

Abstract
Acute promyelocytic leukemia (APL) is a particular type of acute myeloid leukemia with characteristic biological and clinical features, the frequent association at diagnosis of a severe hemorrhagic diathesis. Immune thrombocytopenia (ITP) is a common autoimmune disease characterized by low platelet counts and an increased risk of bleeding. Here we present a patient with the diagnosis of APL who achieved and maintained a remission with an induction consisting of idarubicin and ATRA, and then developed corticosteroid refractory ITP which is successfully treated with laparoscopic splenectomy.
Keywords: Immune thrombocytopenia, ITP, Splenectomy, Acute promyelocytic leukemia, APL
Introduction
Acute promyelocytic leukemia (APL) is a particular type of acute myeloid leukemia (AML) with characteristic biological and clinical features, which include the presence of a specific t(15;17) chromosome translocation in the leukemic blasts, the frequent association at diagnosis of a severe hemorrhagic diathesis, and sensitivity to the differentiating agent all-trans retinoic acid (ATRA) [1, 2]. Induction consisting of anthracycline-based chemotherapy and ATRA are the mainstay of APL treatment [2]. A cure rate of approximately 90 % of patients who survive induction and achieve complete remission (CR) can be expected [3].
Immune thrombocytopenia (ITP) is a common autoimmune disease characterized by low platelet counts and an increased risk of bleeding. Antibody-mediated platelet destruction has been the prevailing hypothesis to explain ITP pathogenesis, also the recent success of thrombopoietin (TPO) receptor agonists lends support to the notion that platelet production is also insufficient. Up until the last decade, corticosteroid treatment, other immunosuppressive drugs, and splenectomy were considered as the mainstay of treatment in patients with chronic ITP. 10 years ago, rituximab, a monoclonal antibody against CD20, started to be used for patients with ITP, which resulted in platelet count increases that were often sustained for months [4]. A new class of drugs, called TPO receptor agonists, was shown to produce a dose-dependent increase in platelet counts even in some patients with refractory ITP [5].
Here we present a patient with the diagnosis of APL who achieved and maintained a remission with an induction consisting of idarubicin and ATRA, and then developed corticosteroid refractory ITP which is successfully treated with laparoscopic splenectomy (LS).
Case Report
A 22-year-old female otherwise healthy patient was admitted to our department with gum bleeding and pancytopenia in November 2009. White blood cell (WBC) count was 0.96 × 109/L, hemoglobin (Hb) and hematocrit (HCT) levels were 9.1 g/dL and 26.8 %, respectively with a platelet (PLT) count of 69 × 109/L. Prothrombin time was 15.6 s. (normal range, 10.2–13.8 s.), and international normalized ratio was 1.38 (normal range, 0.9–1.2), activated partial thromboplastin time was 43.8 s. (normal range, 29.5–40.8 s.), thrombin time was 29.3 s. (normal, <22 s.). The fibrinogen and D-dimer levels were found to be 50.9 mg/dL (normal range, 150–350 mg/dL) and 34.07 mg/dL (normal range, 0-0.5 mg/dL), respectively. Physical examination revealed paleness of the skin and mucous membranes, with petechiae and ecchymosis at the lower extremities. There were no peripheral lymphadenopathies nor organomegaly. The initial diagnosis were disseminated intravascular coagulation (DIC) with pancytopenia. The peripheral blood smear showed marked leukopenia with 4 % bands, 49 % neutrophils, 2 % eosinophils, 43 % lymphocytes, 2 % monocytes, and thrombocytopenia. A bone marrow aspiration and biopsy was done which revealed, a hypercellular bone marrow (100 % cellularity) and a diffuse infiltration of blasts with Auer rods, showing the typical morphology of a promyelocyte (Picture 1) which were myeloperoxidase (MPO) positive, CD34 and CD117 negative. Since the diagnosis was APL on the basis of clinical and bone marrow findings, we started an induction chemotherapy consisting of 45 mg/m2/daily ATRA and 12 mg/m2 idarubicin (on days 2, 4, 6, and 8) immediately [6]. The flow-cytometric evaluation was also consistent with the bone marrow aspiration and biopsy findings, and t(15;17) was detected both by conventional cytogenetics and fluorescence in situ hybridization (FISH) from the bone marrow. Fresh frozen plasma and platelet transfusions were given in order to prevent major bleeding, and there were no bleeding complications during the induction therapy. After the induction, a bone marrow aspiration and biopsy was performed which was consistent with CR, and the complete blood count (CBC) was normal [WBC: 4.43 × 109/L (neutrophils: 3.84 × 109/L), Hb: 11.8 g/dL, HCT: 33.7 %, PLT: 154 × 109/L] in February 2010. The consolidation chemotherapy courses were initiated in March 2010 which included; idarubicin 5 mg/m2/day × 4 (course no. 1), mitoxantrone 10 mg/m2/day × 5 (course no. 2), and idarubicin 12 mg/m2/day × 1 (course no. 3), with 45 mg/m2/daily ATRA on the first fifteen days of every course. After the second course of consolidation, the platelet counts remained low (28 × 109/L) in the absence of consumption coagulopathy. The patient had a score less than 5 suggestive of non-overt DIC according to International Society on Thrombosis and Haemostasis DIC scoring system [7]. Serological testing for hepatitis B&C and human immunodeficiency virus (HIV) were negative, anti-nuclear antibody, lupus anticoagulant and anti-cardiolipin antibodies were found to be negative. Helicobacter pylori antigen was negative. Serum immunoglobulin levels were normal as well as a direct Coomb’s test which was negative. She had no splenomegaly, and the leukocyte count and red blood cell indices were within the normal ranges [WBC: 4.03 × 109/L (neutrophils: 2.14 × 109/L), Hb: 11.5 g/dL, HCT: 33.2 %], with a normal reticulocyte percentage (1.3 %). Since the patient maintained her remission, in order to investigate the cause of thrombocytopenia, we performed a bone marrow aspiration and biopsy, and the re-evaluation of her bone marrow in June 2010 confirmed CR for acute leukemia with a normal number of megakaryocytes (Picture 2). The PML/RARA was negative from the bone marrow aspirate by real-time polymerase chain reaction (RT-PCR). The diagnosis was immune thrombocytopenia, and high dose dexamethasone (HDD) treatment (40 mg/day × 4, every 4 weeks) was initiated in June 2010. After the HDD treatment the platelet count raised up to 64 × 109/L. The third consolidation chemotherapy was administered afterwards, in order to maintain the remission state. Following each HDD treatment, the corticosteroid dosage was tapered slowly, and after 2 cycles of HDD, the thrombocytopenia persisted. We made another bone marrow aspiration and biopsy following the 3rd consolidation course, which showed similar findings with the previous one. Intravenous immunoglobulin (IVIg) with a total dose of 2 g/kg was administered in August 2010 when the platelet count was 32 × 109/L. After IVIg, the platelet count raised (109 × 109/L), and the patient was discharged from the hospital. Maintenance therapy for APL consisting of 90 mg/m2/day mercaptopurine orally, 15 mg/m2/weekly methotrexate orally, and, intermittently, 45 mg/m2/day ATRA for 15 days every 3 months was started in September 2010. During the follow-up, the thrombocytopenia persisted, and the patient had received 3 more cycles of HDD with conventional dose of methylprednisolone administered between the HDD courses. Despite these treatments, the platelet counts remained below 30 × 109/L, so splenectomy was planned in March 2011. A control bone marrow aspiration and biopsy was done, which revealed a remission state for APL and increased megakaryocytes. Haemophilus influenzae type b, pneumococcal and meningococcal vaccines were performed before the operation. The splenic scintigraphy showed no accessory spleen. Before the surgery, IVIg was administered when the platelet count was 26 × 109/L, and the platelet count raised up to 89 × 109/L. A LS procedure was performed, without any perioperative complications, and the histopathological evaluation of the splenectomy material revealed no abnormal findings. After splenectomy, the platelet count raised-up to 1,114 × 109/L, then decreased and remained within the normal ranges after a 6-month of follow-up duration. The platelet counts and the treatment modalities during the follow-up are displayed in Fig. 1. At this writing, the patient is still in remission for both APL and ITP, t(15;17) is negative by RT-PCR, the platelet count is 186 × 109/L, the leukocyte count is 4.33 × 109/L with Hb and HCT levels of 13.8 g/dL and 42.3 %, respectively in September 2011.
Picture 1.
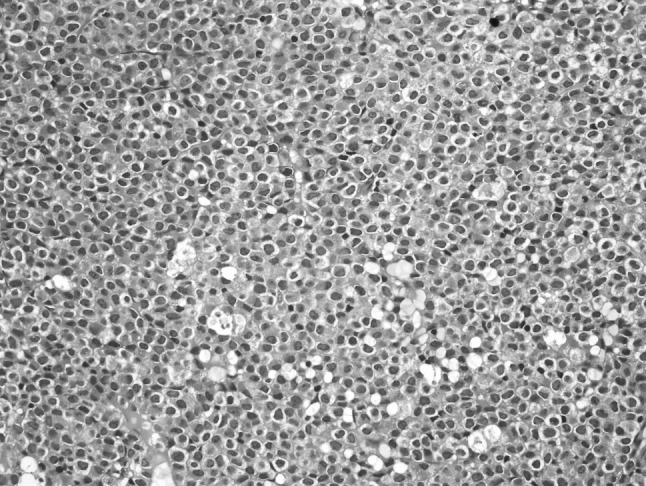
Bone marrow biopsy consistent with 100 % cellularity and a diffuse infiltration of blasts with Auer rods, showing the typical morphology of a promyelocyte (Hematoxylin and Eosin, ×400)
Picture 2.
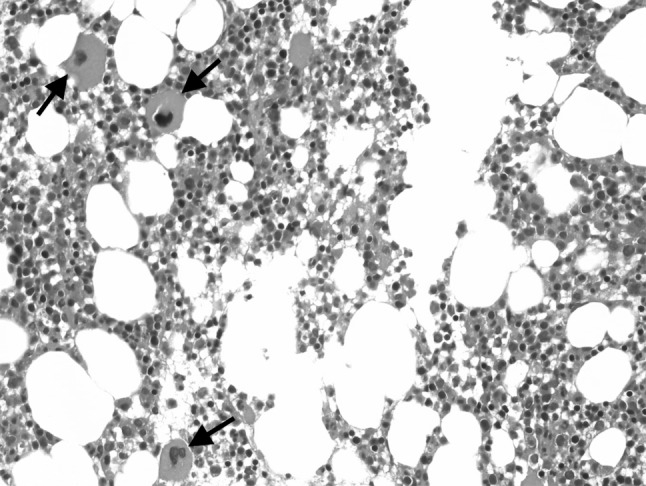
Bone marrow biopsy showing a normal number of megakaryocytes (arrows) with blasts less than 5 % at the time of the initial diagnosis of immune thrombocytopenia (Hematoxylin and Eosin, ×400)
Fig. 1.

The platelet counts and the treatment modalities during the follow-up of the patient
Discussion
Thrombocytopenia is a recognized side effect of treatment with a wide range of medications such as chemotherapeutic and immunosuppressive agents, which tend to produce pancytopenia by suppressing hematopoiesis. Only a few drugs preferentially inhibit megakaryocytopoiesis to produce isolated thrombocytopenia, however, many medications lower platelet levels by accelerating platelet clearance through immune and (less often) non-immune mechanisms. In patients who experience an acute drop in platelet levels, usually within a week or two of starting a new medication, antibody-mediated platelet destruction should be suspected.
In the differential diagnosis of thrombocytopenia in our patient, drug-induced immune thrombocytopenia (DITP) can be a possible explanation. DITP can be triggered by a wide range of medications [8]. During the course of her disease, we did not use heparin in our patient, but we administered broad-spectrum antibiotics due to febrile neutropenia. Hapten-dependent antibody generation after antibiotic administration can play a role in the mechanisms of DITP [9], and many patients with DITP have only petechial hemorrhages and occasional ecchymoses and require no specific treatment other than discontinuation of the sensitizing medication. After the discontinuation of antibiotics in our patient, during the follow-up, the thrombocytopenia persisted which is unlikely in patients with DITP. For these reasons, the most probable cause of thrombocytopenia in our patient was ITP.
Another clinical condition that has to be taken into account in our patient is, posttransfusion purpura (PTP). PTP is a rare disorder which typically presents as severe thrombocytopenia resulting within 7–10 days after an immunogenic blood transfusion. It usually affects women who have been sensitized to the alloantigen during previous pregnancy [10, 11]. Thrombocytopenia can result from the transfusion of any blood product, and is most commonly seen after the infusion of packed red cells. Fever and chills are often observed during the transfusion which is followed by petechiae and ecchymoses once thrombocytopenia develops. Most probably in our patient, PTP may not be the explanation of thrombocytopenia, because our patient received packed red cells and platelets after the second consolidation therapy, but more than four weeks prior to the recognition of thrombocytopenia, and we never observed any fever and chills during these transfusions. She did not give childbirth nor experience any pregnancies. Also the thrombocytopenia persisted for more than 6 months, and all these findings make the diagnosis of PTP unlikely in our patient.
Immune thrombocytopenia has been associated with several hematologic malignancies such as Hodgkin and non-Hodgkin lymphomas and chronic lymphocytic leukemia, but it is relatively rare in patients with acute leukemia [12]. The development of ITP in a patient with APL is even rarer, and only 2 such cases are demonstrated in the literature [13, 14]. The diagnosis of ITP was made when the patient was in remission for APL, and we both administered conventional and high-dose corticosteroids in the management of immune thrombocytopenia, but refractory thrombocytopenia prolonged more than 6 months and persisted for approximately twelve months.
Laparoscopic splenectomy has become the gold-standard surgical intervention for the treatment of ITP patients who experienced medical relapse to steroid. Since durable remissions have been achieved with splenectomy in patients with chronic refractory ITP [15, 16], we planned to perform LS in our patient, which induced a durable remission. We did not administer anti-CD20 monoclonal antibody, since response to this treatment modality can be temporary, and we also had concerns about the long-term immunosuppression and reactivation of viral infections that can be caused by rituximab.
In conclusion, although thrombocytopenia is a recognized side effect of chemotherapeutic drugs, also the immune mechanisms should be suspected as a possible cause of thrombocytopenia in the leukemia patient. Splenectomy may induce remissions in patients with chronic ITP, and this procedure can be a good treatment option in the selected cases, even in leukemia patients.
Acknowledgments
Conflict of interest
All authors have no conflict of interest to declare.
References
- 1.Warrell RP, Jr, de The′ H, Wang Z-Y, Degos L. Acute promyelocytic leukemia. N Engl J Med. 1993;329(3):177–189. doi: 10.1056/NEJM199307153290307. [DOI] [PubMed] [Google Scholar]
- 2.Grignani F, Fagioli M, Alcalay M, Longo L, Pandolfi PP, Dotti E, Biondi A, Lo Coco F, Grignani F, Pelicci PG. Acute promyelocytic leukemia: from genetics to treatment. Blood. 1994;83(1):10–25. [PubMed] [Google Scholar]
- 3.Tallman MS, Nabhan C, Feusner JH, Rowe JM. Acute promyelocytic leukemia: evolving therapeutic strategies. Blood. 2002;99(3):759–767. doi: 10.1182/blood.V99.3.759. [DOI] [PubMed] [Google Scholar]
- 4.Arnold DM, Dentali F, Crowther MA, Meyer RM, Cook RJ, Sigouin C, Fraser GA, Lim W, Kelton JG. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med. 2007;146(1):25–33. doi: 10.7326/0003-4819-146-1-200701020-00006. [DOI] [PubMed] [Google Scholar]
- 5.Bussel JB, Kuter DJ, George JN, McMillan R, Aledort LM, Conklin GT, Lichtin AE, Lyons RM, Nieva J, Wasser JS, Wiznitzer I, Kelly R, Chen CF, Nichol JL. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med. 2006;355(16):1672–1681. doi: 10.1056/NEJMoa054626. [DOI] [PubMed] [Google Scholar]
- 6.Sanz MA, Martín G, Rayón C, Esteve J, González M, Díaz-Mediavilla J, Bolufer P, Barragán E, Terol MJ, González JD, Colomer D, Chillón C, Rivas C, Gómez T, Ribera JM, Bornstein R, Román J, Calasanz MJ, Arias J, Alvarez C, Ramos F, Debén G. A modified AIDA protocol with anthracycline-based consolidation results in high antileukemic efficacy and reduced toxicity in newly diagnosed PML/RARalpha-positive acute promyelocytic leukemia PETHEMA group. Blood. 1999;94(9):3015–3021. [PubMed] [Google Scholar]
- 7.Taylor FB, Jr, Toh CH, Hoots WK, Wada H, Levi M. Scientific Subcommittee on Disseminated Intravascular Coagulation (DIC) of the International Society on Thrombosis and Haemostasis (ISTH). Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost. 2001;86(5):1327–1330. [PubMed] [Google Scholar]
- 8.Aster RH, Curtis BR, McFarland JG, Bougie DW. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. J Thromb Haemost. 2009;7(6):911–918. doi: 10.1111/j.1538-7836.2009.03360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med. 2007;357(6):580–587. doi: 10.1056/NEJMra066469. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez CE, Pengetze YM. Post-transfusion purpura. Curr Hematol Rep. 2005;4(2):154–159. [PubMed] [Google Scholar]
- 11.Abramson N, Eisenberg PD, Aster RH. Post-transfusion purpura: immunologic aspects and therapy. N Engl J Med. 1974;291(22):1163–1166. doi: 10.1056/NEJM197411282912205. [DOI] [PubMed] [Google Scholar]
- 12.Horino S, Rikiishi T, Niizuma H, Abe H, Watanabe Y, Onuma M, Hoshi Y, Sasahara Y, Yoshinari M, Kazama T, Hayashi Y, Kumaki S, Tsuchiya S. Refractory chronic immune thrombocytopenic purpura in a child with acute lymphoblastic leukemia. Int J Hematol. 2009;90(4):483–485. doi: 10.1007/s12185-009-0424-0. [DOI] [PubMed] [Google Scholar]
- 13.Ashihara E, Shimazaki C, Hirata T, Okawa K, Oku N, Goto H, Inaba T, Fujita N, Nakagawa M. Autoimmune thrombocytopenia following peripheral blood stem cell autografting. Bone Marrow Transplant. 1993;12(3):297–299. [PubMed] [Google Scholar]
- 14.Fujita H, Togami K, Mori M, Hashimoto H, Nagai K, Nagai Y, Tabata S, Kurata M, Mastusita A, Maeda A, Takahashi T. Successful treatment with azathioprine for autoimmune thrombocytopenia developing after autologous peripheral blood stem cell transplantation. (Article in Japanese) Rinsho Ketsueki. 2007;48(8):637–641. [PubMed] [Google Scholar]
- 15.Toltl LJ, Arnold DM. Pathophysiology and management of chronic immune thrombocytopenia: focusing on what matters. Br J Haematol. 2011;152(1):52–60. doi: 10.1111/j.1365-2141.2010.08412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu Z, Zhou J, Pankaj P, Peng B. Laparoscopic splenectomy for immune thrombocytopenia (ITP) patients with platelet counts lower than 1 × 109/L. Int J Hematol. 2011;94(6):533–538. doi: 10.1007/s12185-011-0962-0. [DOI] [PubMed] [Google Scholar]