

Abstract
Many “non-metastatic” cancers have spawned undetectable metastases prior to diagnosis. Eventual outgrowth of these microscopic lesions causes metastatic relapse and death, yet the events that dictate when and how micrometastases convert to overt metastases are largely unknown. We report that macrophage stimulating protein (MSP) and its receptor, Ron, are key mediators in conversion of micrometastases to bona fide metastatic lesions through immune suppression. Genetic deletion of Ron tyrosine kinase activity specifically in the host profoundly blocked metastasis. Our data show that loss of Ron function promotes an effective anti-tumor CD8+ T cell response, which specifically inhibits outgrowth of seeded metastatic colonies. Treatment of mice with a Ron-selective kinase inhibitor prevented outgrowth of lung metastasis, even when administered after micrometastatic colonies had already been established. Our findings indicate that Ron inhibitors may hold potential to specifically prevent outgrowth of micrometastases in cancer patients in the adjuvant setting.
Keywords: Ron, MST1R, macrophage stimulating protein, MSP, metastasis, anti-tumor immunity, tumor dormancy
INTRODUCTION
Metastatic tumor growth in secondary organs is the main cause of death from cancer. For example, 20–30% of people diagnosed with Stage II-III breast cancer eventually develop metastatic disease, which typically occurs 3–16 years after the initial diagnosis (1). This clinical “dormancy” period followed by late relapse is also frequently observed in cancers of the prostate, kidney, and thyroid, and in B cell lymphomas and melanoma (2), making this a critical issue in clinical cancer biology. The long latency between excision of the primary tumor and development of clinically-detectable distant metastasis suggests that micro-metastatic tumor cells are already seeded at other sites throughout the body at the time of diagnosis and surgery, but only “re-awaken” after a period of inactivity or nonproductive growth.
The ability of micrometastatic tumor cells to convert into overt metastases is a key point in disease progression because, once detected, metastatic cancer is essentially incurable. Identifying pathways that can be targeted to prevent metastatic outgrowth is particularly important to understand from a therapeutic perspective, since prevention of very early tumor dissemination may not be clinically feasible. In fact, it has been suggested that “a new frontier” in cancer therapy will be to identify ways to revert or maintain cancers in an occult, minimal residual disease state (2,3).
How tumor cells maintain and/or escape clinical dormancy is still largely unknown, but both tumor cell-intrinsic and -extrinsic mechanisms appear to contribute. For example, occult cancer cells are often senescent or arrested in G0/G1, a process that may be mediated by the TH1 cytokines interferon-γ (IFNγ) and tumor necrosis factor (TNFα) (4). Failure to achieve sufficient angiogenesis, even in a proliferating lesion, can also induce dormancy (for review see reference 5). Escape from immune-mediated control has also been demonstrated to contribute to outgrowth of micro-metastases (6–8). A recent study of melanoma metastasis demonstrated that dissemination of cancer cells occurs early in tumorigenesis - even before tumors are detectable; however, their outgrowth in metastatic sites was limited by cytostatic CD8+ T lymphocytes (9). T lymphocytes have also been implicated in late metastatic outgrowth in other models (6) and key cytokines that regulate T cell activity can contribute to maintenance of the dormant state (8). However, the pathways by which micrometastatic tumor cells suppress T cell responses in order to facilitate outgrowth and give rise to overt metastases are very poorly understood.
CD8+ cytotoxic T lymphocytes (CTLs) destroy tumor cells using perforin- and granzyme-mediated cell death (10) as well as by secreting TNFα, causing tumor cell apoptosis (11,12). To survive, tumor cells evade the immune system through mechanisms such as downregulation of class I major histocompatibility complex (MHC) molecules, production of anti-inflammatory cytokines, and/or recruitment of inflammatory-suppressor cells (13). However most studies have focused on tumor-immune interactions in established primary tumors rather than in occult metastases. Identifying and targeting key mechanisms by which tumor cells mediate suppression of CTLs during metastatic outgrowth holds potential as a strategy to reduce or prevent escape from dormancy, and thereby block progression of metastasis.
Macrophage Stimulating Protein (MST1; gene product commonly referred to as MSP), one of its activating proteases (ST14; gene product commonly referred to as matriptase), and the MSP receptor Ron (MST1R, gene product commonly referred to as Ron) become aberrantly overexpressed in around 40%, 45% and 50% of human breast cancers, respectively (14), and are upregulated in many other cancers as well (15). We previously reported that overexpression of MSP/matriptase/Ron is a strong, independent, poor prognostic factor for outcome in human breast cancer patients due to metastasis, and that expression of MSP in a mouse model of mammary cancer was sufficient to promote spontaneous metastasis to lung, lymphatics, and bone (14). However, the mechanisms by which MSP promotes metastasis were not understood.
MSP is constitutively secreted from liver into serum as an inactive protein that is subsequently activated locally on macrophages by matriptase (16) or other extracellular serine proteases (17) in response to infection or injury. The processed MSP binds to Ron, which is selectively expressed on a subset of fully differentiated tissue macrophages, and also at low levels on various epithelial cells (18,19). Ron is essential for protection from unregulated inflammation in several models of infection or injury; MSP/Ron signaling is responsible for regulation of several inflammatory mediators such as TNFα, interleukin-12, IFNγ, arginase, and inducible nitric oxide synthase (20–25). It is unknown, however, whether the role of MSP/Ron in inflammation also contributes to its function in cancer metastasis.
Here, we utilized both genetic and pharmacological approaches to interrogate the mechanism by which MSP drives metastasis, and determined that MSP facilitates metastasis by suppressing anti-tumor immunity. Blocking MSP/Ron signaling, specifically in the host, selectively prevents conversion of pulmonary micrometastases to metastatic colonies by eliciting an effective CD8+ CTL response. We found that inhibition of Ron with a selective tyrosine kinase inhibitor reduced outgrowth of metastasis, even when treatment was delayed until after metastatic colonies were established. Thus, inhibition of MSP/Ron signaling holds promise as an exciting new therapeutic approach to manage the problem of metastatic outgrowth in the adjuvant setting.
RESULTS
Loss of host Ron signaling blocks metastasis
We previously described a highly metastatic transgenic mouse model of breast cancer in which MSP expression drives widespread spontaneous metastasis to clinically relevant sites (PyMT-MSP; (14)). MSP is a secreted protein, and both the tumor cells and host tissues express endogenous Ron, so the pro-metastatic function of MSP could be attributed to direct effects on tumor cells or to indirect effects on the host tumor microenvironment. To determine whether MSP/Ron promotes metastasis through cell-autonomous or non cell-autonomous mechanisms, we transplanted PyMT-MSP tumor cells or PyMT-pMIG control tumor cells into cleared mammary fat pads of immune-competent syngeneic wild type (WT) mice or syngeneic mice lacking Ron activity through targeted deletion of the intracellular kinase domain (Ron TK−/−; 21)(Figure 1A). We found that knocking out host Ron function had no significant effect on primary tumor development, growth rates, proliferation, or apoptosis (Table 1 and Figure S1A–B). However, loss of host Ron activity nearly eliminated spontaneous lung metastasis (p<0.0001; Table 1 and Figure 1B), suggesting that MSP/Ron functions through the host to promote metastasis. We also noted that, although not statistically significant, control PyMT-MIG tumors were also less metastatic in the Ron TK−/− hosts, suggesting that host Ron may promote metastasis even in the absence of overexpressed MSP from the tumor (Supplementary Table 1). In fact, MSP is constitutively produced by hepatocytes and present in the serum, where it can then be activated by macrophages in response to tissue injury or remodeling (31,32).
Figure 1. Loss of host Ron signaling attenuates metastasis specifically during the conversion of seeded micro-metastasis to overt metastasis.
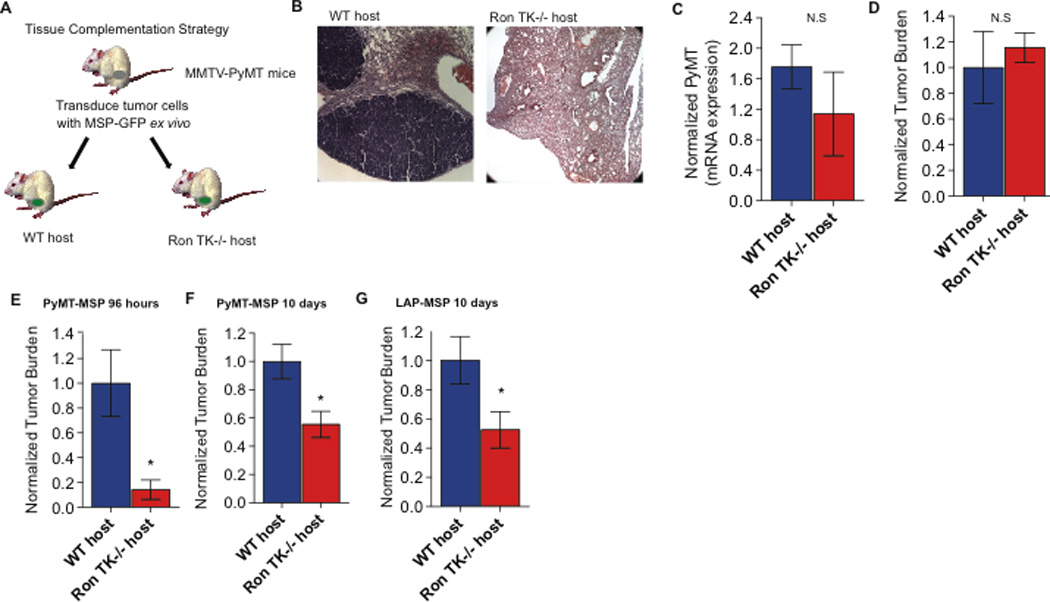
A. Schematic of the experimental strategy to determine whether host MSP/Ron signaling plays a role in mammary tumor development, initiation, and/or metastasis. B. Representative image of spontaneous metastasis in lung sections from tumor-bearing WT and Ron TK−/− hosts. C. PyMT mRNA expression from peripheral blood (normalized to GAPDH) as a surrogate marker for circulating tumor cells in WT and Ron TK−/− mice (n=6 and n=5, respectively). D. Quantification of tumor cell seeding in the lung 36 hours after intravenous injection into WT or Ron TK−/− hosts (n=3 per group). E. Quantification of the metastatic tumor burden in the lung per field of vision 96 hours following intravenous tumor cell injection into WT or Ron TK−/− hosts (n=4 per group). F. Quantification of metastatic tumor burden in the lung per field of vision 10 days following intravenous tumor cell injection into WT or Ron TK−/− hosts (n= 5 and 4, respectively). G. Quantification of DiI labeled tumor cells in lung 10 days following intravenous injection of LAP-MSP lung cancer into WT or Ron TK−/− hosts (n=5). Data are depicted as mean +/− s.e.m. *p<0.05 (unpaired, two-sided t-test). N.S (not statistically significant)
Table 1.
Summary of the effect of host Ron on PyMT-MSP tumor growth, spontaneous metastasis, and survival following experimental metastasis.
| Host animal | Days to palpable tumor |
Days to reach 2cm |
Spontaneous metastasis frequency |
Survival (days to ethical endpoint)1 |
|---|---|---|---|---|
| FVB wild type (n=15) | 35 | 66 | 13/15 (87%) | 40 |
| FVB Ron TK−/− (n=15) | 35 | 53 | 1/15 (6.7%)** | 52* |
| FVB Ron TK−/−; Prkdcscid (n=7) | 41 | 71 | 5/7 (71.4%)*** | ND2 |
p<0.05 (Mantel-Cox test)
p<0.0001 vs. wild type (Fisher’s exact test)
p<0.005 vs. FVB Ron TK−/− (Fisher’s exact test)
experimental metastasis assay; mice were euthanized when in respiratory distress
not done
Metastasis involves multiple steps: cell detachment from the primary tumor mass, local tissue invasion, entry into the circulation, extravasation into new tissues, colonization, and growth at the distant site (33). To test whether lack of metastasis in the Ron TK−/− hosts was due to a defect with invasion or intravasation, we analyzed the relative numbers of circulating tumor cells (CTCs) in both groups of mice. CTCs were measured in blood from tumor bearing WT and Ron TK−/− mice by quantifying levels of tumor-specific PyMT mRNA as a surrogate measure. Evidence for CTCs was found in both groups of mice, but we detected no significant difference between WT and Ron TK−/− tumor-bearing hosts (Figure 1C). To determine whether host Ron plays a role in the later steps of metastasis, such as metastatic cell extravasation, seeding and/or colonization of lungs, we performed experimental metastasis assays. We injected equal numbers of identical tumor cells into the tail veins of WT or Ron TK−/− mice, and examined the ability of the cells to extravasate and seed the lung (36 hours later) and the ability of the cells to form colonies (5 days later; time points are based on (34)). Tumor burden was calculated using two different methods, which both consistently supported the same conclusions (see Materials and Methods for details). Both WT and Ron TK−/− hosts were equally competent for extravasation and metastatic seeding (Figure 1D). However, Ron TK−/− hosts were defective in supporting the conversion of the seeded micrometastatic cells into metastatic colonies, resulting in less tumor burden in the lungs 5 days after injection (Figure 1E and Figure S1C). This effect was sustained; tumor burden in the lungs was still significantly reduced 10 days after injection (Figure 1F and Figure S1D), and Ron TK−/− hosts were able to survive about 50% longer than WT hosts before reaching the experimental endpoint of respiratory distress (Table 1). Similar results were obtained using syngeneic mouse lung cancer cells (LAP-MSP; 26) (Figure 1G), as well as with PyMT-MIG control mammary tumor and LAP-MIG control lung tumor cells (Figure S2A–B). Thus, host Ron activity specifically facilitates the transition from micrometastasis to overt metastasis in multiple models of metastasis.
Ron TK−/− hosts mount a robust CTL response to tumors, which is critical for preventing metastasis
The expression pattern and known function of Ron (15) led us to postulate that the Ron-dependent host role in metastasis would be related to immune function. We first assessed splenic leukocyte populations in tumor-bearing WT or Ron TK−/− mice. We observed no significant differences between cohorts with respect to the proportion of splenic CD11b+ macrophages; Gr-1+ granulocytes; CD11b+/Gr-1+ myeloid-derived suppressor cells; CD11c+ dendritic cells; CD4+ T cells; or CD4+CD25+ T cells (Figure S3A–F). However, we detected a significant (∼2-fold) increase in the proportion of splenic CD8+ T cells in Ron TK−/− compared to WT hosts (Figure 2A). Large clusters of CD8+ T cells were also detected around the margin of primary tumors in Ron TK−/− hosts compared to a general lack of CD8+ T cells around tumors in WT hosts (Figure 2B). We could not detect CD8+ T cells within the core of the primary tumor in either group of mice (data not shown). WT and Ron TK−/− mice without tumors had similar numbers and proportions of splenic CD8+ T cell populations (Figure 2A; naïve hosts), indicating that the expansion of the CD8+ T cells in Ron TK−/− hosts is tumor-dependent.
Figure 2. Loss of host Ron signaling results in expansion of CD8+ T cells and promotes production of pro-inflammatory cytokines.
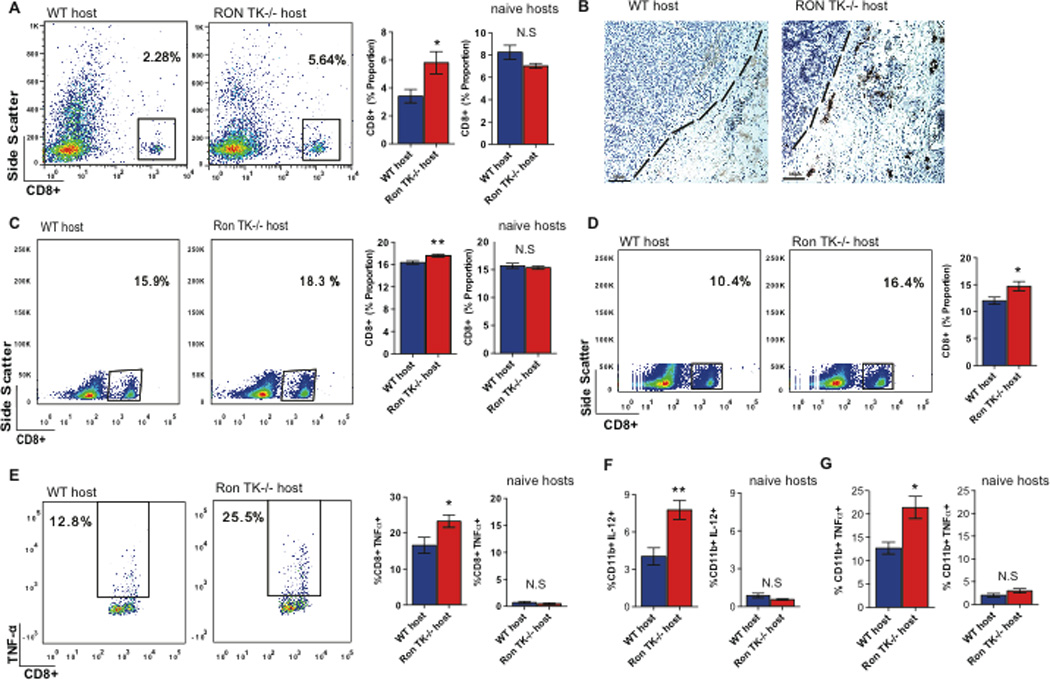
A. Representative flow cytometric analysis (left) and graph (right) depicting the population of CD8+ T cells in spleens of tumor bearing WT and Ron TK−/− mice (n=6 and n=7, respectively), as well as naïve hosts. B. Representative immunohistochemical analysis of CD8+ T cell infiltration into the primary tumors of WT (left panel) and Ron TK−/− hosts (right panel). The dashed lines denote the tumor-stroma border. C. Representative flow cytometric analysis (left) and graph (right) depicting the population of CD8+ T cells in blood of tumor bearing WT and Ron TK−/− mice 72 hours post PyMT-MSP tumor cell injection (n=5 per group) as well as naïve hosts. D. Representative flow cytometric analysis (left) and graph (right) showing the proportion of CD8+ T cells in the blood of tumor bearing WT and Ron TK−/− mice 72 hours post LAP-MSP injection (n=5 per group). E. Representative flow cytometric analysis (left) and graph (right) depicting the population of TNFα expressing CD8+ T cells in lungs of tumor bearing WT and Ron TK−/− mice 72 hours post PyMT-MSP tumor cell injection (n=5 per each group) as well as naïve hosts. F. Graph depicting the population of IL-12 expressing CD11b+ cells in lungs of tumor bearing WT and Ron TK−/− hosts 72 hours post PyMT-MSP tumor cell injection (n=5 per group) as well as naïve hosts (n=4). G. Graph depicting the population of TNF-α expressing CD11b+ cells in the spleen of tumor bearing WT and Ron TK−/− hosts 72 hours post PyMT-MSP tumor cell injection (n=5 per group) as well as naïve hosts (n=4). Data are depicted as mean+/− s.e.m. *p<0.05; ** p<0.01 (unpaired, two-sided t-test). N.S (not statistically significant)
To specifically determine whether CD8+ T cells were responding to the tumor challenge in the context of an experimental metastasis assay, we analyzed the immune milieu in the Ron TK−/− hosts and WT hosts in more detail. The initial stages of T cell activation involve expansion of CD8+ T cells in response to antigen stimulation (35), so we postulated that an expansion of CD8+ T cells in the spleen or peripheral blood would precede an anti-tumor cytotoxic response in the lungs, which occurs between 36 and 96 hours after tumor injection (Figure 1D–E) . Therefore, we analyzed the CD8+ T cell response at an intermediate time point (72 hours after tumor injection). Ron TK−/− hosts had an expanded CD8+ T cell pool in the peripheral blood relative to WT hosts, in both the mammary and lung cancer models (Figure 2C–D). Again, non-tumor bearing Ron TK−/− and WT mice had similar levels of CD8+ T cells in the blood (Figure 2C; naïve hosts).
Despite increased expansion of CD8+ T cells in the periphery of Ron TK−/− mice 72 hours following tumor injection, we did not observe differences in the overall proportion of CD8+ T cells infiltrating the lungs at this time point (data not shown). To determine whether the CD8+ T cells were active, we profiled the inflammatory cytokines produced by CD8+ T cells isolated from both the lungs and peripheral blood of tumor-bearing animals. We found that the CD8+ T cells in the lungs of Ron TK−/− hosts produced more TNF-α than those from WT hosts (Figure 2E), suggesting a stronger pro-inflammatory immune milieu in the lungs of the Ron TK−/− hosts specifically following tumor challenge. CD8+ T cells from non-tumor bearing WT and Ron TK−/− hosts had similar low levels of TNF-α (Figure 2E; naïve hosts). TNF-α is a potent anti-tumor factor secreted by immune cells that induces tumor cell apoptosis (12), and canonical MSP/Ron signaling is known to downregulate IL-12 and TNF-α to drive the switch from classical to alternative macrophage activation (20,22–23). Consistent with this, macrophages derived from lungs of Ron TK−/− mice 72 hours after tumor injection expressed increased IL-12 compared to macrophages isolated from WT hosts at the same time point (Figure 2F and Figure S4). Moreover, macrophages from the spleen of Ron TK−/− hosts also expressed more TNF-α (Figure 2G and Figure S4). Macrophages from non-tumor bearing WT and Ron TK−/− hosts had similar, low levels of IL-12 (Figure 2F; naïve hosts) and TNF-α (Figure 2G; naïve hosts). Thus, loss of host Ron activity enhanced tumor-dependent production of pro-inflammatory cytokines by macrophages, allowed expansion of the peripheral CD8+ T cell population, and promoted infiltration of TNFα-producing CD8+ T cells into the lungs 72 hours following tumor challenge. These events preceded the diminished tumor burden in lungs of Ron TK−/− hosts (at the 96 hour time point; Figure 1E) time point, suggesting that enhanced anti-tumor immunity could be the cause of reduced metastasis in Ron TK−/− mice.
To test whether the improved CD8+ T cell response in Ron TK−/− hosts was directly related to inhibition of metastasis, we asked if loss of T cells would restore metastasis in Ron TK−/− hosts. We crossed Ron TK−/− mice with Prkdcscid mice, which lack functional lymphocytes. The double mutants, versus control Ron TK+/+;Prkdcscid littermates (all backcrossed to the FVB background), were used as hosts for orthotopically-transplanted PyMT-MSP tumors. Tumors developed and grew with similar rates in both hosts; however, Ron TK−/−;Prkdcscid hosts displayed normal (restored) metastasis compared to the almost complete lack of metastasis in immune-competent Ron TK−/− hosts (Table 1; p=0.0043).
To specifically determine whether CD8+ T cells were required to inhibit metastasis in Ron TK−/− hosts, we selectively depleted CD8+ T cells using antibodies against CD8+ T cells in the context of a 10-day lung colonization assay (as in Figure 1F). Successful depletion of CD8+ T cells was confirmed by flow cytometry on splenic, lung, and peripheral blood cell populations (Figure S5A–C). Depletion of CD8+ T cells resulted in a significant, ∼2-fold increase in metastatic tumor burden in the lungs of Ron TK−/− mice (Figure 3A and Figure S5D).
Figure 3. The CD8+ cytotoxic T cell response in Ron TK−/− hosts in response to tumors is necessary and sufficient to block metastasis.
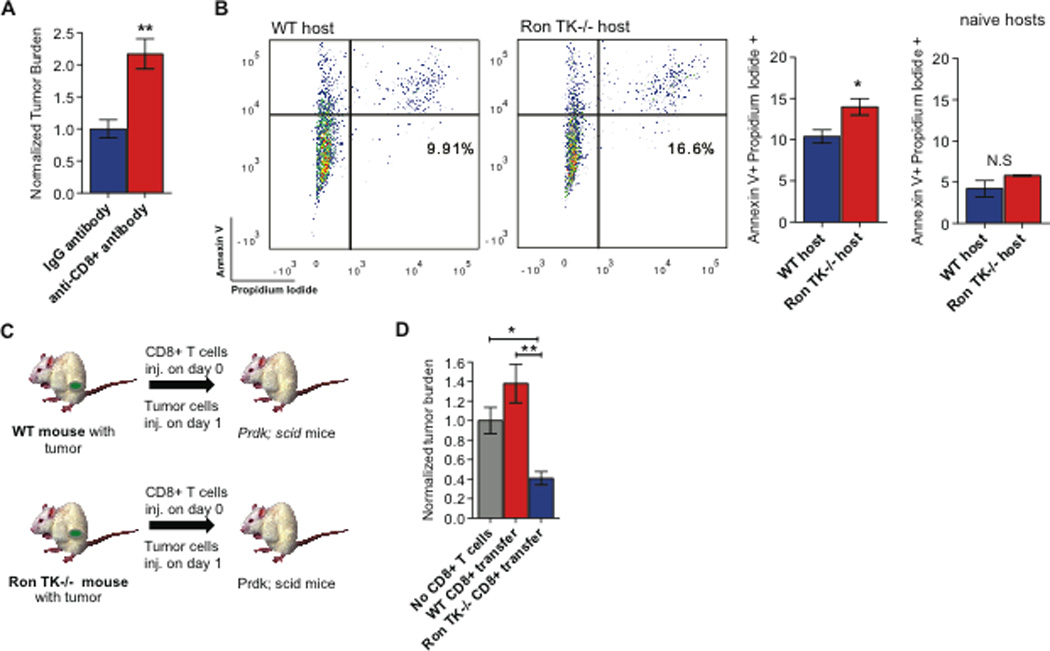
A. Quantification of the tumor burden in the lungs 10 days following intravenous tumor cell injection in animals treated with anti-CD8 antibody or IgG control (n=5 per group).
B. Representative flow cytometric analysis (left) and graph (right) depicting tumor cell apoptosis 24 hours following co-culture of PyMT-MSP tumor cells and CD8+ T cells isolated from the blood of tumor bearing WT and Ron TK−/− hosts (n=3 per group) and naïve hosts (n=3 per group). C. Schematic of the experimental strategy to determine whether tumor-educated CD8+ T cells from Ron TK −/− mice were sufficient to inhibit metastatic lung colonization. D. Quantification of the metastatic tumor burden in mice 10 days following intravenous injection of tumor cells (11 days following T cell adoptive transfer) (n=4 for adoptive transfer of CD8+ T cells from WT or Ron TK −/− mice, and n=10 for controls receiving no T cells). Data are depicted as mean +/− s.e.m. *p<0.05; **p<0.005; ***p<0.0001 (unpaired, two-sided t-test). N.S (not statistically significant).
We next sought to determine if the tumor-induced expansion of CD8+ T cells also resulted in increased cytotoxic ability. We isolated CD8+ T cells from the blood of Ron TK−/− and WT hosts 96 hours following i.v injection of PyMT-MSP tumor cells. We co-cultured PyMT-MSP tumor cells with CD8+ T cells (1:1 ratio) for 24 hours. We observed that CD8+ T cells isolated from the blood of Ron TK−/− hosts had increased cytotoxic ability in vitro, as evidenced by the increased proportion of Annexin V+ PI+ double positive tumor cells (Figure 3B). This was tumor specific; CD8+ T cells isolated from naïve WT and Ron TK−/− hosts had similar, low levels of cytotoxicity (Figure 3B; naïve hosts).
We next wanted to determine whether CD8+ T cells were sufficient to block metastasis in tumor-bearing Ron TK−/− hosts in vivo. We isolated tumor-educated CD8+ T cells from the spleens of tumor-bearing mice and performed adoptive transfer of these cells into tumor-naïve, syngeneic Ron TK+/+;Prkdcscid mice, which lack endogenous lymphocyte function. One day later, we injected the tumor cells (derived from the same donor mice as the CD8+ T cells) into the tail veins (Figure 3C). This strategy allowed us to determine if the CD8+ T cells that were educated and activated in tumor-bearing WT or Ron TK−/− mice were sufficient to affect metastasis in a naïve host in the absence of other functional lymphocytes. Adoptive transfer of CD8+ T cells from WT tumor-bearing mice did not have a significant effect on metastasis, while adoptive transfer of the same number of CD8+ T cells from tumor-bearing Ron TK−/− mice significantly reduced metastatic tumor burden in the lungs (Figure 3D and Figure S5E). Collectively, these results demonstrated that the expanded CD8+ T cell population in tumor-bearing Ron TK−/− mice was both necessary and sufficient to reduce metastasis, while the CD8+ T cells in tumor-bearing WT mice were incapable of anti-metastatic activity. Our data shed light on how MSP/Ron signaling causes metastasis of breast cancer, at least in these animal models: through suppression of an effective anti-tumor CD8+ T cell response. Blocking host Ron activity relieved this immunosuppression and effectively inhibited metastasis. Our next question centered on the potential clinical relevance of our findings.
Pharmacologic inhibition of Ron diminishes metastatic outgrowth
To test whether pharmacologic inhibition of Ron could decrease metastatic outgrowth in WT mice we utilized BMS-777607/ASLAN002, a small molecule inhibitor selective for Ron and, to a lesser extent, its homolog Met (36). We validated the ability of BMS-777607/ASLAN002 to inhibit mouse Ron activity by treating PyMT tumor cells, which express endogenous Ron, with MSP in the presence or absence of BMS-777607/ASLAN002. As expected from published data (36), this compound was effective in diminishing MSP-induced phosphorylation of Ron at sub-micromolar concentrations (IC50 <500 nM; Figure S6A)).
To establish whether BMS-777607/ASLAN002 treatment could reduce metastatic colonization in a manner comparable to that seen in Ron TK−/− hosts, we first tested Ron inhibition in the prophylactic setting. WT mice were treated orally with 50mg/kg BMS-777607/ASLAN002 (or vehicle control) once a day for two weeks (days 1–14). PyMT-MSP tumor cells were injected into the tail vein on day 3, and on day 14 the lungs were harvested and assessed for tumor colonization. The results showed that prophylactic treatment with BMS-777607/ASLAN002 significantly reduced metastatic outgrowth in the lungs by 2–3-fold (Figure 4A and Figure S6B). To determine if CD8+ T cells mediated the anti-colonization effects of BMS-777607/ASLAN002, WT mice were treated orally with 50mg/kg BMS-777607/ASLAN002 (or vehicle control) once a day for 7 days. We concurrently depleted CD8+ T cells with anti-CD8 antibodies. PyMT-MSP tumor cells were injected into the tail vein on day 3, and 96 hours later the lungs were harvested and assessed for tumor colonization. Treatment with BMS-777607/ASLAN002 resulted in 2–3-fold more TNFα-positive macrophages, similar to the increased pro-inflammatory milieu we had observed in Ron TK−/− (Figure 4B and Figure S6C). However, treatment with BMS-777607/ASLAN002 in the absence of CD8+ T cells did not reduce tumor colonization, indicating that CD8+ T cells are key mediators in the anti-colonization effect of BMS-777607/ASLAN002, phenocopying the genetic loss of Ron. (Figure 4C and Figure S6D).
Figure 4. Treatment with a Ron inhibitor significantly reduces metastatic outgrowth.
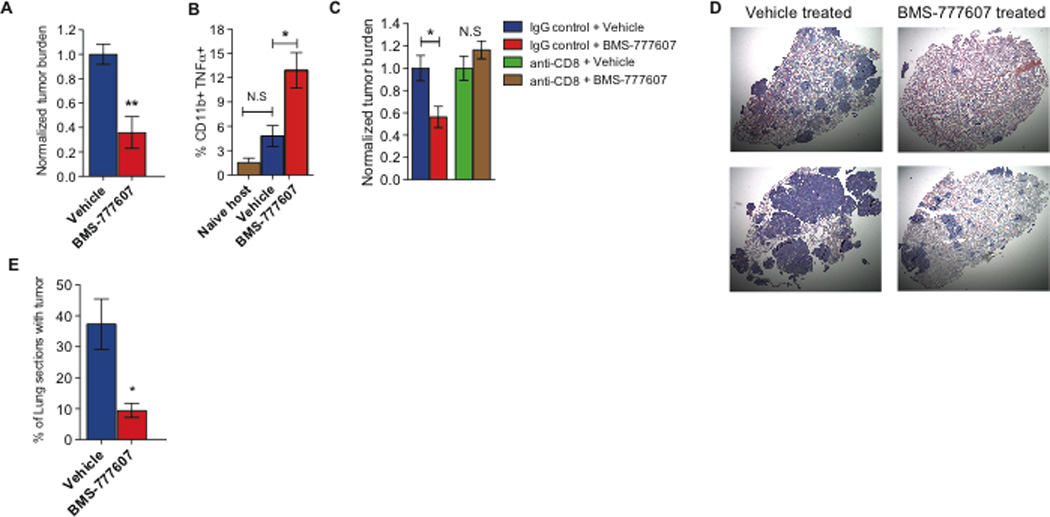
A. Quantification of the metastatic tumor burden in lungs of mice treated prophylactically with vehicle only versus 50mg/kg BMS-777607/ASLAN002 (n = 6 per group). B. Quantification of the population of TNFα expressing CD11b+ cells in the lungs of tumor bearing WT and Ron TK−/− hosts treated with vehicle only versus 50mg/kg BMS-777607/ASLAN002 96 hours post PyMT-MSP tumor cell injection (n=4 and n=3 respectively) and naïve hosts (n=4). C. Quantification of labeled tumor cells 96 hours after intravenous tumor cell injection in animals treated with vehicle or BMS-777607/ASLAN002 following depletion with an anti-CD8 antibody or IgG control. (n=4,3,3,4 respectively). D. Representative H&E staining of metastatic growth in lungs of WT mice treated with vehicle only or 50mg/kg BMS-777607/ASLAN002 (top pictures represent least metastasis observed in each group and bottom pictures represent most metastasis observed in each group). E. Percentage of lung area occupied by metastatic growth in the lungs of mice treated with vehicle only (n=3) versus 50mg/kg BMS-777607/ASLAN002 (n = 4). Data are depicted as mean +/− s.e.m. *p<0.05; **p<0.005 (unpaired, two-sided t-test). N.S (not statistically significant)
To mirror the clinical situation more closely, however, where micro-metastases may have been seeded prior to diagnosis, we next tested Ron inhibition in the “adjuvant” setting. We injected PyMT-MSP tumor cells into the tail veins of WT mice and waited 14 days for metastatic colonies to become fully established, then began daily treatment for 8 days (days 14–22). On day 22, the lungs were harvested and assessed for metastatic outgrowth by determining the percent of lung area that was taken by tumor. Treatment with BMS-777607/ASLAN002 attenuated the formation of metastatic nodules by ∼4-fold, even when administered after metastatic colonization had occurred (Figure 4D–E). Thus, inhibition of Ron kinase activity carries promising potential as a novel therapeutic option to inhibit metastatic outgrowth when given in the adjuvant setting.
DISCUSSION
Ultimately, our ability to reduce cancer mortality depends on identifying ways to prevent or treat distant metastatic disease over long periods of time. The data presented here reveal that the Ron ligand, MSP, which is aberrantly overexpressed in 40% of human breast cancers and many other cancers (14,15), promotes metastasis by inhibiting an effective anti-tumor immune response through activation of Ron signaling in the host. While there are clearly many ways that tumors achieve metastasis, we propose that some tumors upregulate MSP as one way to effectively evade the immune system. Furthermore, our data show that host Ron is also important for immune suppression when tumors themselves do not overexpress MSP, presumably through activation of endogenous serum-derived MSP by macrophage- and/or tumor-derived serine proteases (16). Together, our data suggest that blocking Ron kinase activity allows for reactivation of the anti-tumor immune response and reduces metastatic outgrowth.
It has long been known that infiltration of CD8+ CTLs into tumors is a useful prognostic indicator for various types of tumors (37–39). Recent evidence suggests that immunosurveillance by CD8+ T cells keeps melanoma metastasis in check by promoting tumor dormancy (9). However, suppression of the immune system, also known as immunosubversion, is a critical step in tumor development (40). Tumors downregulate MHC molecules and overproduce arginase-1 and IDO, both of which inhibit CD8+ T cell function. Hypoxia also suppresses T cell activity through expression of hypoxia-inducible factor in macrophages (40). These effects likely cooperate, ultimately leading to a strong immunosuppressive environment in tumors. Such redundancy may explain why primary tumor growth is similar in WT and Ron TK−/− mice despite increased CD8+ T cell infiltration around the periphery of primary tumors in Ron TK−/− hosts. Conversely, tumor cells that have seeded a new environment or are just beginning to effectively colonize the distant organ may be more vulnerable to immune-mediated control. Indeed, our results demonstrate that cells that are in the process of converting from seeded tumor cells to overt metastases are vulnerable to CD8+ T cells. Here, we describe a novel pathway that, when inhibited, is sufficient to activate the CTL response, reducing metastasis and extending life – at least in immune-competent mouse models. These results warrant additional studies focused on whether Ron inhibitors could be tested for anti-metastatic effects in the clinical adjuvant setting.
The precise molecular role for MSP/Ron in suppressing anti-tumor immunity is still unknown and will be the focus of important future studies. Tumors are sites of chronic inflammation and are reminiscent of unhealed wounds, where tumor-associated macrophages (TAMs) appear to be skewed toward an M2 alternative activation state (41,42). Although M2 macrophages are important for wound healing, they are thought to contribute inadvertent advantages to tumors by stimulating angiogenesis and producing polyamines, growth factors, and cytokines that favor tumor growth. Several factors, most notably colony simulating factor 1, have been implicated in the recruitment of macrophages into tumors where they promote metastasis (43,44), but little is known about the specific signaling pathways in tumors that drive the M2 state of TAMs. Based on published studies and our results, it is tempting to speculate that MSP/Ron signaling simply favors conversion of TAMs to an M2 state, resulting in suppression of CTL responses (45). In support of this hypothesis, subcutaneous growth of several mouse tumor types is regulated extrinsically through Ron function in TAMs, which impacts CTL responses (25,46). Additionally, Ron-deficient mice clearly exhibit amplified inflammatory responses upon challenge with infection or injury due to unregulated production of pro-inflammatory cytokines (21,47–48). A similar mechanism could be involved in the tumor setting, whereby increased production of IL-12 and TNF-α from Ron TK−/− macrophages is either causal to or symptomatic of a broad pro-inflammatory cytokine milieu that results in improved CD8+ T cell responses, including production of TNF-α. However, the immune milieu of tumors (and the resulting effects on tumor progression) is extremely complicated; detailed genetic and immunological studies will be required to determine the precise role of Ron in anti-tumor immunity.
Cancer immunotherapy carries strong appeal because the immune response is individualized, it is effective against diverse antigens, and it is potentially able to evolve and retain immunologic memory for long-term control of disease. A major challenge, however, is that by the time tumors are clinically detectable they are already “invisible” to the immune system. Strong natural selection exists to favor tumor cells that can escape immune control by promoting immune tolerance and/or by fostering a strong immunosuppressive environment that renders effector cells inactive (49). These same issues have also been barriers to effective anti-tumor immune therapies, and the clinical results of immunotherapy for breast cancer have been, at most, only moderately effective (50). Drugs that block the inhibitory signals on T cells, such as CTLA-4 inhibitors, are now being used in combination with immunotherapy to generate a more productive anti-tumor immune response (51). Future work will be important to determine whether Ron inhibitors may function in a similar immune-modulatory role to boost the clinical response to immunotherapy and whether CTL activity is a good clinical biomarker of Ron inhibition.
MATERIALS AND METHODS
Mice and cells
All procedures were reviewed and approved by the University of Utah Institutional Animal Care and Use Committee. FVB mice with a deletion in the Ron tyrosine kinase domain (TK−/−) have been described previously (21). Prkdcscid mice (Jackson Labs) and Ron TK−/− or WT mice were backcrossed to generate Ron TK−/−;Prkdcscid mice and Ron TK+/+;Prkdcscid mice on the FVB background. Tumors were generated from MMTV-PyMT transgenic mice engineered to express MSP-IRES-GFP or IRES-GFP (pMIG), and 100,000 GFP+ cells were orthotopically transplanted as described (Welm et al., 2007). LAP-0297 lung cancer cells (26) were engineered to express MSP-IRES-GFP (LAP-MSP) using the same method and 250,000 cells were injected into the tail vein. These cells were obtained from Dr. Peigen Huang (Harvard/Massachusetts General Hospital) without additional authentication.
Immunohistochemistry
Tissues were processed, sectioned (5μm), and stained using standard procedures. Apoptosis was assessed with TUNEL assays (Roche). Antibodies used for immunohistochemistry were phosphohistone H3 (1:100, Cell Signaling) and CD8+ (1:100, Abcam). The Envision+System HRP detection kit (DAKO) and Vector MOM and HRP kits were used according to manufacturers’ instructions.
Lymphocyte isolation and FACS
Splenocytes were isolated by disrupting spleens over a wire mesh, followed by red blood cell (RBC) lysis. Lung lymphocytes were isolated following digestion of lungs in Collagenase IV (Sigma) for 1 hour, followed by Percol (Sigma) separation. Peripheral blood lymphocytes were isolated as previously described (27). Briefly, blood was harvested from WT and Ron TK−/− mice into anti-coagulant citrate dextrose solution Formula A (ACD) followed by incubation with dextran solution for 20 minutes at 37°C. The upper layer of RBC-depleted fluid was harvested, and lymphocytes were collected. Antibodies used for FACS were CD8α-FITC, CD8b–PE, CD4-FITC, CD45-FITC, CD11c–PeCy7, CD11b–PeCy7, Gr-1-APC, CD25-PE, (all 1:400 BD Pharmingen) for cell surface staining. Intracellular FACS staining was done with TNFα-APC and IL-12-eFlour450 (1:400,eBioscience). For cell surface staining, cells were incubated in 2% FBS in PBS for 20 minutes. For intracellular staining, cells were stimulated for 6 hours with PMA/ionomycin in the presence of Brefeldin A (1 μL/mL). Cells were stained with cell surface staining antibodies, permeabilized and stained with anti-cytokine antibodies as per manufacturers’ instructions (BD Biosciences). Cells were analyzed using FACScan and FACS Canto II cytometers (BD) and results analyzed using FlowJo Software (Treestar).
Experimental metastasis assays
Tumor cells were stained with DiI (Invitrogen) and resuspended in HBSS at 106 cells/mL. 250μL (250,000 cells) was injected into the lateral tail veins of Ron TK−/− or WT mice. At experimental endpoints, mice were euthanized, lungs prepared by perfusion with 4% paraformaldyde, and frozen in OCT. Images of 16μm sections were captured using 10× magnification. Fluorescent cells/colonies were quantified using Image J software. Tumor burden was calculated by multiplying the colony count by the colony size for each section. Alternatively, as a secondary quantitative measure, epithelial cells from freshly harvested lungs were collected following Percol (Sigma) separation and analyzed with a FACS Canto II cytometer to calculate percentage of DiI labeled tumor cells.
CD8+ Killing Assays
CD8+ T cells were magnetically sorted from the blood of WT and Ron TK−/− hosts 96 hours after i.v. injection of PyMT-MSP tumor cells and control non-tumor bearing hosts (CD8α microbeads; MACS). Subsequently, the 50,000 CD8+ T cells were cultured with plate-bound anti-CD3 antibody (BD; 5 μg/ml) and co-cultured with 50,000 PyMT-MSP tumor cells (12,28). 24 hours later, cell pellets were collected for apoptosis analysis using Annexin V-APC and propidium iodide as per the manufacturer’s instructions (eBioscience).
CD8+T cell depletion and adoptive transfer
For CD8+ T cell depletion, mice were injected with 100µg anti-CD8 or IgG control antibodies (Bio-X-Cell) once a day, intraperitoneally, for three days prior to tumor injection. 250,000 tumor cells were injected into the tail vein on the fourth day (day 0). Antibodies were re-injected on day 2 and day 7. On day 10, mice were euthanized and metastatic burden quantified as described above. For adoptive transfer experiments, splenocytes from tumor bearing WT and Ron TK−/− mice were stained with CD8α-FITC antibodies and FACS sorted. 500,000 donor CD8+ T cells were injected into the lateral tail veins of recipient Ron TK+/+;Prkdcscid mice. 24 hours later, the mice were injected with 250,000 DiI-labeled tumor cells that were isolated from the same mice as the donor T cells.
Circulating tumor cells
Blood was harvested by cardiac puncture on freshly euthanized WT and Ron TK−/− mice with tumors. Whole blood RNA was isolated using manufacturer’s instructions (Qiagen RNeasy kit). Reverse transcription followed by 35 cycles of PCR for PyMT RNA was performed using the following primers: 5’-CTCCAACAGATACACCCGCACATACT-3’ (forward) and 5’-GCTGGTCTTGGTCGCTTTCTGGATAC-3’ (29). 35 cycles of PCR for GAPDH on the same samples was performed using the following primers: 5′-ATGTTCCAGTATGACTCCACT-3′ and 5′-CCACAATGCCAAAGTTGTCAT-3′) (30) and served as a control for normalization. Ethidium bromide stained gels were quantified according to pixel density analysis using Image J software.
Drug Treatment
For “prophylactic” treatment, mice were administered 50mg/kg BMS-777607/ASLAN002 (or 70% PEG-400 vehicle) orally once a day for 3 days prior to intravenous injection of 250,000 tumor cells (day 0 of the experiment). Treatment continued for 10 more days. On day 11, mice were euthanized and metastasis quantified as described above. For “adjuvant” treatment, 250,000 tumor cells were injected intravenously (day 0 of the experiment). Beginning on day 14, mice were treated with 50mg/kg BMS-777607/ASLAN002 or vehicle orally once a day for 8 days. On day 22, mice were euthanized and lungs were fixed and paraffin-embedded. The extent of metastasis was quantified using Image J and calculated as the average tumor area versus total lung area on each hematoxylin-eosin stained section. In vitro activity of BMS-777607/ASLAN002 against murine Ron was measured by growing mouse tumor cells (MMTV-PyMT) until 80% confluent, incubating overnight in medium with 0.5% serum, and then stimulating the cells with 1ng/mL recombinant human MSP and 0.5μM, 2.0μM, or 5.0 μM BMS-777607/ASLAN002. Cells were harvested 60 minutes later and analyzed by Western with phospho-Ron and total-Ron antibodies (1:400, Santa Cruz).
To determine whether BMS-777607/ASLAN002 mechanism of action was dependent on CD8+ T cells, mice were injected with 100µg anti-CD8 or IgG control antibody (Bio-X-Cell), intraperitoneally, seven days prior to tumor injection and every five days until euthanasia. Tumor cell injection and drug treatment was identical to the “prophylactic” treatment protocol. Epithelial cells from the lungs were collected following Percol (Sigma) separation and analyzed with a FACS Canto II cytometer to calculate percentage of DiI labeled tumor cells.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Our data shed new light on an understudied, yet critically important aspect of metastasis: the conversion of clinically undetectable micrometastatic tumor cells to overt metastases that eventually cause death of the patient. Our work shows that Ron inhibition can significantly reduce metastatic outgrowth, even when administered after metastatic colonies are established.
ACKNOWLEDGEMENTS
We thank the HCI Comparative Oncology Resource Center for assistance with drug administration to mice. We are also grateful to Alexandra Locke for assistance with tissue staining, to Ling Zhao for testing inhibition of Ron by BMS-777607/ASLAN002 in vitro, to Dr. Peigen Huang (Harvard/Massachusetts General Hospital) for providing the published LAP-0297 lung cancer line, and to Dr. David Lum for critical reading of the manuscript.
Financial support: This work was supported by the Department of Defense Breast Cancer Research Program (to A.L.W.), the American Association for Cancer Research and Breast Cancer Research Foundation (to A.L.W.), the Huntsman Cancer Foundation/Institute (to A.L.W.), the Veteran Administration Merit Award (1001BX000803 to S.E.W), and the HHMI Med into Grad Program (to H.E. and A.E). We also utilized the HCI Comparative Oncology Core and University of Utah Flow Cytometry shared resources, supported in part by NCI Cancer Center Support Grant P30-CA042014.
Abbreviations
- MSP
macrophage stimulating protein
- IFN
interferon
- TNF
tumor necrosis factor
- CTL
cytotoxic T lymphocyte
- MHC
major histocompatibility complex
- MMTV-PyMT
mouse mammary tumor virus - polyomavirus middle T antigen
- IRES-GFP
internal ribosome entry site-green fluorescent protein
- TK
tyrosine kinase
- WT
wild type
- RBC
red blood cell
- FACS
fluorescence-activated cell sorting
- CTC
circulating tumor cell
- PI
propidium iodide
- TAM
tumor-associated macrophage
Footnotes
Potential conflicts of interest: ALW received grant funding from OSI Pharmaceuticals/Astellas for work on Met/Ron inhibitors (not included in this manuscript).
REFERENCES
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Uhr JW, Pantel K. Controversies in clinical cancer dormancy. Proc. Natl. Acad. Sci. U.S.A. 2011;108:12396–12400. doi: 10.1073/pnas.1106613108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teng MWL, Swann JB, Koebel CM, Schreiber RD, Smyth MJ. Immune-mediated dormancy: an equilibrium with cancer. J Leukoc Biol. 2008;84:988–993. doi: 10.1189/jlb.1107774. [DOI] [PubMed] [Google Scholar]
- 4.Braumüller H, Wieder T, Brenner E, Aßmann S, Hahn M, Alkhaled M, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature. 2013;494:361–365. doi: 10.1038/nature11824. [DOI] [PubMed] [Google Scholar]
- 5.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer. 2007;7:834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 7.Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target? Nat. Rev. Cancer. 2010;10:871–877. doi: 10.1038/nrc2933. [DOI] [PubMed] [Google Scholar]
- 8.Teng MWL, Vesely MD, Duret H, McLaughlin N, Towne JE, Schreiber RD, et al. Opposing Roles for IL-23 and IL-12 in Maintaining Occult Cancer in an Equilibrium State. Cancer Res. 2012;72:3987–3996. doi: 10.1158/0008-5472.CAN-12-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eyles J, Puaux A-L, Wang X, Toh B, Prakash C, Hong M, et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Invest. 2010;120:2030–2039. doi: 10.1172/JCI42002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trapani JA, Smyth MJ. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002;2:735–747. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- 11.Calzascia T, Pellegrini M, Hall H, Sabbagh L, Ono N, Elford AR, et al. TNF-alpha is critical for antitumor but not antiviral T cell immunity in mice. J. Clin. Invest. 2007;117:3833–3845. doi: 10.1172/JCI32567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sauer KA, Maxeiner JH, Karwot R, Scholtes P, Lehr HA, Birkenbach M, et al. Immunosurveillance of lung melanoma metastasis in EBI-3-deficient mice mediated by CD8+ T cells. J. Immunol. 2008;181:6148–6157. doi: 10.4049/jimmunol.181.9.6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Welm AL, Sneddon JB, Taylor C, Nuyten DSA, van de, Vijver MJ, Hasegawa BH, et al. The macrophage-stimulating protein pathway promotes metastasis in a mouse model for breast cancer and predicts poor prognosis in humans. Proceedings of the National Academy of Sciences. 2007;104:7570–7575. doi: 10.1073/pnas.0702095104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kretschmann KL, Eyob H, Buys SS, Welm AL. The macrophage stimulating protein/Ron pathway as a potential therapeutic target to impede multiple mechanisms involved in breast cancer progression. Curr Drug Targets. 2010;11:1157–1168. doi: 10.2174/138945010792006825. [DOI] [PubMed] [Google Scholar]
- 16.Bhatt AS, Welm A, Farady CJ, Vásquez M, Wilson K, Craik CS. Coordinate expression and functional profiling identify an extracellular proteolytic signaling pathway. Proc Natl Acad Sci U S A. 2007;104:5771–5776. doi: 10.1073/pnas.0606514104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawaguchi M, Orikawa H, Baba T, Fukushima T, Kataoka H. Hepatocyte growth factor activator is a serum activator of single-chain precursor macrophage-stimulating protein. FEBS J. 2009;276:3481–3490. doi: 10.1111/j.1742-4658.2009.07070.x. [DOI] [PubMed] [Google Scholar]
- 18.Kurihara N, Tatsumi J, Arai F, Iwama A, Suda T. Macrophage-stimulating protein (MSP) and its receptor, RON, stimulate human osteoclast activity but not proliferation: effect of MSP distinct from that of hepatocyte growth factor. Exp. Hematol. 1998;26:1080–1085. [PubMed] [Google Scholar]
- 19.Suzuki Y, Funakoshi H, Machide M, Matsumoto K, Nakamura T. Regulation of cell migration and cytokine production by HGF-like protein (HLP) / macrophage stimulating protein (MSP) in primary microglia. Biomed. Res. 2008;29:77–84. doi: 10.2220/biomedres.29.77. [DOI] [PubMed] [Google Scholar]
- 20.Liu Q-P, Fruit K, Ward J, Correll PH. Negative Regulation of Macrophage Activation in Response to IFN-γ and Lipopolysaccharide by the STK/RON Receptor Tyrosine Kinase. The Journal of Immunology. 1999;163:6606–6613. [PubMed] [Google Scholar]
- 21.Waltz SE, Eaton L, Toney-Earley K, Hess KA, Peace BE, Ihlendorf JR, et al. Ron-mediated cytoplasmic signaling is dispensable for viability but is required to limit inflammatory responses. J. Clin. Invest. 2001;108:567–576. doi: 10.1172/JCI11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrison AC, Correll PH. Activation of the stem cell-derived tyrosine kinase/RON receptor tyrosine kinase by macrophage-stimulating protein results in the induction of arginase activity in murine peritoneal macrophages. J. Immunol. 2002;168:853–860. doi: 10.4049/jimmunol.168.2.853. [DOI] [PubMed] [Google Scholar]
- 23.Morrison AC, Wilson CB, Ray M, Correll PH. Macrophage-stimulating protein, the ligand for the stem cell-derived tyrosine kinase/RON receptor tyrosine kinase, inhibits IL-12 production by primary peritoneal macrophages stimulated with IFN-gamma and lipopolysaccharide. J. Immunol. 2004;172:1825–1832. doi: 10.4049/jimmunol.172.3.1825. [DOI] [PubMed] [Google Scholar]
- 24.Wilson CB, Ray M, Lutz M, Sharda D, Xu J, Hankey PA. The RON receptor tyrosine kinase regulates IFN-gamma production and responses in innate immunity. J. Immunol. 2008;181:2303–2310. doi: 10.4049/jimmunol.181.4.2303. [DOI] [PubMed] [Google Scholar]
- 25.Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, et al. Regulation of Macrophage Arginase Expression and Tumor Growth by the Ron Receptor Tyrosine Kinase. J. Immunol. 2011;187:2181–2192. doi: 10.4049/jimmunol.1003460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang P, Duda DG, Jain RK, Fukumura D. Histopathologic findings and establishment of novel tumor lines from spontaneous tumors in FVB/N mice. Comp. Med. 2008;58:253–263. [PMC free article] [PubMed] [Google Scholar]
- 27.Spangrude GJ, Brooks DM, Tumas DB. Long-term repopulation of irradiated mice with limiting numbers of purified hematopoietic stem cells: in vivo expansion of stem cell phenotype but not function. Blood. 1995;85:1006–1016. [PubMed] [Google Scholar]
- 28.Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med. 2009;206:51–59. doi: 10.1084/jem.20081242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaskelioff M, Song W, Xia J, Liu C, Kramer J, Koido S, et al. Telomerase deficiency and telomere dysfunction inhibit mammary tumors induced by polyomavirus middle T oncogene. Oncogene. 2009;28:4225–4236. doi: 10.1038/onc.2009.268. [DOI] [PubMed] [Google Scholar]
- 30.Wu CA, Peluso JJ, Shanley JD, Puddington L, Thrall RS. Murine Cytomegalovirus Influences Foxj1 Expression Ciliogenesis and Mucus Plugging in Mice with Allergic Airway Disease. Am J Pathol. 2008;172:714–724. doi: 10.2353/ajpath.2008.070462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang MH, Skeel A, Leonard EJ. Proteolytic cleavage and activation of pro-macrophage-stimulating protein by resident peritoneal macrophage membrane proteases. J Clin Invest. 1996;97:720–727. doi: 10.1172/JCI118470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nanney LB, Skeel A, Luan J, Polis S, Richmond A, Wang MH, et al. Proteolytic cleavage and activation of pro-macrophage-stimulating protein and upregulation of its receptor in tissue injury. J. Invest. Dermatol. 1998;111:573–581. doi: 10.1046/j.1523-1747.1998.00332.x. [DOI] [PubMed] [Google Scholar]
- 33.Gupta GP, Massagué J. Cancer Metastasis: Building a Framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. A Distinct Macrophage Population Mediates Metastatic Breast Cancer Cell Extravasation, Establishment and Growth. 2009 doi: 10.1371/journal.pone.0006562. PLoS ONE 4, e6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev Immunol. 2002;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 36.Schroeder GM, An Y, Cai Z-W, Chen X-T, Clark C, Cornelius LAM, et al. Discovery of N-(4-(2-Amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carboxamide (BMS-777607), a Selective and Orally Efficacious Inhibitor of the Met Kinase Superfamily. Chem. J. Med. 2009;52:1251–1254. doi: 10.1021/jm801586s. [DOI] [PubMed] [Google Scholar]
- 37.Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58:3491–3494. [PubMed] [Google Scholar]
- 38.Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J Med. 2005;353:2654–2666. doi: 10.1056/NEJMoa051424. [DOI] [PubMed] [Google Scholar]
- 39.Swann JB, Smyth MJ. Immune surveillance of tumors. J. Clin. Invest. 2007;117:1137–1146. doi: 10.1172/JCI31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715–727. doi: 10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- 41.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 42.Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. Journal of Leukocyte Biology. 2009;86:1065–1073. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 43.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022–7029. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 44.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discovery. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lepique AP, Daghastanli KRP, Cuccovia IM, Villa LL. HPV16 tumor associated macrophages suppress antitumor T cell responses. Clin. Cancer Res. 2009;15:4391–4400. doi: 10.1158/1078-0432.CCR-09-0489. [DOI] [PubMed] [Google Scholar]
- 46.Gurusamy D, Gray JK, Pathrose P, Kulkarni RM, Finkleman FD, Waltz SE. Myeloid-Specific Expression of Ron Receptor Kinase Promotes Prostate Tumor Growth. Cancer Res. 2013;73:1752–1763. doi: 10.1158/0008-5472.CAN-12-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lentsch AB, Pathrose P, Kader S, Kuboki S, Collins MH, Waltz SE. The Ron receptor tyrosine kinase regulates acute lung injury and suppresses nuclear factor kappaB activation. Shock. 2007;27:274–280. doi: 10.1097/01.shk.0000239755.82711.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McDowell SA, Mallakin A, Bachurski CJ, Toney-Earley K, Prows DR, Bruno T, et al. The role of the receptor tyrosine kinase Ron in nickel-induced acute lung injury. Am. J. Respir. Cell Mol. Biol. 2002;26:99–104. doi: 10.1165/ajrcmb.26.1.4621. [DOI] [PubMed] [Google Scholar]
- 49.Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z, et al. Tumor-specific CD8+ T cells expressing IL-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010;70:6725–6734. doi: 10.1158/0008-5472.CAN-10-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Curigliano G, Spitaleri G, Pietri E, Rescigno M, de Braud F, Cardillo A, et al. Breast cancer vaccines: a clinical reality or fairy tale? Ann. Oncol. 2006;17:750–762. doi: 10.1093/annonc/mdj083. [DOI] [PubMed] [Google Scholar]
- 51.Zhou Q, Xiao H, Liu Y, Peng Y, Hong Y, Yagita H, et al. Blockade of programmed death-1 pathway rescues the effector function of tumor-infiltrating T cells and enhances the antitumor efficacy of lentivector immunization. J. Immunol. 2010;185:5082–5092. doi: 10.4049/jimmunol.1001821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.