

Abstract
Non-coding RNAs (ncRNAs) are increasingly recognized as central players in diverse biological processes. DNA damage response (DDR) elicits a complex signaling cascade, which includes the induction of several ncRNA species. Recent studies indicate that ncRNAs are major regulators of the DDR. DNA-damage induced ncRNAs contribute to regulation of cell cycle, apoptosis and DNA repair, and thus play a key role in maintaining genome stability. This review summarizes the role of ncRNAs in DNA damage and repair.
Keywords: noncoding RNAs, DNA damage, Repair, miRNAs, Long noncoding RNAs, Genome Integrity
Introduction
1. DNA Damage and Repair
Genomes continually face the challenge of DNA damage caused by environmental and endogenous insults. To combat the potential adverse effects from DNA damage, cells have developed a sophisticated signaling cascade to sense and repair DNA damage and maintain genome integrity. Depending on the source of damage, DNA may experience a wide variety of lesions such as modification of bases, single strand breaks (SSBs), or double strand breaks (DSBs) [1,2].
Repair of DNA lesions by the DDR pathway comprises three major steps: (i) detection of damage by sensors, (ii) recruitment of repair factors to sites of damage by signal transducers and (iii) repair by effectors [3]. Repair of different types of DNA damage is carried out by specific repair pathways: DNA mismatches are corrected by mismatch repair (MMR) whereas chemical modifications of DNA bases are repaired by base excision repair (BER) [4,5]; the nucleotide excision repair (NER) pathway corrects more complex lesions such as pyrimidine dimers and intrastrand crosslinks [6,7] and SSBs are repaired by single-strand break repair (SSBR) [8]; DSBs which are the most toxic and difficult to repair DNA lesions are corrected either by nonhomologous end joining (NHEJ) or homologous recombination (HR). NHEJ occurs primarily during pre-replicative (G0 and G1) phases of the cell cycle and does not require a template DNA sequence since broken DNA ends are directly rejoined. On the other hand, HR requires a homologous DNA template sequence for error-free repair and predominates in S phase of the cell cycle. For repair of DSBs by HR, DNA ends are resected to yield 3′ single-strand DNA overhangs and the resected DNA, with help of HR proteins, permits strand invasion of a partner homologous sequence to form a nascent D-loop structure [9]. This is followed by synthesis of DNA by synthesis-dependent strand annealing (SDSA) pathway or the double-strand break repair (DSBR) pathway [9]. Repair of DSBs is mediated by proteins of the phosphatidylinositol 3-kinase-like protein kinase (PIKKs) family, particularly ATM, ATR, and DNA-PKcs and the choice of repair pathway is influenced by type of lesion and the cell cycle phase [10]. In NHEJ, DSBs are recognized by Ku70–Ku80 heterodimers leading to activation of DNAPKcs, which then stabilize DSBs through phosphorylation of the repair protein Artemis, the histone variant H2AX, and by recruitment of XRCC4/LIG4 ligase complex for religation of the broken ends with the help of the stimulatory factor XLF [7] For repair by HR in S-phase, DSBs are detected by the MRE11-RAD50-NBS1 (MRN) complex, which promotes the activation of ATM by autophosphorylation [11]. This is followed by the rapid phosphorylation by ATM of various DNA repair factors like H2AX, CtIP, BRCA1 and exonuclease EXO1 [9]. The phosphorylation cascade forms chromatin domains flanking the DSB site that contain phosphorylated histone H2AX (γH2AX), which is bound by mediator protein MDC1 with high affinity. MDC1 in turn triggers the recruitment of chromatin remodeling- and modification-complexes, which allow the association of downstream factors, such as 53BP1 and BRCA1. 53BP1 is an inhibitor of BRCA1 accumulation at DSB sites in the G1 phase of the cell cycle and promotes NHEJ, whereas BRCA1 promotes end resection and HR [12–14]. Single-stranded DNA generated by resection due to the activities of MRE11, CtIP, EXO-1 and BRCA 1, is rapidly coated by replication protein A (RPA) and is subsequently replaced by RAD51 in the presence of BRCA2 [9]. The single-strand ends bound by repair proteins can subsequently invade the homologous template to prime DNA synthesis, which copies and restores genetic information disrupted by the DSB. Although ATM is the primary responder to DSBs in S-phase, recent evidence suggests that ATR, which responds to ssDNA and stalled replication forks, is also activated upon ionizing radiation (IR)-induced DSBs in a cell-cycle dependent manner. ATR activation in response to DSB occurs during S- and G2-phase of the cell cycle and requires ATM, MRN and CtIP [15,16].
In response to damage and activation of the DDR, cells may undergo cell cycle arrest until repair is complete or, if the damage is irreparable, cells undergo apoptosis or move into senescence. While the primary response to DNA damage is very fast and mediated through posttranslational modifications, such as phosphorylation by kinases of the PIKKs family, the decision to induce cell-cycle arrest or apoptosis is mediated through the slower transcriptional responses largely mediated by p53, which is regulated by ATM and CHK2 in response to DSBs [17]. Following DSBs, p53 is activated by the ATM kinase through a transcriptional circuit involving the WIP1 phosphatase and the MDM2 E3 ubiquitin ligase, which are induced by p53 and negatively regulate ATM and p53, respectively [18].
The DNA repair process manifests itself in the form of stable cytological structures called DNA-repair foci, generated by recruitment and accumulation of DNA-repair factors at the site of DNA damage [19,20]. These foci are intrinsically dynamic in nature, formed by the continuous exchange of DNA-repair factors between the chromatin-bound pool and the freely diffusing nucleoplasmic pool at the site of DSBs [19,20]. Details of the structural organization of repair foci have not been elucidated, but some of the factors involved in the formation of foci are the sensor complex MRN, the DDR mediator MDC1, ATM kinase and the downstream factors γ-H2AX, 53BP1 and BRCA1. It is not known whether the accumulation of repair factors in foci is essential for efficient repair or is merely a byproduct of the repair process, but it is possible that they play a key role in the repair process by concentrating essential factors and/or by keeping the broken ends in spatial proximity, enhancing the efficiency of repair [19]. Chromatin modifications and remodeling events, most prominently the phosphorylation of H2AX, around the DNA lesions are thought to contribute to the fine-tuning of damage signaling and repair [19,21–23]. In order to facilitate repair and checkpoint signaling, chromatin undergoes rapid local and global decondensation in response to DNA damage [24,25]. Apart from phosphorylation of the C-terminal tail of H2AX, several modifications of multiple core histones through acetylation, methylation, ubiquitination and phosphorylation, have been linked to various aspects of DNA damage and repair [19,22,26].
2. Noncoding RNAs (ncRNAs)
During the last decade our understanding of genome organization has significantly increased and it has been recognized that large stretches of once assumed non-transcribed intergenic regions in fact code for a large number of noncoding transcripts [27–30]. Noncoding RNAs are generally defined as RNA species that do not have protein coding potential. With the exception of ncRNAs such as ribosomal RNAs (rRNAs), small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), and transfer RNAs (tRNAs), the remaining ncRNAs are arbitrarily grouped into short (< 200 nt) and long (> 200 nt) ncRNAs. Short RNAs can be further subdivided into microRNAs (miRNAs), piwi interacting RNAs (piRNAs) and small interfering RNAs (siRNAs). Short and Long ncRNAs differ in their origin, processing and mode of action. Increasing evidence suggests that various ncRNAs play a pivotal role in DDR. This was not unexpected, as changes in transcription and chromatin structure that are an integral part of DDR are also modulated by ncRNAs [31–33]. This review highlights the emerging roles of ncRNAs in DDR.
2 (a) Micro RNAs
miRNAs are short (~19–24 nt), single-stranded, ncRNAs, that regulate gene expression at the post-transcriptional level either by cleavage of target mRNA or by repressing translation [34,35]. miRNAs probably contribute to the regulation of most major gene pathways as more than half of the human transcriptome is predicted to be under miRNA regulation [36,37]. miRNAs biogenesis and maturation is a complex multi-step process and initiates with their transcription by RNA polymerase II into primary miRNA (pri-miRNAs) from intergenic or intronic/exonic loci, often during transcription of their host genes. The pri-miRNAs are then cleaved in the nucleus by the DROSHA-DGCR8 microprocessor to generate approximately 70-nt long hairpin-shaped precursors called pre-miRNAs [38]. The transport of pre-miRNAs from the nucleus to the cytoplasm is mediated by exportin-5, a RanGTP-binding nuclear transporter [39,40]. In the cytoplasm, the RNAse III-like enzyme DICER and TARBP2 (TAR binding protein 2) cleaves pre-miRNAs into a transient duplex of around 20–24 nt in size made up of the functional miRNA strand and the passenger strand [39,40]. The mature miRNA binds to Argonaute (Ago) proteins to form an miRNA-induced silencing complex termed RISC, which mediates gene silencing by mRNA degradation or translation inhibition [41,42]. Target recognition by miRNA generally depends on base-pairing between miRNA seed sequence (nt 2–8 at the 5′ end of miRNA) and sequences in the 3′ UTR of the target mRNA. The choice of gene silencing by mRNA degradation or translation inhibition appears to be determined by degree and nature of complementary sites between miRNA and the mRNA target [37,43–45]. Interestingly, it has been recently shown that translational inhibition precedes mRNA degradation and is necessary for mRNA degradation by miRNAs [46]. Expression and biogenesis of several miRNAs is affected by DNA damage whereas, in turn, some miRNAs regulate DNA repair factors (Fig. 1).
Figure 1.
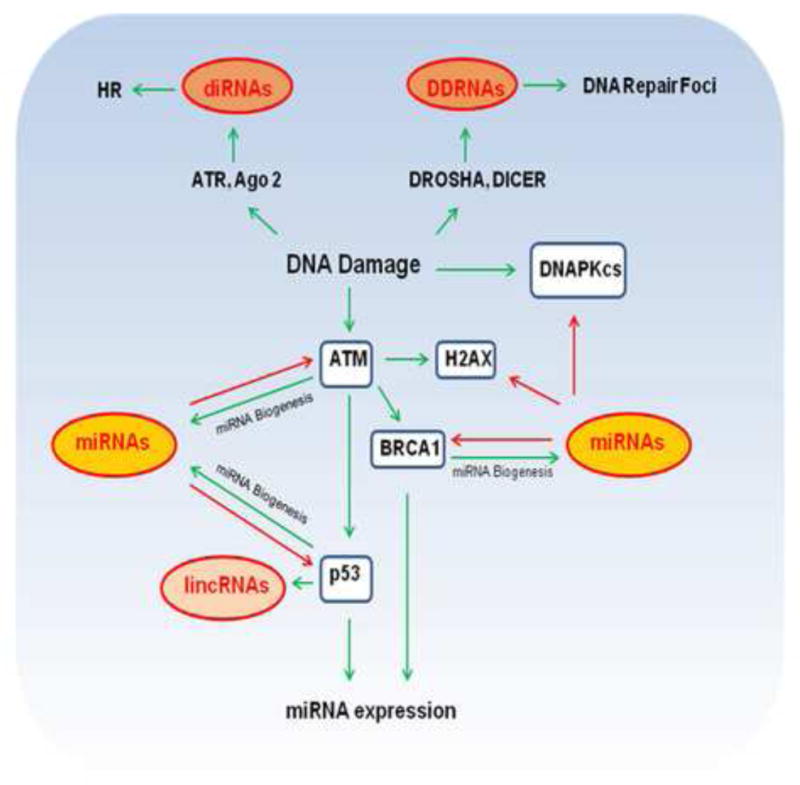
The complex interplay of ncRNAs and DNA Damage. Activation of the DNA Damage Response (DDR) induces multiple kinds of noncoding RNAs (ncRNAs) such as microRNAs (miRNAs), DROSHA and DICER dependent (ddRNAs), DSB-induced small RNAS (diRNAs), and long intergenic noncoding RNAs (lincRNAs). ATM, BRCA1 and p53 are involved in regulation of miRNA biogenesis. P53 and BRCA1 also regulate miRNA expression. Misregulated miRNAs expression such as in cancer may also contribute to direct inhibition of key proteins in DDR such as ATM, DNAPKcs, H2AX, BRCA1 and p53. DDRNAs produced from sites of DNA damage contribute to formation of DNA repair foci. diRNAs contribute to (Homologous Recombination) HR mediated repair. p53 also induces expression of lincRNAs which modulate gene expression to regulate cell cycle and apoptosis. Activating effects (green), inhibitory effects (red).
2 (b) Long noncoding RNAs
Long noncoding RNAs (lncRNAs) are defined as RNA species longer than 200 nt that do not appear to have protein coding potential [33]. Use of tiling arrays have revealed that a vast majority of the genome is transcribed and transcription is not limited to protein-coding regions, but is instead pervasive and the genome probably codes for as many, if not more, lncRNAs genes as protein coding genes [47–50]. lncRNAs have been shown to play functional roles in numerous biological processes ranging from cell cycle control, pluripotency, differentiation, to disease. They can act as both cis- and trans-regulators of gene activity and as modulators of the epigenome [33,51–53]. lncRNAs have been classified on the basis of their position with respect to protein coding genes : (i) antisense lncRNAs arise from the antisense strand of known protein-coding genes, (ii) intronic lncRNAs are encoded within introns of protein-coding genes, (iii) long intergenic noncoding RNAs (lincRNAs) are transcribed from intergenic regions between protein coding genes, and (iv) bidirectional lncRNAs that initiate in the opposite direction from the promoter of a protein coding gene [33]. The molecular mechanisms by which lncRNAs exert their function is not fully understood, but emerging studies indicate that lncRNAs associate with proteins in chromatin or ribonucleoprotein complexes and act as signals, decoys, guides or scaffolds for these protein complexes to execute their activities in the genome [33,54]. Some examples of lncRNA expression in response to DNA damage have recently emerged (Fig. 1), and will be discussed below.
3. miRNAs in DNA damage
3 (a). miRNAs expression upon DNA damage
A role for miRNAs in the cellular response to DNA damage is supported by the fact that knockdown of key miRNA processing proteins, particularly DICER or Ago2, reduces survival and checkpoint response after UV damage and is accompanied by cell-cycle-dependent relocalization of Ago2 into cellular stress granules [55]. In addition, a large number of miRNAs are transcriptionally induced upon DNA damage and the level of induction is variable depending on cell type and the nature and the intensity of DNA damage [55–63].
The transcription of a subset of miRNAs in the DDR is dependent on the tumor suppressor p53, which is a well-studied transcription factor that is activated upon DNA damage and induces growth arrest or apoptosis. The study of miRNA expression profiles in wild-type and p53 null cells revealed that expression of three miRNAs, miRNA-34a, miRNA-34b and miRNA-34c, is precisely correlated with p53 status [64]. Over-expression of miRNA-34a induces apoptosis and G1 arrest [64–66]. miRNA-34c overexpression suppresses c-Myc expression and is thought to prevent inappropriate replication which may promote genomic instability [67]. Induction of miR-34c also occurs independently of p53 via the alternative ATM dependent p38MAPK signaling pathway [67]. Apart from the miR-34 family, a p53-dependent up regulation of miRNA-192, miRNA-194 and miRNA-215 upon genotoxic stress also induces cell cycle arrest [68–71]. Conversely, p53-mediated transcriptional repression of miRNAs has also been reported. In cells subjected to hypoxia, levels of miRNA-17-92 cluster were reduced in cells containing wild-type p53, but were unchanged in p53-deficient cells [72] and over expression of the miRNA-17-92 cluster inhibits hypoxia-induced apoptosis [72]. Upon irradiation, a decrease in expression of the let-7a and let-7b family of miRNA is observed in human and mice cells [73]. This decrease in expression of let-7a and let-7b miRNAs is dependent on p53 and ATM [73]. These studies suggest that regulation of miRNA expression by p53 modulates the cellular response to DNA damage via regulation of cell cycle checkpoints and apoptosis. In fact, promoters of a number of components of the miRNA processing machinery, including Dicer, contain p53 response elements, suggesting that they could be direct transcriptional targets of p53 [74]. P53-induced miRNAs also play regulatory roles in other cancer-associated processes such as tumorigenesis, metastasis, epithelial-mesenchymal transition and metabolic adaptation [75,76]. However, it is currently not clear whether p53-induced miRNAs actually control any genes directly involved in DNA repair. Multiple physiological stress conditions such as oncogenic stress can activate p53-mediated transcriptional programs, but miRNA expression profiles in response to direct activation of p53 by DNA damage have not been studied in detail. More studies are required to specifically identify miRNAs induced by direct p53 activation upon DNA damage and to elucidate the role of these miRNAs in regulating proteins important in DDR. In addition to p53, other proteins in the DNA repair pathway also regulate miRNA expression. Besides its role in miRNA biogenesis, BRCA1 represses transcription of miRNA-155 [77]. BRCA1 epigenetically represses miRNA-155 expression via its association with the histone deacetylase HDAC2, which in turn deacetylates histones H2A and H3 on the miR-155 promoter [77].
3 (b). Regulation of miRNA biogenesis by DDR factors
The key DNA repair factors ATM and BRCA1 have been shown to modulate miRNA biogenesis by phosphorylating and interacting with components of the DROSHA microprocessor complex [78,79]. About 25% of the miRNAs induced upon DNA damage depend on ATM for upregulation [79] and ATM specifically regulates processing and biogenesis of these miRNAs by phosphorylating splicing regulatory protein KSRP without affecting their transcription. KSRP is a component of both the DROSHA and DICER complexes and has been previously shown to promote biogenesis of a subset of miRNAs [80]. KSRP phosphorylation by ATM leads to enhanced interaction between KSRP and terminal loops of pri-miRNAs which in turn allows for increased recruitment of these pri-miRNAs for processing by DROSHA and DICER[81].
BRCA1 also regulates miRNA biogenesis (Fig. 1). However, unlike ATM, BRCA1 directly binds to both specific pri-miRNAs and DROSHA [78]. BRCA1 binds to specific pri-miRNAs via its DNA-binding domain due to its ability to recognize a stem-loop in the secondary structure of pri-miRNAs [78]. More studies are required to understand how regulation of miRNA biogenesis by ATM and BRCA1 contributes to maintenance of genomic stability.
p53 also facilitates the processing of specific pri-miRNAs into pre-miRNAs independently of transcription by associating with DDX5, a component of the DROSHA/DGCR 8 microprocessor complex [82]. This association leads to an increase in the levels of the mature miRNAs, such as miR-16-1, miR-143 and miR-145 [82]. Use of computational approaches to identify molecules that regulate miRNA processing also suggest that p53 and its related family members p63 and p73 regulate components of miRNA processing [74].
3 (c). miRNA regulation of proteins involved in DDR
While some DDR proteins appear to regulate miRNA expression, miRNAs in turn also influence DDR protein expression (Fig. 1). Key DNA repair proteins such as ATM, H2AX and BRCA1 are subjected to direct inhibition by miRNAs. ATM is targeted by miRNA-421, miRNA-18a, miRNA 26b, miRNA-101, miRNA-181 and miRNA100 [83–88]. miRNA-421 suppresses ATM expression by targeting the 3′ UTR of the ATM transcript [83]. Ectopic expression of miR-421 in cells results in increased sensitivity to IR [83,89] and over expression of other miRNAs that target ATM also reduces ATM expression, alters cell cycle checkpoints and leads to hypersensitivity to IR. Interestingly, apart from ATM, miRNA-101 also inhibits DNA-PKcs via binding to the 3′- UTR of DNA-PKcs transcripts [87]. These observations suggest a feedback loop between miRNAs and ATM (Fig. 1).
H2AX, which plays a key role in DNA damage signaling via phosphorylation of its C-terminus, is a target of miRNA-24 [90]. Up-regulation of miRNA-24 in post-replicative cells reduces H2AX and thereby renders cells highly vulnerable to DNA damage [90]. Screening of a library of human miRNA-mimics in osteosarcoma cells revealed several miRNAs that inhibit γH2AX foci formation [91]. Among them, miR-138 was shown to directly target the histone H2AX 3′-untranslated region, to reduce histone H2AX expression, and to induce chromosomal instability after DNA damage [91].
BRCA1 is an important player in homologous recombination and also regulates miRNA processing. BRCA1 is a target of miRNA-182 [92]. Down regulation of miRNA-182 increases BRCA1 protein levels and protects cells from IR-induced cell death [92]. Consistent with this, overexpression of miRNA-182 reduces BRCA1 protein levels, impairs homologous recombination-mediated repair, and renders cells hypersensitive to IR [92]. Pull-down experiments with synthetic miRNA indicate that apart from BRCA1, miRNA-182 also targets a set of other genes involved in the DDR pathway [93]. Interestingly, miRNA-96, which is expressed as a polycistronic transcript with miRNA-182, targets RAD51, which, together with BRCA1, is involved in homologous recombination [94]. miRNA-146a and miRNA-146-5p bind to the same site in the 3′UTR of BRCA1 and down-regulate its expression [95]. In breast tumors, levels of these miRNAs are inversely correlated with that of the BRCA1 protein and these miRNAs are overexpressed in triple negative breast cancers, the most common type of breast cancer in women with BRCA1 mutations [95]. miRNA-1, a candidate prognostic marker of prostate cancer and miRNA-1245, a c-myc induced miRNA, also regulate DNA repair by targeting BRCA-1 and BRCA-2, respectively [96,97]. Interestingly, it has been shown that overexpression of miR-99a and miR-100, which target SNF2H, a SWI/SNF chromatin remodeling factor, leads to reduced localization of BRCA1 and RAD51 to sites of DNA damage [98], suggesting that miRNA regulation occurs at many steps in the DNA repair and signaling cascade.
Several miRNAs including miR-125b, miR-504, miR-33, miR-380-5p, miR-1285, miR-30d and miR-25 have also been shown to down regulate p53 in a context-dependent manner [99–104]. Ectopic expression of these miRNAs induced phenotypes that are associated with the loss of p53.
CU1276 is a miRNA derived from tRNA which was first identified during screening of miRNA expression in human B cells [105,106]. CU1276 is derived from tRNA, by DICER dependent biogenesis and associates with Ago proteins, it has been shown to represses in a sequence-specific manner transcripts of RPA1, which is a key gene in DNA replication and repair [106].
Finally, using computational approaches miRNA binding sites have been found in several DSB sensors with long 3′ UTRs such as NBS1 and Ku80, and they have been predicted to be regulated by miRNAs, but these predictions have not yet been validated experimentally [107,108].
4. Non canonical small RNAs in DNA damage response
DNA damage also induces DROSHA and DICER dependent small RNAs called DDRNAs (Fig. 1), which are distinct from the canonical miRNAs [109]. These transcripts are produced from sequences transcribed from the damaged site and control DDR foci formation in cultured human and mouse cells and in zebrafish [109]. In support of an active role of DDRNAs in DDR, transient inactivation of DICER or DROSHA in human cells exposed to IR impaired formation of pATM, 53BP1, and MDC1 foci, but not γ-H2AX foci, without decreasing the level of these proteins, suggesting that DICER and DROSHA RNA products control DDR activation and act independently from canonical miRNA-mediated repression of DDR factors [109]. Moreover, RNase A treatment reduces 53BP1, pATM, and MDC1 foci formation in DNA damaged cells [109]. Interestingly, it has been previously shown that 53BP1 associates with RNA and that RNase A treatment dissociates 53BP1 from IR-induced foci [110]. Furthermore, RNase A inhibition or addition of exogenous total RNA purified from the same cells, but not tRNA, can rapidly restore DDR foci formation in DNA damaged cells treated with RNase A [109]. Restoration of DDR foci upon RNase A inhibition in RNase-treated cells is prevented by α-amanitin, suggesting that DDR foci stability requires RNA polymerase II-dependent transcription [109]. Use of a site-specific chromosomally integrated DNA damage reporter system [111] and deep sequencing indicates that DDRNAs originate from the damaged genomic locus [109].
The production of small RNA species from near the site of DSB has also been described in Arabidopsis thaliana and these RNAs have been termed DSB-induced small RNAs (diRNAs) (Fig. 1) [112]. These diRNAs require the PI3 kinase ATR, RNA polymerase IV, and DICER-like (DCL) proteins for their biogenesis and they are recruited by AGO2 to mediate DSB repair [112]. Interestingly, while diRNAs were generated from sites in the immediate vicinity of the DNA break in Arabidopsis, deep sequencing in human cells revealed that diRNAs are generated from sense- and antisense-strands within a 5kb region of the damage site [112]. These diRNAs appear to regulate HR mediated repair of DSBs in Arabidopsis and humans [112].
Induction of small RNAs upon DNA damage seems to be conserved across species, such as the production small RNAs termed qiRNAs, from the rDNA locus in response to DNA damage in the fungus Neurospora crassa[113]. The exact function of these qiRNAs is unclear but Neurospora strains with mutations in any proteins involved in qiRNA biogenesis show heightened sensitivity to DNA damage [113]. In Drosophila, analogous to DDRNAs and diRNAs, transfection of linearised plasmid DNA mimicking DSB ends elicits induction of small RNAs known as endo-siRNAs [114]. This response is specific to DSBs, depends on Drosophila endo-siRNA factors such as Dcr-2, and has the capacity to silence transcripts with homologous sequence in trans[114].
Chowdhury et al. have speculated on the possible roles these small ncRNAs could serve in DNA repair. They may act (i) as a template for DNA polymerase to fill in for resected DNA in HR, (ii) as guides for recruiting DNA repair factors or chromatin modifying complexes at DSBs, (iii) in siRNA pathways to degrade nascent RNA from the damaged loci to prevent its aberrant expression or (iv) the ncRNA and Ago complex may act as scaffold for maintaining repair foci [107]. Overall these studies suggest that small RNAs generated from the site of DSB or the regions flanking a DSB are important for DSB repair. The precise mechanisms of action and function of these ncRNAs remain unclear, additional studies dissecting the exact molecular and biochemical function of these unique classes of small RNAs in DSB repair are required.
5. Long noncoding RNA in response to genotoxic stress
The first indication that lncRNAs are induced in response to DNA damage was the identification of noncoding, > 200nt, low copy number, pol II-regulated, polyadenylated, uncapped transcripts generated upstream of the CCND1 promoter in response to DNA damage [115]. These transcripts were shown to bind to TLS, an RNA-binding protein that has been suggested to play roles in DNA repair and is an inhibitor of histone acetyl transferase CBP/p300 [115,116]. Upon upregulation by DNA damage, these ncRNAs bind to TLS to activate it and promote its interaction with CBP/p300 to cause repression of CCND1 transcription, a cell cycle regulator [115].
Subsequently several p53-dependent lincRNAs induced upon genotoxic stress were identified [117]. One of these, lincRNAp21, has been shown to play an important role in p53-dependent gene repression [117]. LincRNAp21 is located 15Kb upstream of the gene encoding the cell cycle regulator p21. The p53-dependent transcriptional repression by lincRNA-p21 is mediated through its physical association with the transcription- and RNA processing-factor hnRNP-K. This interaction is required for proper genomic localization of hnRNP-K at p53-repressed genes [117]. Interestingly, another lncRNA TUG1 is also induced by p53, binds to PRC2, and has a role in repressing specific genes involved in cell-cycle regulation[29].
To identify functional ncRNAs in the promoter region of 56 human cell-cycle genes, Hung et al. used high resolution tiling arrays to probe polyadenylated transcripts in response to diverse perturbations including DNA damage [118]. They identified one lncRNA, named PANDA (P21 associated ncRNA DNA damage activated), which is induced in a p53-dependent manner. Similar to lincRNAp21, PANDA is located 5kb upstream of the cell cycle regulator p21. PANDA interacts with the transcription factor NF-YA to impede induction of pro-apoptotic genes by NF-YA and PANDA knockdown sensitizes cells to DNA-damage induced apoptosis [118].
Finally, in response to genotoxic agents such as the DSBs-inducers mitomycin C or the topoisomerase II inhibitor doxorubicin, mammalian cells induce distinct nuclear long ncRNAs [119]. These lncRNAs are not likely to be transcriptional noise as they are ubiquitously expressed in various human tissues. Specific functions for these lncRNAs, however, remain unknown [119].
Given the fact that DNA repair factors like 53BP1, KU80 and BRCA1 associate with RNA [110,120,121], certain RNA binding proteins like RBMX and hRNPU are recruited to sites of DSBs [122,123] and association of RNAs such as telomeric repeat-containing RNA TERRA with DNA repair proteins [124,125], it is likely that several of the DNA damage-induced lncRNAs play a role in DDR. As of now, information about the function of lncRNAs in DNA damage is only available for lincRNAp21 and PANDA. lncRNAs may function in various ways in DDR pathway such as (i) by acting as guides or signals for recruitment of repair proteins or chromatin modifying complexes to sites of DNA damage, (ii) acting as scaffolds for DNA repair proteins or the chromatin remodeling machinery at the site of DNA repair foci, (iii) lncRNA may prevent the action of negative regulators of DNA repair at the site of DNA damage by acting as decoys, or (iv) lncRNAs may act as regulators of DNA damage sensitive gene expression programs like lincRNAp21 and PANDA.
6. Conclusions and future perspectives
DNA damage leads to the induction of several ncRNA species (Fig. 1). The majority of studies on ncRNAs in the DNA damage response have so far focused on the role of miRNAs. It is evident that miRNA induction after DNA damage modulates cell cycle progression and alters the sensitivity of cells to DNA damage by targeting downstream gene expression. Since different cell types activate non-overlapping sets of miRNAs upon DNA damage and the miRNA response varies depending on the nature of the DNA damaging agent, it is tempting to speculate that different miRNAs play distinct roles in different repair pathways. The ability of miRNAs to repress key DDR factors has been demonstrated, however, most of these studies have been done in cancer lines and it is not evident how miRNAs contribute to DNA repair in normal cells and whether these responses reflect cancer-specific pathways. It will also be important to examine whether molecules like 53BP1, NBS1 and Ku 80, which have been predicted to have miRNA binding sites, are actually regulated by miRNAs in vivo. Further studies are also required to delineate the role of miRNAs in different repair pathways such as BER and NER. Considering the fact that many miRNAs are late responders in DDR, it is also probable that miRNAs play a role in repressing DDR proteins after completion of repair.
The recent discovery of DDRNAs and diRNAs [109,112] has raised several intriguing questions. To start with, the exact origin of these small RNAs remains unclear. Francia et al. suggest that they arise directly from the site of DSBs [109] whereas Wei et al. suggest that small RNAs are produced from regions around the DSBs [112]. It remains to be seen what is the reason for these differences. Furthermore, the observed requirement for RNA polymerase II-dependent transcription for DDR foci stability needs to be reconciled with the notion that DNA damage inhibits transcription [126,127]. These observations also raise the important question of whether DNA repair foci formation mechanisms are different in transcriptionally silent heterochromatin regions compared to transcriptionally active euchromatin. It is also not clear whether these non-canonical small ncRNAs are induced by other kinds of DNA damage in addition to DSB. Furthermore, it would be important to study the localization of these small ncRNAs by FISH.
In addition to the miRNAs and other short ncRNAs, numerous lncRNAs have been discovered, which are induced upon DNA damage. However, for most of these lncRNAs there is no experimental evidence available to indicate a functional role in DDR and further studies are required to evaluate their role in DDR. Since it is now believed that a large number of intergenic regions are transcribed into lncRNAs, it is possible that lncRNAs may be the RNA source for DICER and DROSHA processing of short RNAs required for DNA repair upon DNA damage at these loci.
Efforts are still needed to identify and further characterize additional ncRNA species involved in DDR. Use of RNA Immunoprecipitation followed by high-throughput sequencing (RIP-seq) to characterize RNA binding properties of repair proteins and chromatin complexes important in DNA repair should be a powerful approach to achieve this goal. The study of ncRNA function in the cellular response to different types of DNA damage is still in its infancy. But it is already evident that ncRNAs are important players in maintaining genomic stability. Given the complexity suggested by the few identified players to date, it is likely that many additional DNA damage-relevant ncRNAs with various functions will be identified in the near future and will increase our understanding of mechanisms of maintaining genome stability.
Acknowledgments
VS is supported by an NIH Khorana-Nirenberg Fellowship. Work in the Misteli laboratory is supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The authors would like to thank Misteli laboratory members for helpful feedback on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–46. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 5.Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000;65:127–33. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 6.Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–85. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 7.Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 8.Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619–31. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 9.San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–57. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 10.Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- 11.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–21. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bunting SF, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Escribano-Diaz C, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872–83. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 2013;339:700–4. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM-and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 16.Sartori AA, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–14. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–9. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 18.Batchelor E, Loewer A, Lahav G. The ups and downs of p53: understanding protein dynamics in single cells. Nat Rev Cancer. 2009;9:371–7. doi: 10.1038/nrc2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Misteli T, Soutoglou E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat Rev Mol Cell Biol. 2009;10:243–54. doi: 10.1038/nrm2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lukas C, Bartek J, Lukas J. Imaging of protein movement induced by chromosomal breakage: tiny ‘local’ lesions pose great ‘global’ challenges. Chromosoma. 2005;114:146–54. doi: 10.1007/s00412-005-0011-y. [DOI] [PubMed] [Google Scholar]
- 21.Downs JA, Nussenzweig MC, Nussenzweig A. Chromatin dynamics and the preservation of genetic information. Nature. 2007;447:951–8. doi: 10.1038/nature05980. [DOI] [PubMed] [Google Scholar]
- 22.Soria G, Polo SE, Almouzni G. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell. 2012;46:722–34. doi: 10.1016/j.molcel.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 23.Dinant C, Houtsmuller AB, Vermeulen W. Chromatin structure and DNA damage repair. Epigenetics Chromatin. 2008;1:9. doi: 10.1186/1756-8935-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Muller WG, McNally JG, Bazett-Jones DP, Nussenzweig A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol. 2006;172:823–34. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ziv Y, et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–6. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 26.Jackson SP, Durocher D. Regulation of DNA Damage Responses by Ubiquitin and SUMO. Mol Cell. 2013;49:795–807. doi: 10.1016/j.molcel.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 27.Alexander RP, Fang G, Rozowsky J, Snyder M, Gerstein MB. Annotating non-coding regions of the genome. Nat Rev Genet. 2010;11:559–71. doi: 10.1038/nrg2814. [DOI] [PubMed] [Google Scholar]
- 28.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 29.Khalil AM, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci ULS A. 2009;106:11667–72. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–27. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lejeune E, Allshire RC. Common ground: small RNA programming and chromatin modifications. Curr Opin Cell Biol. 2011;23:258–65. doi: 10.1016/j.ceb.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 32.Magistri M, Faghihi MA, St Laurent G, 3rd, Wahlestedt C. Regulation of chromatin structure by long noncoding RNAs: focus on natural antisense transcripts. Trends Genet. 2012;28:389–96. doi: 10.1016/j.tig.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 35.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–34. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 36.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012;13:271–82. doi: 10.1038/nrg3162. [DOI] [PubMed] [Google Scholar]
- 38.Lee Y, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–9. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 39.Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–8. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 40.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011–6. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rand TA, Petersen S, Du F, Wang X. Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell. 2005;123:621–9. doi: 10.1016/j.cell.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 42.Diederichs S, Haber DA. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell. 2007;131:1097–108. doi: 10.1016/j.cell.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 43.Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol. 2012;19:586–93. doi: 10.1038/nsmb.2296. [DOI] [PubMed] [Google Scholar]
- 44.Czech B, Hannon GJ. Small RNA sorting: matchmaking for Argonautes. Nat Rev Genet. 2011;12:19–31. doi: 10.1038/nrg2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 46.Meijer HA, et al. Translational Repression and eIF4A2 Activity Are Critical for MicroRNA-Mediated Gene Regulation. Science. 2013;340:82–5. doi: 10.1126/science.1231197. [DOI] [PubMed] [Google Scholar]
- 47.Bertone P, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–6. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- 48.Carninci P, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–63. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 49.Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, Fodor SP, Gingeras TR. Large-scale transcriptional activity in chromosomes 21 and 22. Science. 2002;296:916–9. doi: 10.1126/science.1068597. [DOI] [PubMed] [Google Scholar]
- 50.Birney E, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23:1494–504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsai MC, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–93. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flynn RA, Chang HY. Active chromatin and noncoding RNAs: an intimate relationship. Curr Opin Genet Dev. 2012;22:172–8. doi: 10.1016/j.gde.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–14. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pothof J, et al. MicroRNA-mediated gene silencing modulates the UV-induced DNA-damage response. EMBO J. 2009;28:2090–9. doi: 10.1038/emboj.2009.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Josson S, Sung SY, Lao K, Chung LW, Johnstone PA. Radiation modulation of microRNA in prostate cancer cell lines. Prostate. 2008;68:1599–606. doi: 10.1002/pros.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maes OC, An J, Sarojini H, Wu H, Wang E. Changes in MicroRNA expression patterns in human fibroblasts after low-LET radiation. J Cell Biochem. 2008;105:824–34. doi: 10.1002/jcb.21878. [DOI] [PubMed] [Google Scholar]
- 58.Templin T, Paul S, Amundson SA, Young EF, Barker CA, Wolden SL, Smilenov LB. Radiation-induced micro-RNA expression changes in peripheral blood cells of radiotherapy patients. Int J Radiat Oncol Biol Phys. 2011;80:549–57. doi: 10.1016/j.ijrobp.2010.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cha HJ, et al. Identification of ionizing radiation-responsive microRNAs in the IM9 human B lymphoblastic cell line. Int J Oncol. 2009;34:1661–8. doi: 10.3892/ijo_00000297. [DOI] [PubMed] [Google Scholar]
- 60.Wagner-Ecker M, Schwager C, Wirkner U, Abdollahi A, Huber PE. MicroRNA expression after ionizing radiation in human endothelial cells. Radiat Oncol. 2010;5:25. doi: 10.1186/1748-717X-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simone NL, et al. Ionizing radiation-induced oxidative stress alters miRNA expression. PLoS One. 2009;4:e6377. doi: 10.1371/journal.pone.0006377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu Y, Lu X. Non-coding RNAs in DNA damage response. Am J Cancer Res. 2012;2:658–75. [PMC free article] [PubMed] [Google Scholar]
- 63.Metheetrairut C, Slack FJ. MicroRNAs in the ionizing radiation response and in radiotherapy. Curr Opin Genet Dev. 2013 doi: 10.1016/j.gde.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He L, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, Meister G, Hermeking H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–93. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- 66.Chang TC, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–52. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cannell IG, et al. p38 MAPK/MK2-mediated induction of miR-34c following DNA damage prevents Myc-dependent DNA replication. Proc Natl Acad Sci U S A. 2010;107:5375–80. doi: 10.1073/pnas.0910015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Braun CJ, et al. p53-Responsive micrornas 192 and 215 are capable of inducing cell cycle arrest. Cancer Res. 2008;68:10094–104. doi: 10.1158/0008-5472.CAN-08-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Georges SA, et al. Coordinated regulation of cell cycle transcripts by p53-Inducible microRNAs, miR-192 and miR-215. Cancer Res. 2008;68:10105–12. doi: 10.1158/0008-5472.CAN-08-1846. [DOI] [PubMed] [Google Scholar]
- 70.He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network--another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7:819–22. doi: 10.1038/nrc2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pichiorri F, et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell. 2010;18:367–81. doi: 10.1016/j.ccr.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72.Yan HL, et al. Repression of the miR-17–92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. 2009;28:2719–32. doi: 10.1038/emboj.2009.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saleh AD, et al. Cellular stress induced alterations in microRNA let-7a and let-7b expression are dependent on p53. PLoS One. 2011;6:e24429. doi: 10.1371/journal.pone.0024429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boominathan L. The tumor suppressors p53, p63, and p73 are regulators of microRNA processing complex. PLoS One. 2010;5:e10615. doi: 10.1371/journal.pone.0010615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hermeking H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer. 2012;12:613–26. doi: 10.1038/nrc3318. [DOI] [PubMed] [Google Scholar]
- 76.Landau DA, Slack FJ. MicroRNAs in mutagenesis, genomic instability, and DNA repair. Semin Oncol. 2011;38:743–51. doi: 10.1053/j.seminoncol.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang S, et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat Med. 2011:17, 1275–82. doi: 10.1038/nm.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kawai S, Amano A. BRCA1 regulates microRNA biogenesis via the DROSHA microprocessor complex. J Cell Biol. 2012;197:201–8. doi: 10.1083/jcb.201110008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang X, Wan G, Berger FG, He X, Lu X. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol Cell. 2011;41:371–83. doi: 10.1016/j.molcel.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A, Gherzi R, Rosenfeld MG. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature. 2009;459:1010–4. doi: 10.1038/nature08025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han C, Wan G, Langley RR, Zhang X, Lu X. Crosstalk between the DNA damage response pathway and microRNAs. Cell Mol Life Sci. 2012;69:2895–906. doi: 10.1007/s00018-012-0959-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–33. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 83.Hu H, Du L, Nagabayashi G, Seeger RC, Gatti RA. ATM is down-regulated by N-Myc-regulated microRNA-421. Proc Natl Acad Sci U S A. 2010;107:1506–11. doi: 10.1073/pnas.0907763107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ng WL, Yan D, Zhang X, Mo YY, Wang Y. Over-expression of miR-100 is responsible for the low-expression of ATM in the human glioma cell line: M059J. DNA Repair (Amst) 2010;9:1170–5. doi: 10.1016/j.dnarep.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 85.Song L, Lin C, Wu Z, Gong H, Zeng Y, Wu J, Li M, Li J. miR-18a impairs DNA damage response through downregulation of ataxia telangiectasia mutated (ATM) kinase. PLoS One. 2011;6:e25454. doi: 10.1371/journal.pone.0025454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu CW, Dong YJ, Liang QY, He XQ, Ng SS, Chan FK, Sung JJ, Yu J. MicroRNA-18a Attenuates DNA Damage Repair through Suppressing the Expression of Ataxia Telangiectasia Mutated in Colorectal Cancer. PLoS One. 2013;8:e57036. doi: 10.1371/journal.pone.0057036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yan D, et al. Targeting DNA-PKcs and ATM with miR-101 sensitizes tumors to radiation. PLoS One. 2010;5:e11397. doi: 10.1371/journal.pone.0011397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang Y, Yu Y, Tsuyada A, Ren X, Wu X, Stubblefield K, Rankin-Gee EK, Wang SE. Transforming growth factor-beta regulates the sphere-initiating stem cell-like feature in breast cancer through miRNA-181 and ATM. Oncogene. 2011;30:1470–80. doi: 10.1038/onc.2010.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hu H, Gatti RA. MicroRNAs: new players in the DNA damage response. J Mol Cell Biol. 2011;3:151–8. doi: 10.1093/jmcb/mjq042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lal A, et al. miR-24-mediated downregulation of H2AX suppresses DNA repair in terminally differentiated blood cells. Nat Struct Mol Biol. 2009;16:492–8. doi: 10.1038/nsmb.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang Y, et al. MicroRNA-138 modulates DNA damage response by repressing histone H2AX expression. Mol Cancer Res. 2011;9:1100–11. doi: 10.1158/1541-7786.MCR-11-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Moskwa P, et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell. 2011;41:210–20. doi: 10.1016/j.molcel.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Krishnan K, et al. MicroRNA-182-5p targets a network of genes involved in DNA repair. RNA. 2013;19:230–42. doi: 10.1261/rna.034926.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Y, Huang JW, Calses P, Kemp CJ, Taniguchi T. MiR-96 downregulates REV1 and RAD51 to promote cellular sensitivity to cisplatin and PARP inhibition. Cancer Res. 2012;72:4037–46. doi: 10.1158/0008-5472.CAN-12-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Garcia AI, et al. Down-regulation of BRCA1 expression by miR-146a and miR-146b-5p in triple negative sporadic breast cancers. EMBO Mol Med. 2011;3:279–90. doi: 10.1002/emmm.201100136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hudson RS, et al. MicroRNA-1 is a candidate tumor suppressor and prognostic marker in human prostate cancer. Nucleic Acids Res. 2012;40:3689–703. doi: 10.1093/nar/gkr1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Song L, et al. Up-regulation of miR-1245 by c-myc targets BRCA2 and impairs DNA repair. J Mol Cell Biol. 2012;4:108–17. doi: 10.1093/jmcb/mjr046. [DOI] [PubMed] [Google Scholar]
- 98.Mueller AC, Sun D, Dutta A. The miR-99 family regulates the DNA damage response through its target SNF2H. Oncogene. 2013;32:1164–72. doi: 10.1038/onc.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Le MT, Teh C, Shyh-Chang N, Xie H, Zhou B, Korzh V, Lodish HF, Lim B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23:862–76. doi: 10.1101/gad.1767609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hu W, et al. Negative regulation of tumor suppressor p53 by microRNA miR-504. Mol Cell. 2010;38:689–99. doi: 10.1016/j.molcel.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Herrera-Merchan A, Cerrato C, Luengo G, Dominguez O, Piris MA, Serrano M, Gonzalez S. miR-33-mediated downregulation of p53 controls hematopoietic stem cell self-renewal. Cell Cycle. 2010;9:3277–85. doi: 10.4161/cc.9.16.12598. [DOI] [PubMed] [Google Scholar]
- 102.Tian S, Huang S, Wu S, Guo W, Li J, He X. MicroRNA-1285 inhibits the expression of p53 by directly targeting its 3′ untranslated region. Biochem Biophys Res Commun. 2010;396:435–9. doi: 10.1016/j.bbrc.2010.04.112. [DOI] [PubMed] [Google Scholar]
- 103.Swarbrick A, et al. miR-380-5p represses p53 to control cellular survival and is associated with poor outcome in MYCN-amplified neuroblastoma. Nat Med. 2010;16:1134–40. doi: 10.1038/nm.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kumar M, Lu Z, Takwi AA, Chen W, Callander NS, Ramos KS, Young KH, Li Y. Negative regulation of the tumor suppressor p53 gene by microRNAs. Oncogene. 2011;30:843–53. doi: 10.1038/onc.2010.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Basso K, et al. Identification of the human mature B cell miRNome. Immunity. 2009;30:744–52. doi: 10.1016/j.immuni.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Maute RL, Schneider C, Sumazin P, Holmes A, Califano A, Basso K, Dalla-Favera R. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:1404–9. doi: 10.1073/pnas.1206761110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chowdhury D, Choi YE, Brault ME. Charity begins at home: non-coding RNA functions in DNA repair. Nat Rev Mol Cell Biol. 2013;14:181–9. doi: 10.1038/nrm3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–17. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 109.Francia S, et al. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature. 2012;488:231–5. doi: 10.1038/nature11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pryde F, Khalili S, Robertson K, Selfridge J, Ritchie AM, Melton DW, Jullien D, Adachi Y. 53BP1 exchanges slowly at the sites of DNA damage and appears to require RNA for its association with chromatin. J Cell Sci. 2005;118:2043–55. doi: 10.1242/jcs.02336. [DOI] [PubMed] [Google Scholar]
- 111.Soutoglou E, Misteli T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science. 2008;320:1507–10. doi: 10.1126/science.1159051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wei W, et al. A role for small RNAs in DNA double-strand break repair. Cell. 2012;149:101–12. doi: 10.1016/j.cell.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 113.Lee HC, Chang SS, Choudhary S, Aalto AP, Maiti M, Bamford DH, Liu Y. qiRNA is a new type of small interfering RNA induced by DNA damage. Nature. 2009;459:274–7. doi: 10.1038/nature08041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Michalik KM, Bottcher R, Forstemann K. A small RNA response at DNA ends in Drosophila. Nucleic Acids Res. 2012;40:9596–603. doi: 10.1093/nar/gks711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang X, et al. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–30. doi: 10.1038/nature06992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kuroda M, et al. Male sterility and enhanced radiation sensitivity in TLS(−/−) mice. EMBO J. 2000;19:453–62. doi: 10.1093/emboj/19.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huarte M, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–19. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hung T, et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011;43:621–9. doi: 10.1038/ng.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mizutani R, et al. Identification and characterization of novel genotoxic stress-inducible nuclear long noncoding RNAs in mammalian cells. PLoS One. 2012;7:e34949. doi: 10.1371/journal.pone.0034949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yoo S, Dynan WS. Characterization of the RNA binding properties of Ku protein. Biochemistry. 1998;37:1336–43. doi: 10.1021/bi972100w. [DOI] [PubMed] [Google Scholar]
- 121.Ganesan S, et al. BRCA1 supports XIST RNA concentration on the inactive X chromosome. Cell. 2002;111:393–405. doi: 10.1016/s0092-8674(02)01052-8. [DOI] [PubMed] [Google Scholar]
- 122.Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat Cell Biol. 2012;14:318–28. doi: 10.1038/ncb2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Polo SE, et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol Cell. 2012;45:505–16. doi: 10.1016/j.molcel.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science. 2007;318:798–801. doi: 10.1126/science.1147182. [DOI] [PubMed] [Google Scholar]
- 125.Schoeftner S, Blasco MA. Chromatin regulation and non-coding RNAs at mammalian telomeres. Semin Cell Dev Biol. 2010;21:186–93. doi: 10.1016/j.semcdb.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 126.Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2010;141:970–81. doi: 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pankotai T, Bonhomme C, Chen D, Soutoglou E. DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat Struct Mol Biol. 2012;19:276–82. doi: 10.1038/nsmb.2224. [DOI] [PubMed] [Google Scholar]