter construct by the Renilla luciferase activity from the pRL-TK vector. All experiments were performed in triplicate.
Drosophila strains.
Full-length human NLK (WT or mutant [KN] form, 515 amino acids) cDNAs were cloned into the pUAST-dest vector using the Gateway cloning system (Invitrogen) to generate UAS-NLK (WT) or UAS-NLK (KN) transgenic fly lines. The Drosophila model of SCA1 has been described (Fernandez-Funez et al., 2000). Other strains were obtained from the Bloomington Stock Center (Flybase; www.flybase.org).
Results
NLK physically interacts with ATXN1
We discovered NLK as an ATXN1 interactor using a yeast two-hybrid screen as part of our efforts to develop an interactome for inherited ataxias (Lim et al., 2006). Because kinases, such as NLK, are often potential therapeutic targets, we were interested in investigating the role of NLK in SCA1 pathogenesis. To obtain independent evidence for the interaction between NLK and ATXN1, we performed coaffinity purification assays using lysates from HEK293T cells transfected with GST-tagged ATXN1 and Flag-tagged Nlk. We found that WT ATXN1(30Q) pulled down both WT NLK and the kinase-inactive NLK-KN (Fig. 1A), suggesting that the two proteins interact physically in mammalian cells. NLK-KN is a lysine to methionine substitution at residue 155 (K155M) and is defective for kinase activity (Ishitani et al., 2003; Ishitani et al., 2011). We also performed coimmunoprecipitation assays on mouse brain extracts using a specific NLK antibody. We were not able to detect a strong physical interaction between Nlk and Atxn1 (mouse homologs of NLK and ATXN1, respectively) from brain extracts (data not shown), suggesting that the NLK and ATXN1 interaction may be transient in vivo. Thus, to gain confidence in the interaction between the two proteins, we next determined the domains responsible for the interaction using a series of ATXN1 deletion constructs and found that the ATXN1 N-terminal region (amino acids 1–575) interacted best with NLK (Fig. 1B). Given that the ATXN1 N-terminal region contains the polyglutamine tract and that the glutamine expansion is the central cause of SCA1 pathogenesis (Orr et al., 1993), we tested the effect of polyglutamine tract length on the ATXN1-NLK interaction. We found that NLK interacted equally well with both WT and polyglutamine-expanded mutant ATXN1 by coaffinity purification assays in mammalian cells (data not shown). We also explored the possibility that the interaction between NLK and ATXN1 may depend on the phosphorylation status at serine 776 (S776) of ATXN1, a key feature that is absolutely required for SCA1 pathogenesis (Emamian et al., 2003; Duvick et al., 2010). Again, NLK interacted well with ATXN1 regardless of the phosphorylation status of S776 in mammalian cells (data not shown).
Figure 1.

NLK interacts with ATXN1. A, Coaffinity purification (Co-AP) assay shows that ATXN1(30Q) interacts with both WT and kinase activity mutant (KN) NLK. KN is a lysine to methionine substitution at residue 155 (K155M). Co-AP assay was performed in HEK293T cells. Top, The presence of Flag-NLK after affinity purification on glutathione-Sepharose 4B beads, demonstrating the ATXN1-NLK interaction. GST-empty vector (−) was used as a control. GFP expression was used as a transfection and loading control. B, The N-terminal region (amino acids 1–575) of ATXN1 interacted most strongly with NLK. Bottom, Schematic representation of the ATXN1 constructs.
NLK phosphorylates ATXN1
Because NLK is a kinase and interacts with ATXN1, it is possible that ATXN1 may be a substrate for NLK-mediated phosphorylation. If this is the case, then NLK will phosphorylate ATXN1 at serine (S) or threonine (T) residues followed by proline (P) because it is an MAPK-like kinase (Brott et al., 1998) and therefore targets the (S/T-P) consensus motif for phosphorylation. To test whether NLK phosphorylates ATXN1, we transfected HEK293T cells with N-terminal-tagged myc-ATXN1(30Q) together with either the WT or kinase-inactive form of Nlk. Myc-tagged ATXN1 was immunoprecipitated with anti-myc antibody and immunoblotted with either anti-myc or the phospho-specific MPM2 (specific to phosphorylated S/T-P) antibody to reveal total and phosphorylated ATXN1, respectively. We found that WT NLK strongly induced the phosphorylation of ATXN1 (Fig. 2A, lane 2, arrow), whereas the kinase-inactive mutant did not (Fig. 2A, lane 3). In addition, we found that transfection of the full-length ATXN1 also led to the production of an ∼55 kDa truncated ATXN1 fragment within cells, and the phosphorylation of this N-terminal fragment was also increased by WT, but not kinase-inactive NLK (Fig. 2A, arrowhead). This suggests that NLK can phosphorylate ATXN1 at S/T-P site(s) in our cell culture system.
Figure 2.

NLK phosphorylates ATXN1. A, NLK increases phosphorylation of ATXN1 at S/T-P sites. B, The S239 residue in ATXN1 is an NLK phosphorylation site. HEK293T cells were transfected with N-terminal-tagged myc-ATXN1(30Q) and WT, kinase-inactive mutant (KN) Nlk, or a control plasmid (−). The phosphorylation of full-length (arrows) or an N-terminal fragment (∼55 kDa in size, arrowheads) of WT ATXN1 is strongly increased by WT NLK, but not by the KN mutant. *Immunoglobin heavy chain used for immunoprecipitation (IP). S239A is a serine to alanine substitution at residue 239. Normalized levels of MPM2 signal from the full-length ATXN1 are shown (A, right). Mean relative levels (control plasmid (−) = 100%) and SE are shown (n = 2). *p < 0.05 (t test).
Next, we sought to identify the phosphorylation site(s) of ATXN1 that are targeted by NLK. ATXN1 has several putative NLK phosphorylation sites that match the consensus (S/T-P) motif for MAPK phosphorylation. One of these sites is serine 239 (S239), which was identified as an in vivo ATXN1 phosphorylation site in multiple tissues, including brain, by mass spectrometry analysis (Vierra-Green et al., 2005; Huttlin et al., 2010). To test whether S239 is an NLK phosphorylation site, we mutated it to alanine (S239A, phosphorylation-defective) and examined the phosphorylation status of ATXN1. We found that the S239A mutation strongly reduced NLK-mediated ATXN1 phosphorylation (Fig. 2B, compare lane 3 with lane 7), suggesting that the S239 residue is an NLK phosphorylation site. Furthermore, whereas the phosphorylation of full-length ATXN1-S239A was decreased (Fig. 2B, arrow), the phosphorylation of the N-terminal fragment of ATXN1-S239A was completely abolished (Fig. 2B, arrowhead). This suggests that the S239 residue is likely the only NLK phosphorylation site in these N-terminal ATXN1 fragments, but there may be other putative targets for NLK phosphorylation in the C-terminal region of ATXN1. Indeed, the observation that the full-length ATXN1-S239A protein still migrated slightly slower when it was cotransfected with WT NLK (Fig. 2B, compare lane 6 with lane 7, arrow) further supports the idea of the presence of a second NLK-mediated phosphorylation site at the C-terminal region of ATXN1.
NLK modulates polyglutamine-expanded ATXN1 toxicity in a Drosophila model of SCA1
To determine whether NLK could affect SCA1 pathology in vivo, we used a Drosophila model of SCA1 (Fernandez-Funez et al., 2000) in which targeted expression of polyglutamine-expanded mutant ATXN1 in the eye causes retinal depigmentation, disruption of the structured eye pattern, and retinal degeneration (Fig. 3B,F,J). Light microscopic analysis of SCA1 flies shows a yellow-colored eye (compare Fig. 3A with Fig. 3B) and, occasionally, necrotic patches on the external surface of the eyes (Fig. 3B, arrow). To test whether NLK could affect SCA1 pathology in Drosophila, we first checked for a genetic interaction between SCA1 flies and nmo (Drosophila homolog of NLK, nemo) flies. A heterozygous loss of one nmo allele strongly suppressed retinal depigmentation and ommatidial disorganization phenotypes of SCA1 flies when raised at 27.5°C (Fig. 3C,D,G,H,M), a temperature at which polyglutamine-expanded mutant ATXN1(82Q) causes only moderate retinal degeneration (Fig. 3B,F). Next, to verify the specificity of the genetic interaction and to test the effects of NLK kinase activity on the Drosophila model of SCA1, we generated UAS constructs with either the WT or kinase-inactive form of human NLK. Expression of human NLK alone in the Drosophila eye did not cause any major external morphological phenotypes at 27.5°C (data not shown). However, coexpression of WT NLK with ATXN1(82Q) enhanced the ommatidial disorganization and retinal degeneration phenotypes in SCA1 flies (Fig. 3K,N). In particular, coexpression of WT NLK led to a dramatic increase in the number and size of the areas of necrotic cell death seen on the external eyes of flies expressing ATXN1(82Q) (Fig. 3K,N, arrow). In contrast, kinase-inactive NLK had little effect when coexpressed with ATXN1(82Q) (Fig. 3L,N). Together, these studies indicate that NLK exacerbates the toxicity of polyglutamine-expanded ATXN1 in a Drosophila model of SCA1 in a kinase activity-dependent manner.
Figure 3.
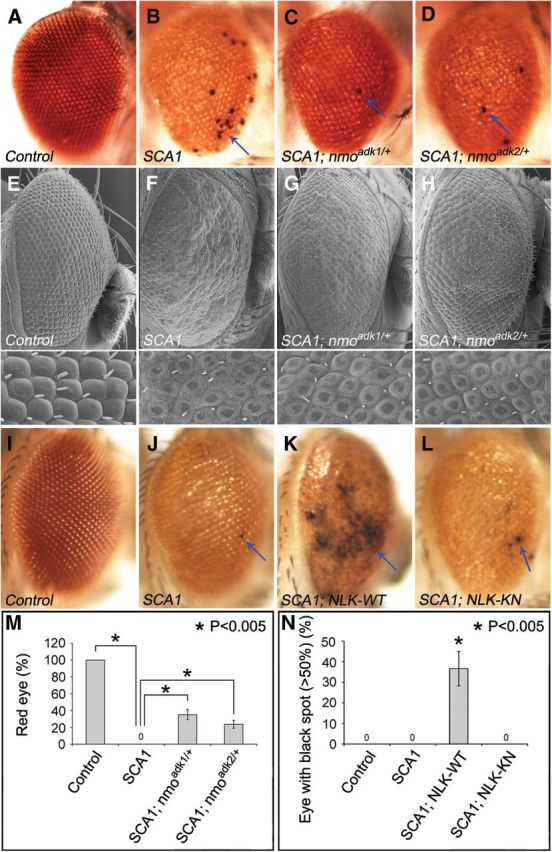
NLK modulates polyglutamine-expanded ATXN1 disease phenotypes in a Drosophila model of SCA1. A–H, Loss of one nmo allele suppressed ATXN1(82Q)-mediated retinal phenotypes. Both light microscopy (A–D) and scanning electron microscopy (E–H) of adult Drosophila eyes are shown, and magnified images are on the bottom of each panel. Two independent mutant alleles (adk1 and adk2) of nmo showed the same results. Light microscopic analysis clearly shows the depigmentation and necrotic death (marked by arrows) phenotypes of the external eyes. Scanning electron microscopy shows the retinal degeneration phenotypes of SCA1 with better resolution. Flies were raised at 27.5°C, and genotypes are as follows: A, E, GMR-Gal4/+; UAS-EGFP/+; B, F, UAS-ATXN182Q/+; GMR-Gal4/+; C, G, UAS-ATXN182Q/+; GMR-Gal4/+; nmoadk1/+; and D, H, UAS-ATXN182Q/+; GMR-Gal4/+; nmoadk2/+. I–L, Coexpression of NLK-WT, but not kinase-inactive mutant NLK (NLK-KN), worsened retinal degenerative phenotypes induced by polyglutamine-expanded mutant ATXN1(82Q). Flies were raised at 27.5°C, and genotypes are as follows: I, GMR-Gal4/UAS-EGFP; UAS-EGFP/+; J, UAS-ATXN182Q/+; GMR-Gal4/UAS-EGFP; K, UAS-ATXN182Q/+; GMR-Gal4/UAS-NLKWT; and L, UAS-ATXN182Q/+; GMR-Gal4/UAS-NLKKN. M, Quantification of the genetic interaction study between loss-of-nmo-function and SCA1 (A–D). A total of 100% of SCA1 flies showed a yellow eye color (thus, 0% showed a red color) and a heterozygous loss of one nmo allele strongly suppressed SCA1 phenotypes, resulting in a red eye color. For this genetic interaction study, flies were raised at 27.5°C and >100 adult eyes per genotype were examined at day 2 after eclosion. Four independent experiments were performed (p < 0.005, t test). N, Quantification of the genetic interaction study between gain-of-NLK-function and SCA1 (I–L). SCA1 flies show patches of necrotic death on the external surface of the eyes, and coexpression of the WT NLK strongly enhances this phenotype. For this genetic interaction study, flies were raised at 27.5°C, collected at day 1, and kept separately at 27.5°C. At day 5 after eclosion, eyes with >50% necrotic black spots were scored. More than 100 adult eyes per genotype were examined, and five independent experiments were performed. *p < 0.005 (t test).
Decreased NLK expression improves the SCA1 motor phenotype and cerebellar neuropathology in mice
Studies using biochemical approaches as well as a Drosophila model system demonstrate that NLK enhances SCA1 neuropathology in a kinase activity-dependent manner. Next, we wished to test whether the effect of NLK on SCA1 pathology is conserved in mammals. We reasoned that, if the relationship between NLK and SCA1 is preserved in the mammalian system, then it is likely that suppressing NLK enzymatic activity or decreasing NLK protein levels would ameliorate the neurotoxicity induced by polyglutamine-expanded ATXN1 in SCA1 mouse models. Thus, we decided to test this idea in vivo by examining whether loss of one functional Nlk allele could suppress the neurodegenerative phenotypes of SCA1 in mice. To perform the genetic interaction study in mammals, we first generated Nlk mutant mice. We obtained two independent gene trap insertion lines of murine ES cells that target the Nlk locus from BayGenomics (now available from the International Gene Trap Consortium). The first ES cell line (ID RRJ297) has the targeting cassette inserted into the second intron of the Nlk gene, and the second line (ID XN619) has the targeting cassette in intron 1 (Fig. 4A). We determined the exact site of the gene trap within Nlk in each line by conducting PCR with primer pairs spanning the length of the intron (Fig. 4B). Both RRJ297 and XN619 alleles have uncharacterized genomic rearrangement, which precludes simple PCR genotyping between the heterozygous and homozygous allele (data not shown). Splicing of the trap into Nlk results in an mRNA transcript that contains only the first one or two exons of Nlk followed by the βgeo (β-galactosidase-neomycin) sequence, thereby excluding the majority of the NLK protein, including the kinase domain. We also determined the level of NLK protein in whole brain or cerebellar extracts from WT (Nlk+/+), heterozygous (NlkRRJ297/+ and NlkXN619/+), and compound heterozygous mutant (NlkRRJ297/XN619) mice. Correct targeting of the gene trap alleles was confirmed by Western blot, which showed that NLK expression was strongly reduced in both Nlk gene trap mouse lines (Fig. 4C). Compared with WT cerebellum, NlkRRJ297/XN619 cerebellum shows an ∼90% reduction in NLK expression (Fig. 4C), suggesting that RRJ297 and XN619 are strong hypomorphic alleles. Our Nlk gene trap mice display phenotypes that vary depending on the genetic background, which is consistent with a previously reported Nlk knock-out mouse (Kortenjann et al., 2001). We then backcrossed these gene trap mice onto pure C57BL/6J or 129S6/SvEv genetic backgrounds over 10 generations. NlkRRJ297/XN619 mice die at birth on the pure C57BL/6J background, but they survive up to several months of age on the pure 129S6/SvEv background with various neurological and non-neurological abnormalities (data not shown).
Figure 4.
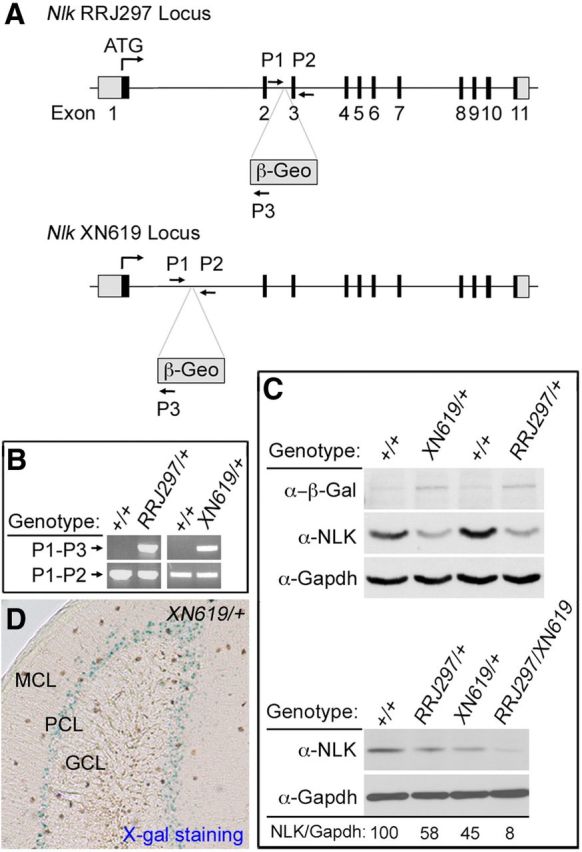
Generation and evaluation of Nlk gene trapped mice. A, Schematic representation of the gene trapped Nlk locus. The Nlk locus is disrupted by insertion of the gene trap within the first or the second intron. Exons are depicted as numbered boxes. Primers used for genotyping are shown as lettered arrows (P1, P2, and P3). B, PCR genotyping of tail tip genomic DNA using the primer set shown in A. Primers P1 and P2 create a product in the absence of the gene trap, whereas primers P1 and P3 create a product in the presence of the gene trap. C, Western blot analysis of cerebellar extracts from WT (Nlk+/+), heterozygous (NlkRRJ297/+ and NlkXN619/+), and compound heterozygous mutant (NlkRRJ297/XN619) mice. NLK expression level relative to Gapdh is shown (%). D, Strong LacZ expression of the Nlk gene trap is observed within the Purkinje cell layer from 6-week-old NlkXN619/+ mouse cerebellum. MCL, Molecular cell layer; PCL, Purkinje cell layer; GCL, granular cell layer.
Because NLK expression can be detected by Western blot analysis in cerebellar extracts (Fig. 4C), we next explored the expression pattern of NLK within the various cell types of the cerebellum. We performed immunofluorescence staining using currently available NLK antibodies on the WT (positive control) and NlkRRJ297/XN619 (negative control) mice but failed to detect specific endogenous Nlk expression patterns (data not shown). Thus, alternatively, we analyzed Nlk expression by LacZ staining and found that LacZ staining was strongest in the Purkinje cell layer (Fig. 4D), consistent with the Nlk mRNA expression pattern shown in the Allen Brain Atlas (Lein et al., 2007). To determine whether Nlk gene dosage affects the SCA1 phenotype, we crossed Nlk heterozygote (NlkRRJ297/+) animals with SCA1 knock-in (Atxn1154Q/+) mice (Watase et al., 2002) and evaluated the progeny using behavioral and pathological analyses. We first intended to assess mouse motor coordination by performing the accelerating rotating rod (“rotarod”) analysis. We found that Nlk heterozygote (NlkRRJ297/+ or Nlk+/−) mice displayed an impaired performance on the rotarod on the pure C57BL/6J background (data not shown), which precluded a genetic interaction study with SCA1 knock-in mice. Thus, as an alternative test for motor coordination and balance defects, we performed the dowel rod walking test at 9–10 weeks of age (Fig. 5A). We measured the latency to reach the side (latency first side touch) and the frequency of walking off the rod (number of side touches in 120 s). All four genotypes tested remained on the dowel for the maximum time (data not shown). The latency of Atxn1154Q/+ mice to reach the side for the first time was dramatically increased compared with littermate control mice; consequently, they walked off the rod much fewer times. The Nlk heterozygote mice did not show any obvious difference on the dowel rod test compared with their WT littermates. Interestingly, loss of just one Nlk allele almost completely rescued the phenotypes of Atxn1154Q/+ mice back to WT and Nlk heterozygote levels. The Atxn1154Q/+; Nlk+/− mice took much less time to walk off the dowel and also crossed the dowel more times in 120 s than Atxn1154Q/+ littermates. Thus, decreasing NLK expression rescues the dowel phenotype caused by polyglutamine-expanded mutant ATXN1.
Figure 5.

Deletion of one Nlk allele rescues SCA1 cerebellar phenotypes in mice. A, In the dowel rod walking test, Atxn1154Q/+ mice showed increased latency to reach the sides of the dowel compared with WT and Nlk+/− littermates (left). They also walked off fewer times in the 120 s interval (right). In contrast, Atxn1154Q/+; Nlk+/− mice exhibited marked reduction in the time for the first side touch and increased the number of side touches compared with Atxn1154Q/+ littermates. Genotypes are color-coded as indicated. Error bars indicate SEM. B–D, Cerebellar neuropathology is partially rescued in Purkinje cells of Atxn1154Q/+ mice lacking one allele of Nlk. B, Representative confocal images showing Purkinje cell morphology stained with anti-calbindin antibody from age-matched littermates. C, Quantitative analysis of calbindin immunofluorescence is an indirect measure of cerebellar Purkinje cell soma and dendritic integrity. Mean fluorescence intensity of optical rectangular subsections from the same folia of 38- to 39-week-old animals was plotted as the distance from the perikaryon center. Nlk heterozygosity partially rescues the Atxn1154Q/+ loss of dendritic arborization phenotype. Error bars indicate SEM. D, Quantification of the cerebellar molecular layer thickness at the primary fissure from 38- to 39-week-old mice (shown by arrow in B). Atxn1154Q/+ mice (four sections per animal; n = 3) significantly decreased molecular layer thickness compared with WT littermates. *p = 0.015 (ANOVA). Loss of one Nlk allele (four sections per animal; n = 2) rescued defects of the molecular layer thickness in Atxn1154Q/+ mice. *p = 0.004 (ANOVA). Error bars indicate SEM.
We also tested whether Nlk haploinsufficiency would affect the cerebellar neuropathology of Atxn1154Q/+ mice. The dendritic arborization defect of the Purkinje cells in the cerebellum is the most prominent pathological sign in aging Atxn1154Q/+ animals (Watase et al., 2002; Jafar-Nejad et al., 2011). Consistent with the behavioral improvement, Atxn1154Q/+ mice that lack one functional copy of Nlk displayed an improved complexity of fine dendritic arbors and Purkinje cell soma at 38–39 weeks of age, as examined by measuring calbindin immunofluorescence (Fig. 5B,C). We also measured the molecular layer thickness in mice at 38–39 weeks of age and found that Atxn1154Q/+; Nlk+/− mice showed a significant improvement in the molecular layer thickness, compared with Atxn1154Q/+ mice alone (Fig. 5D). Together, the gene dosage-dependent genetic interaction between NLK and mutant ATXN1 in mice strongly suggests that reducing NLK expression rescues the ataxic behavioral phenotype and cerebellar pathology caused by polyglutamine expansion in SCA1.
NLK incorporation in toxic ATXN1 large complexes is low in Atxn1154Q/+; Nlk+/− mice
We investigated potential molecular mechanisms by which NLK may affect ATXN1 biology and SCA1 pathology. We first assessed whether NLK affects the expression levels of polyglutamine-expanded mutant (as well as WT) ATXN1 and its associated proteins. We found that there was no notable change in the expression of these proteins in mouse cerebellum when NLK is expressed at 50% of WT levels (Fig. 6A). Having established the importance of a large ATXN1 protein complex for the pathogenesis of SCA1 (Lam et al., 2006; Bowman et al., 2007; Lim et al., 2008; Jafar-Nejad et al., 2011), we next investigated the effects of decreasing NLK protein level by 50% on the formation and maintenance of ATXN1 protein complexes. Consistent with the previous studies (Lam et al., 2006; Bowman et al., 2007; Lim et al., 2008; Jafar-Nejad et al., 2011), all proteins elute in a broad range with at least two peaks at fractions 7–9 (red box, large protein complexes) and 11–12 (blue box, small protein complexes) (Fig. 6B). The elution profile of Nlk covers those of Atxn1 and its associated proteins, CIC and RNA-binding motif protein 17 (Rbm17). We found that the relative ratio of polyglutamine-expanded (as well as WT) Atxn1 incorporation into the toxic large protein complexes was not significantly different between Atxn1154Q/+; Nlk+/− and Atxn1154Q/+ mouse cerebellum (Fig. 6B). Furthermore, the elution profiles of known Atxn1-interacting proteins, including CIC and Rbm17, were also not altered in Atxn1154Q/+; Nlk+/− mice compared with Atxn1154Q/+ mice. Instead, it was noted that the level of Nlk incorporation in toxic large complexes was low in Atxn1154Q/+; Nlk+/− mice compared with Atxn1154Q/+ mice (Fig. 6B, red box). Interestingly, we also found that a high level of the Nlk protein was detected in much smaller complexes in fractions 14–15 in Atxn1154Q/+; Nlk+/− mice compared with Atxn1154Q/+ mice (Fig. 6B, small blue box). Together, these data suggest that NLK may not play a significant role in the degradation of ATXN1 or in the formation and maintenance of the large toxic ATXN1 complexes in mice in vivo.
Figure 6.

Expression levels and ATXN1-associated protein complex formation in Atxn1154Q/+; Nlk+/− mice. A, Nlk heterozygosity does not affect the expression levels of components in Atxn1 native complexes in cerebellar extracts of SCA1 knock-in mice. Expression of Capicua (CIC-L and CIC-S, long and short isoforms of CIC, respectively), WT (Atxn1[2Q]), and polyglutamine-expanded mutant (Atxn1[154Q]) Atxn1 and Rbm17 remained the same in cerebellar extracts from 20-week-old Atxn1154Q/+ or Atxn1154Q/+; Nlk+/− mice. Nlk and Gapdh were used as controls. B, The level of NLK incorporation in toxic large complexes is low in Atxn1154Q/+; Nlk+/− mice compared with Atxn1154Q/+ mice. Elution profiles of CIC, Atxn1 (both WT and mutant Atxn1), Rbm17, and Nlk proteins in Atxn1154Q/+ or Atxn1154Q/+; Nlk+/− mouse cerebellum using gel-filtration chromatography. Representative Western blots of 1.0 ml gel-filtration fractions of Atxn1154Q/+ or Atxn1154Q/+; Nlk+/− mouse cerebellar extracts analyzed for CIC, Atxn1, Rbm17, and Nlk. The column void volume (Vo), gel-filtration standards thyroglobulin (669 kDa) and ADH (150 kDa), and elution volume (ml) of each collected fraction are indicated. Ex, Extract.
NLK affects the transcriptional activity of the ATXN1/CIC complex
The ATXN1 protein binds to the CIC family of transcription factors and regulates the transcription of target genes in vitro in cell culture systems (Lam et al., 2006) as well as in vivo in mice (Crespo-Barreto et al., 2010; Fryer et al., 2011). This transcriptional activity is important for the pathogenesis of SCA1 (Lin et al., 2000; Serra et al., 2004; Lam et al., 2006; Cvetanovic et al., 2007, 2011; Crespo-Barreto et al., 2010; Fryer et al., 2011). We then investigated the possibility of whether NLK affects the transcriptional activity of the ATXN1/CIC complex. To do this, we took advantage of the established CIC-responsive luciferase reporter, which is repressed by the ATXN1/CIC complex (Fig. 7). We first established conditions in which there is only a moderate transcriptional repression of the luciferase activity when HEK293T cells are cotransfected with CIC-S (short isoform of CIC) and ATXN1(82Q). Next, to examine whether NLK modulates the ATXN1/CIC activity, we coexpressed NLK with ATXN1/CIC and found that NLK strongly abolished the ATXN1/CIC-mediated repression activity in HEK293T cells (Fig. 7). The effect of NLK on the ATXN1/CIC complex required NLK's kinase activity. These data provide evidence for the role of NLK in modulating the ATXN1/CIC transcription complex activity.
Figure 7.

NLK relieves repression by the ATXN1/CIC transcription complex in a kinase activity-dependent manner. HEK293T cells were transfected with the pGL3-luciferase reporter construct containing six copies of CIC binding sites, the pRL-TK control vector, and expression plasmids as indicated. The luciferase data are expressed as mean percentage of luciferase activity relative to the reporter alone. All experiments were performed in triplicate. Error bars indicate SEM. *p < 0.001 (ANOVA, Scheffé's test).
Discussion
In this study, we tested the idea that NLK may be implicated in the pathogenesis of SCA1 and explored the potential contribution of NLK to this disease. By combining cell biological studies and genetic approaches, we show that NLK is a key protein contributing to the pathogenesis of SCA1. Experimental evidence from two different SCA1 model systems, Drosophila and mice, strongly supports the idea that reduced enzymatic activity or decreased expression of NLK can have beneficial effects against polyglutamine-expanded ATXN1 protein toxicity. In particular, we show that Nlk haploinsufficiency in the SCA1 knock-in (Atxn1154Q/+) mice rescues SCA1 cerebellar pathology and motor phenotypes. It is very intriguing that modulating kinase activity or genetically altering expression levels can indeed suppress behavioral and pathological phenotypes of polyglutamine diseases in mice, as this is a critical step before clinical trials in patients.
The importance of expression levels and/or the formation of a large ATXN1 protein complex for the pathogenesis of SCA1 have been well established (Lam et al., 2006; Bowman et al., 2007; Lim et al., 2008; Jafar-Nejad et al., 2011). Our finding that there was no notable change in the expression levels of Atxn1 and its known associated proteins in Nlk heterozygous mouse cerebellum (Fig. 6A) suggests that NLK may not impact the stability of these proteins in mice. In addition, the fact that the formation and maintenance of ATXN1 protein complexes were not altered in an Nlk heterozygote background compared with Nlk WT mice (Fig. 6B) suggests that NLK may not play a substantial role in the formation and maintenance of the large toxic ATXN1 complexes in vivo. However, it is worth mentioning that the level of NLK incorporation in large complexes is low in the Nlk heterozygote background (Fig. 6B). This may simply be the result of the reduced NLK expression in the heterozygote background. Alternatively, there are N-terminal fragments of NLK produced as fusion proteins to β-galactosidase in both RRJ297 and XN619 allele-bearing mice (Fig. 4C). Therefore, these fragments could function as dominant-negatives against WT full-length NLK by displacing it from the ATXN1 complexes. It would also be interesting to know what other proteins in addition to NLK exist in fractions 14/15. Identifying proteins and testing their relevance to NLK function and SCA1 disease pathogenesis would be an intriguing and important study to be addressed in the future.
Our cell culture-based luciferase assay indicates that NLK can affect the transcriptional activity of the ATXN1/CIC protein complex (Fig. 7). Because alterations in gene expression is the earliest pathological finding in SCA1 disease (Lin et al., 2000; Serra et al., 2004; Cvetanovic et al., 2007, 2011; Crespo-Barreto et al., 2010; Fryer et al., 2011) and the CIC family proteins play a key role in modulating ATXN1 functions and SCA1 pathogenesis (Lam et al., 2006; Bowman et al., 2007; Lim et al., 2008; Fryer et al., 2011; Lee et al., 2011), this finding may explain in part the mechanistic role of NLK in SCA1 pathogenesis. Consistent with the genetic interaction study in Drosophila (Fig. 3), the effect of NLK in altering ATXN1/CIC activity is also dependent on its kinase activity (Fig. 7). Therefore, it will be very important to know what residues may be target sites of NLK. Our biochemical studies suggest that NLK is able to induce the phosphorylation of ATXN1, either directly or indirectly, on several conserved MAPK consensus sites, including S239, at least in the cell culture system (Fig. 2). In neurodegenerative diseases, phosphorylation of polyglutamine disease proteins plays important roles in disease pathogenesis by affecting diverse aspects of pathogenicity, including proteolytic cleavage, induction of a toxic conformational change, nuclear transport, and gene transcription (Bauer and Nukina, 2009; Pennuto et al., 2009). In SCA1, several phosphorylation sites have been identified so far in mouse brain in vivo, including S776 and S239 (Emamian et al., 2003; Vierra-Green et al., 2005; Huttlin et al., 2010). The phosphorylation of S776 in ATXN1 is extensively studied and is known to be crucial for the initiation and progression of SCA1 (Emamian et al., 2003; Duvick et al., 2010). Interestingly, the phosphorylation level of ATXN1 at residue S776 was not altered by NLK in cell culture and in mouse in vivo (data not shown), indicating that the beneficial effects of decreased expression of NLK against SCA1 toxicity is not mediated by decreased S776-ATXN1 phosphorylation. Instead, we found that NLK could phosphorylate ATXN1 at different sites, including the residue S239 in a cell culture system (Fig. 2). Further studies are needed to validate whether NLK is a kinase for ATXN1-S239 phosphorylaton in vivo and to understand how S239 phosphorylation affects the function and regulation of ATXN1 and its role in SCA1 pathogenesis.
In conclusion, our study clearly shows that decreased expression of Nlk in a Drosophila SCA1 model and in SCA1 knock-in (Atxn1154Q/+) mice rescues pathological and behavioral phenotypes of SCA1. In addition, overexpression of NLK enhances disease phenotypes of polyglutamine-expanded ATXN1 in the Drosophila model in a kinase activity-dependent manner. Therefore, finding a factor capable of inhibiting NLK enzymatic activity or decreasing NLK expression levels will provide a new avenue for developing therapeutic approaches to SCA1.
Footnotes
This work was supported by the National Institutes of Health Grant NS064146 to J.L. and Grant NS027699 to H.Y.Z., the National Ataxia Foundation (to J.L.), Alfred P. Sloan Foundation (to J.L.), and Yale Scholar Award Program (to J.L.). H.Y.Z. is an investigator with the Howard Hughes Medical Institute. We thank Gabriele Schuster for mouse ES cell microinjection work for Nlk gene trap mice; Dr. Hugo Bellen and Yuchun He for Drosophila embryo microinjection for NLK transgenic flies; Dr. Kwang-Wook Choi for nmo mutant flies; Dr. Kunihiro Matsumoto and Dr. Tohru Ishitani for the Nlk constructs; and members of the J.L. and H.Y.Z. laboratories for comments on the manuscript.
The authors declare no competing financial interests.
References
- Banfi S, Borsani G, Rossi E, Bernard L, Guffanti A, Rubboli F, Marchitiello A, Giglio S, Coluccia E, Zollo M, Zuffardi O, Ballabio A. Identification and mapping of human cDNAs homologous to Drosophila mutant genes through EST database searching. Nat Genet. 1996;13:167–174. doi: 10.1038/ng0696-167. [DOI] [PubMed] [Google Scholar]
- Bauer PO, Nukina N. The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J Neurochem. 2009;110:1737–1765. doi: 10.1111/j.1471-4159.2009.06302.x. [DOI] [PubMed] [Google Scholar]
- Bowman AB, Lam YC, Jafar-Nejad P, Chen HK, Richman R, Samaco RC, Fryer JD, Kahle JJ, Orr HT, Zoghbi HY. Duplication of Atxn1l suppresses SCA1 neuropathology by decreasing incorporation of polyglutamine-expanded ataxin-1 into native complexes. Nat Genet. 2007;39:373–379. doi: 10.1038/ng1977. [DOI] [PubMed] [Google Scholar]
- Brott BK, Pinsky BA, Erikson RL. Nlk is a murine protein kinase related to Erk/MAP kinases and localized in the nucleus. Proc Natl Acad Sci U S A. 1998;95:963–968. doi: 10.1073/pnas.95.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KW, Benzer S. Rotation of photoreceptor clusters in the developing Drosophila eye requires the nemo gene. Cell. 1994;78:125–136. doi: 10.1016/0092-8674(94)90579-7. [DOI] [PubMed] [Google Scholar]
- Crespo-Barreto J, Fryer JD, Shaw CA, Orr HT, Zoghbi HY. Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet. 2010;6:e1001021. doi: 10.1371/journal.pgen.1001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvetanovic M, Rooney RJ, Garcia JJ, Toporovskaya N, Zoghbi HY, Opal P. The role of LANP and ataxin 1 in E4F-mediated transcriptional repression. EMBO Rep. 2007;8:671–677. doi: 10.1038/sj.embor.7400983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvetanovic M, Patel JM, Marti HH, Kini AR, Opal P. Vascular endothelial growth factor ameliorates the ataxic phenotype in a mouse model of spinocerebellar ataxia type 1. Nat Med. 2011;17:1445–1447. doi: 10.1038/nm.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvick L, Barnes J, Ebner B, Agrawal S, Andresen M, Lim J, Giesler GJ, Zoghbi HY, Orr HT. SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron. 2010;67:929–935. doi: 10.1016/j.neuron.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, Clark HB, Orr HT. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38:375–387. doi: 10.1016/S0896-6273(03)00258-7. [DOI] [PubMed] [Google Scholar]
- Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, McCall A, Canal I, Orr HT, Zoghbi HY, Botas J. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- Fryer JD, Yu P, Kang H, Mandel-Brehm C, Carter AN, Crespo-Barreto J, Gao Y, Flora A, Shaw C, Orr HT, Zoghbi HY. Exercise and genetic rescue of SCA1 via the transcriptional repressor Capicua. Science. 2011;334:690–693. doi: 10.1126/science.1212673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishitani S, Inaba K, Matsumoto K, Ishitani T. Homodimerization of Nemo-like kinase is essential for activation and nuclear localization. Mol Biol Cell. 2011;22:266–277. doi: 10.1091/mbc.E10-07-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishitani T, Ninomiya-Tsuji J, Nagai S, Nishita M, Meneghini M, Barker N, Waterman M, Bowerman B, Clevers H, Shibuya H, Matsumoto K. The TAK1-NLK-MAPK-related pathway antagonizes signalling between β-catenin and transcription factor TCF. Nature. 1999;399:798–802. doi: 10.1038/21674. [DOI] [PubMed] [Google Scholar]
- Ishitani T, Ninomiya-Tsuji J, Matsumoto K. Regulation of lymphoid enhancer factor 1/T-cell factor by mitogen-activated protein kinase-related Nemo-like kinase-dependent phosphorylation in Wnt/beta-catenin signaling. Mol Cell Biol. 2003;23:1379–1389. doi: 10.1128/MCB.23.4.1379-1389.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafar-Nejad P, Ward CS, Richman R, Orr HT, Zoghbi HY. Regional rescue of spinocerebellar ataxia type 1 phenotypes by 14-3-3epsilon haploinsufficiency in mice underscores complex pathogenicity in neurodegeneration. Proc Natl Acad Sci U S A. 2011;108:2142–2147. doi: 10.1073/pnas.1018748108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortenjann M, Nehls M, Smith AJ, Carsetti R, Schüler J, Köhler G, Boehm T. Abnormal bone marrow stroma in mice deficient for nemo-like kinase, Nlk. Eur J Immunol. 2001;31:3580–3587. doi: 10.1002/1521-4141(200112)31:12<3580::AID-IMMU3580>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD, Hyun ED, Duvick LA, Orr HT, Botas J, Zoghbi HY. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–1347. doi: 10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- Lee Y, Fryer JD, Kang H, Crespo-Barreto J, Bowman AB, Gao Y, Kahle JJ, Hong JS, Kheradmand F, Orr HT, Finegold MJ, Zoghbi HY. ATXN1 protein family and CIC regulate extracellular matrix remodeling and lung alveolarization. Dev Cell. 2011;21:746–757. doi: 10.1016/j.devcel.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lim J, Hao T, Shaw C, Patel AJ, Szabó G, Rual JF, Fisk CJ, Li N, Smolyar A, Hill DE, Barabási AL, Vidal M, Zoghbi HY. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–814. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- Lim J, Crespo-Barreto J, Jafar-Nejad P, Bowman AB, Richman R, Hill DE, Orr HT, Zoghbi HY. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature. 2008;452:713–718. doi: 10.1038/nature06731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Antalffy B, Kang D, Orr HT, Zoghbi HY. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci. 2000;3:157–163. doi: 10.1038/72101. [DOI] [PubMed] [Google Scholar]
- Meneghini MD, Ishitani T, Carter JC, Hisamoto N, Ninomiya-Tsuji J, Thorpe CJ, Hamill DR, Matsumoto K, Bowerman B. MAP kinase and Wnt pathways converge to downregulate an HMG-domain repressor in Caenorhabditis elegans. Nature. 1999;399:793–797. doi: 10.1038/21666. [DOI] [PubMed] [Google Scholar]
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- Orr HT, Chung MY, Banfi S, Kwiatkowski TJ, Jr, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LP, Zoghbi HY. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- Pennuto M, Palazzolo I, Poletti A. Post-translational modifications of expanded polyglutamine proteins: impact on neurotoxicity. Hum Mol Genet. 2009;18:R40–R47. doi: 10.1093/hmg/ddn412. [DOI] [PubMed] [Google Scholar]
- Rocheleau CE, Yasuda J, Shin TH, Lin R, Sawa H, Okano H, Priess JR, Davis RJ, Mello CC. WRM-1 activates the LIT-1 protein kinase to transduce anterior/posterior polarity signals in C. elegans. Cell. 1999;97:717–726. doi: 10.1016/S0092-8674(00)80784-9. [DOI] [PubMed] [Google Scholar]
- Ross CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron. 2002;35:819–822. doi: 10.1016/S0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Röttinger E, Croce J, Lhomond G, Besnardeau L, Gache C, Lepage T. Nemo-like kinase (NLK) acts downstream of Notch/Delta signalling to downregulate TCF during mesoderm induction in the sea urchin embryo. Development. 2006;133:4341–4353. doi: 10.1242/dev.02603. [DOI] [PubMed] [Google Scholar]
- Serra HG, Byam CE, Lande JD, Tousey SK, Zoghbi HY, Orr HT. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet. 2004;13:2535–2543. doi: 10.1093/hmg/ddh268. [DOI] [PubMed] [Google Scholar]
- Verheyen EM, Mirkovic I, MacLean SJ, Langmann C, Andrews BC, MacKinnon C. The tissue polarity gene nemo carries out multiple roles in patterning during Drosophila development. Mech Dev. 2001;101:119–132. doi: 10.1016/S0925-4773(00)00574-8. [DOI] [PubMed] [Google Scholar]
- Vierra-Green CA, Orr HT, Zoghbi HY, Ferrington DA. Identification of a novel phosphorylation site in ataxin-1. Biochim Biophys Acta. 2005;1744:11–18. doi: 10.1016/j.bbamcr.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Watase K, Weeber EJ, Xu B, Antalffy B, Yuva-Paylor L, Hashimoto K, Kano M, Atkinson R, Sun Y, Armstrong DL, Sweatt JD, Orr HT, Paylor R, Zoghbi HY. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron. 2002;34:905–919. doi: 10.1016/S0896-6273(02)00733-X. [DOI] [PubMed] [Google Scholar]