

Abstract
Acetyl coenzyme A (AcCoA) is the central biosynthetic precursor for fatty acid synthesis and protein acetylation. In the conventional view of mammalian cell metabolism, AcCoA is primarily generated from glucose-derived pyruvate through the citrate shuttle and adenosine triphosphate citrate lyase (ACL) in the cytosol1-3. However, proliferating cells that exhibit aerobic glycolysis and those exposed to hypoxia convert glucose to lactate at near stoichiometric levels, directing glucose carbon away from the tricarboxylic acid cycle (TCA) and fatty acid synthesis4. Although glutamine is consumed at levels exceeding that required for nitrogen biosynthesis5, the regulation and utilization of glutamine metabolism in hypoxic cells is not well understood. Here we show that human cells employ reductive metabolism of alpha-ketoglutarate (αKG) to synthesize AcCoA for lipid synthesis. This isocitrate dehydrogenase 1 (IDH1) dependent pathway is active in most cell lines under normal culture conditions, but cells grown under hypoxia rely almost exclusively on the reductive carboxylation of glutamine-derived αKG for de novo lipogenesis. Furthermore, renal cell lines deficient in the von Hippel-Lindau (VHL) tumor suppressor protein preferentially utilize reductive glutamine metabolism for lipid biosynthesis even at normal oxygen levels. These results identify a critical role for oxygen in regulating carbon utilization in order to produce AcCoA and support lipid synthesis in mammalian cells.
Although hypoxic cells exhibit a shift toward aerobic glycolysis4, a functional electron transport chain and glutamine-derived carbon are required for proliferation of most transformed cells6. In line with these studies, we observed increased glucose consumption and lactate secretion when A549 cells were cultured at ~1% oxygen (Fig. S1a). Notably, glutamine consumption also increased while glutamate secretion remained unchanged, indicating that net glutamine consumption was elevated and suggesting that glutamine carbon is used for biosynthesis. Consistent with this observation in both normoxic and hypoxic cells, we found that proliferating cells incorporate glutamine-derived carbon into lipids (Figure S1b). Glutamine can contribute carbon to lipogenic AcCoA through two distinct pathways. Cells can oxidatively metabolize glutamine-derived αKG in the TCA cycle and generate pyruvate from malate via glutaminolysis5. Alternatively, some tissues can reductively carboxylate αKG to generate citrate7,8, and recent studies have indicated that the IDH reaction is highly reversible in various cell types9-11. To determine which pathway cells use to incorporate glutamine carbon into lipids we utilized stable isotopic tracers12-14. We first cultured several cancer cell lines with [1-13C]glutamine under normoxia and quantified the isotopic label present in metabolite pools along this pathway (Fig. 1a, red carbon atoms). All cells tested with this tracer retained significant label from [1-13C]glutamine in citrate and metabolites downstream of the irreversible ACL reaction, indicating that the reductive flux contributes to the cytosolic AcCoA pool (Fig. S2). Additional evidence for activity along this pathway was obtained by using a uniformly 13C labeled ([U-13C5]) glutamine tracer in cell cultures (Fig. S3).
Figure 1.

Reductive carboxylation is the primary route of glutamine to lipids. a) Schematic of carbon atom (circles) transitions and tracers used to detect reductive glutamine metabolism. Isotopic label from [1-13C]glutamine (red) is lost during oxidative conversion to succinate (Suc) but retained on citrate (Cit), oxaloacetate (Oac), aspartate (Asp), malate (Mal), and fumarate (Fum) in the reductive pathway (green arrows). [5-13C]glutamine (blue) transfers label to AcCoA through reductive metabolism only. Molecular symmetry is shown for oxidative metabolism. b) Contribution of [5-13C]glutamine and [U-13C5]glutamine to lipogenic AcCoA in cell lines. c) IDH1 levels in A549 cells expressing IDH1-specific (IDH1a and IDH1b) or control shRNAs. d) Metabolite labeling from [1-13C]glutamine from cells in (c). e) IDH flux estimates from 13C MFA model in control or IDH1-knockdown A549 cells cultured with [U-13C5]glutamine. f) Cell proliferation of A549 cells expressing IDH1-shRNAs. Error bars indicate 95% confidence interval (CI) for (b, e) and s.e.m. (n=3) for (d, f). * denotes p < 0.05.
To quantify the specific contributions of oxidative and reductive glutamine metabolism to fatty acid synthesis, we cultured cells in the presence of tracers for several days and performed Isotopomer Spectral Analysis (ISA; Fig. S4)15. Use of [5-13C]glutamine specifically allows estimation of the flux of glutamine to lipids through reductive carboxylation (Fig. 1a, blue carbon atoms). Virtually all cell lines cultured with this tracer generated labeled fatty acids, metabolizing glutamine reductively in the TCA cycle to supply 10 – 25% of their lipogenic AcCoA (Figs. 1b, blue bars; S5). Consistent with these data, we were able to detect 98 +/− 5 cpm/106 cells in hexane extracts of A549 cells cultured with [5-14C]glutamine. Next we directly compared the contribution of glutamine to fatty acids via the reductive flux as a fraction of the total, the latter determined by using [U-13C5]glutamine. In all cell lines tested, including those derived from lung, mammary, colon, and squamous cell carcinoma as well as melanoma, glioblastoma, and leukemia, [5-13C5]glutamine labeled the majority of AcCoA derived from glutamine (Fig. 1b, white bars),highlighting the general use of reductive carboxylation as the primary route through which glutamine, glutamate, and αKG carbon are converted to lipids in cultured cells. (Figs. 1b, S2, S3, S5). The glutaminolysis pathway is also an important means of glucose catabolism in many cells and can be characterized by quantifying the contribution of glutamine carbon to lactate. Consistent with published reports5, glutamine-derived 13C label was also detected in lactate, and the amount of 13C-labeled lactate produced was highest in glioblastoma-derived cells compared to other cells cultured with [U-13C5]glutamine (Fig. S6).
Mammalian cells express three IDH enzymes encoded by separate genes: IDH1 (cytosolic, NADP+-dependent), IDH2 (mitochondrial, NADP+-dependent), and the multi-subunit enzyme IDH3 (mitochondrial, NAD+-dependent). IDH3 is allosterically regulated and assumed to operate in the oxidative direction16. The NADP+-dependent isozymes are capable of catalyzing the reductive reaction; however, the specific enzyme responsible for this flux is not definitively known17. As measurements of metabolite pools in subcellular compartments and labeling therein cannot yet be reliably obtained, we employed RNA interference to selectively knock down expression of IDH1 and IDH2 in A549 cells. Using labeling from [1-13C]glutamine as a readout, we measured a significant and robust decrease in reductive carboxylation when IDH1 mRNA was targeted using shRNA (Fig. 1c, 1d). These changes were consistent with results using [U-13C5]glutamine (Fig. S7) and reproduced using several cell lines (Fig S8). Finally, we employed 13C Metabolic Flux Analysis (MFA)18,19 to quantify intracellular fluxes using [U-13C5]glutamine in A549 cells. The fitted data suggested that reductive IDH flux significantly decreased when IDH1 protein levels were decreased, and this change was the primary alteration observed in the network (Fig 1e; see Tables S1-5 for complete results and MFA model description).
Our results suggest that IDH1 can convert NADP+, αKG and CO2 to isocitrate and NADPH in the cytosol. Enzymatic analysis using recombinant protein indicated that this IDH1 is indeed capable of consuming NADPH and is responsive to physiological levels of CO2 (Fig. S9). Importantly, the proliferation rate of all cell lines with IDH1 knockdown was impaired (Fig. 1f, S8), indicating that reductive metabolism of αKG in the cytosol may be necessary for robust growth. In contrast to our results with IDH1 shRNAs, we detected no significant change in reductive flux when targeting IDH2 mRNA in A549, MDA-MB-231, and HCT116 cells (Fig. S10). Although IDH2 may promote reductive carboxylation in some tissues or conditions, these results are consistent with the interpretation of IDH2 as an oxidative TCA cycle enzyme proposed by Hartong et al.20.
Intriguingly, we detected a significant increase in reductive carboxylation activity when culturing cells under hypoxia (Fig. S11). As glucose is usually the primary carbon source for mammalian tissues1-3, we next compared the contributions of reductive glutamine metabolism and glucose oxidation to fatty acid synthesis by culturing cells with either [5-13C]glutamine or uniformly labeled [U-13C6]glucose under normal tissue culture conditions or hypoxia. Strikingly, oxygen levels influenced fatty acid labeling from both tracers (Fig. 2a, 2b). Cells preferentially utilized glucose carbon for palmitate synthesis under normoxic conditions; however, fatty acids produced under hypoxia were primarily synthesized from glutamine carbon via the reductive pathway. In fact, the reductive carboxylation of glutamine-derived αKG accounted for approximately 80% of the carbon used for de novo lipogenesis in A549 cells growing under hypoxia (Fig. 2c). Conversely, we detected a concomitant decrease in the contribution of [U-13C6]glucose to fatty acid synthesis under this condition. Knockdown of IDH1 protein mitigated the use of reductive glutamine metabolism for lipogenesis under hypoxia (Fig 2d). Significant increases in the relative utilization of this pathway were observed in all cell lines tested, including non-transformed cells (Fig. S12). In addition, T lymphocytes freshly isolated from a mouse spleen preferentially used reductive glutamine metabolism over glucose oxidation for fatty acid synthesis when activated under hypoxia (Fig. S13). Although proliferation rates and relative de novo lipogenesis were lower under hypoxia (Fig. S14), the net flux of reductive glutamine metabolism to palmitate synthesis was significantly increased in hypoxic cultures (Fig. 2e). While most human cells require glutamine for nucleotide and hexosamine biosynthesis, some cell lines can grow in the absence of exogenous sources of glutamine21. Remarkably, we found that hypoxia increases the dependence of such cells on glutamine, as evidenced by decreased proliferation in the absence of glutamine and increased reductive glutamine metabolism under hypoxia when glutamine is present (Fig. 2f, S15).
Figure 2.

Hypoxia reprograms cells to rely on reductive glutamine metabolism for lipid synthesis. a,b) Labeling of palmitate extracts from A549 cells cultured under normoxia or hypoxia with [5-13C]glutamine (a) or [U-13C6]glucose (b). Similar results were observed in myristate, oleate and stearate pools (not shown). c,d) Relative contribution of glucose oxidation or glutamine reduction to lipogenic AcCoA in A549 cells under normoxia and hypoxia (c) or A549 cells expressing control or IDH1-targeting shRNAs under hypoxia (d). e) Absolute fluxes of [U-13C6]glucose and [5-13C]glutamine to palmitate in A549 cells. Error bars indicate 95% CI from model for c – e; * denotes p < 0.05. f) Huh7 cell proliferation after 4 days in the presence or absence of glutamine. Error bars indicate s.e.m. (n=3) for a, b, and f. ** denotes p < 0.005 comparing glutamine-free cultures. *** denotes p < 0.001 comparing normoxia and hypoxia.
To gain insight into the mechanisms controlling this switch to reductive glutamine metabolism we analyzed changes in the labeling and abundances of TCA cycle metabolites in cells grown under hypoxia. Using [U-13C6]glucose, we observed a significant decrease in relative flux through the pyruvate dehydrogenase (PDH) complex (Fig. 3a, S16a, S16b). In addition, the citrate pool became depleted, which would be expected to increase reductive carboxylation flux through mass action (Fig. 3b, S16c). On the other hand, we detected increased amounts of isotopic label in TCA cycle intermediates when using labeled glutamine tracers under hypoxia (Fig. 3c, 3d, S16d). Reductively metabolized glutamine accounted for as much as 40 – 70% of the intracellular citrate, aspartate, malate, and fumarate pools when cells were cultured in low oxygen (Fig. 3d). Given the marked reduction in PDH flux observed in hypoxia, we tested the ability of dichloroacetate (DCA) to restore PDH activity and mitigate the contribution of reductive metabolism to lipogenesis. DCA inhibits pyruvate dehydrogenase kinases (PDKs)22, and PDK1 is a known target of HIF-1α that inhibits the activity of PDH through phosphorylation23,24. While DCA treatment had no observable effect on carbon utilization under normoxia, reductive glutamine metabolism was inhibited and glucose oxidation was partially restored in A549 cells cultured with DCA under hypoxia (Fig. 3e, S16e), suggesting that hypoxia-induced PDK1 contributes to the use of reductive carboxylation for fatty acid synthesis.
Figure 3.
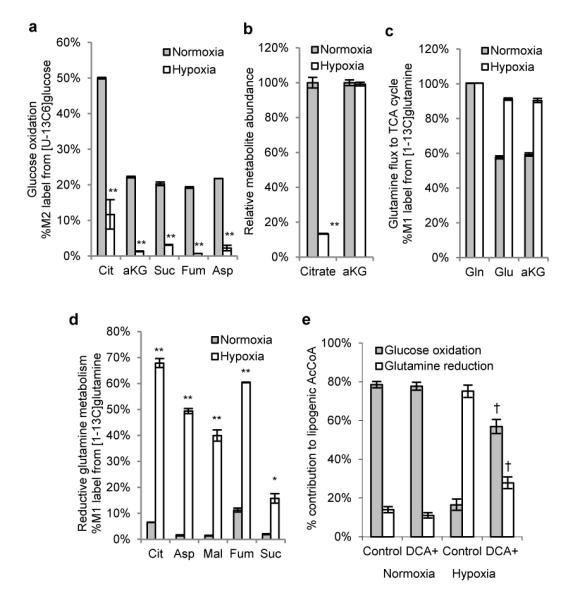
Reductive TCA metabolism increases under hypoxia. MRC5 cells were cultured under normoxia or hypoxia for 3 days in the presence of tracer. A) Relative level of glucose oxidation as determined by M2 labeling from [U-13C6]glucose (see Fig. S16A for atom transition map). M2 isotopologues were the most abundant labeled metabolites in mass spectra. B) Relative abundance of citrate and αKG. c,d) Relative contribution of reductive glutamine metabolism to TCA metabolites, determined by M1 labeling from [1-13C]glutamine. M1 isotopologues were the only species with significant abundance. e) Contribution of glucose oxidation and glutamine reduction to lipogenesis in A549 cells cultured with or without 5 mM DCAError bars indicate s.e.m. for (a-d; n=3) and 95% CI for (e). * denotes p < 0.01 and ** denotes p < 0.001 comparing normoxia to hypoxia. † denotes p < 0.05 comparing control to DCA in hypoxia.
The VHL tumor suppressor protein is frequently lost in renal cell carcinoma (RCC) and results in a state of “pseudohypoxia” by activating HIF signaling25-27. To further understand the role of this pathway in promoting the switch to reductive TCA metabolism we tested RCC cells deficient in VHL using ISA. Remarkably, VHL-deficient RCC cell lines preferentially utilized reductive glutamine metabolism for lipogenesis, even when cultured under normal oxygen levels, while those expressing wild-type (WT) VHL behaved similarly to other carcinoma cell lines (Fig. 4a, S17). Re-expression of WT VHL in previously VHL-deficient cell lines resulted in a switch back to oxidative glucose metabolism as the source of carbon for lipid synthesis (Fig. 4b), reduced extracellular fluxes of glucose, lactate, and glutamine (Fig. S18A), and increased the pool of intracellular citrate relative to αKG (Fig. S18b) under normoxia. Furthermore, shRNA-mediated knockdown of HIF-2α partially restored glucose-mediated lipogenesis in 786-O cells (Fig. 4c, 4d). Consistent with VHL and HIF influencing the switch to reductive glutamine metabolism during hypoxia, glucose entry into the TCA cycle via PDH was increased under normoxia upon introduction of WT VHL or knockdown of HIF-2α in 786-O cells (Fig. S18c). Similar changes were observed following knock down of the HIFα dimerization partner ARNT (aryl hydrocarbon receptor nuclear translocator) in VHL-deficient normoxic UMRC2 cells, which express both HIF-1α and HIF-2α28, or following ARNT knock down in hypoxic A549 and 143B cells (Fig. S19).
Figure 4.

HIF/ARNT/VHL signaling regulate carbon utilization for lipogenesis. A-C) Contribution of glucose oxidation ([U-13C6]glucose) and glutamine reduction ([5-13C]glutamine) to lipogenesis in RCC lines (A), parental control (PRC3) and VHL+ (WT8) cells derived from 786-O line (B), or vector control (pTV) or HIF2α shRNA (pTR) cells derived from 786-O line (C). D) Western blot to determine HIF2α levels for cells in (B-C). Error bars indicate 95% CIs obtained from ISA model. * indicates p < 0.05. E) Model depicting the metabolic reprograming of mammalian cells by hypoxia or VHL loss to employ reductive glutamine metabolism for lipogenesis. HIF stabilization drives transcription of PDK1, which decreases PDH activity and subsequently intracellular citrate levels. IDH1 and ACO1 reductively generate lipogenic citrate from glutamine-derived αKG. DCA can inhibit PDKs, forcing increased glucose oxidation in hypoxic cells.
Our results highlight an important role for reductive TCA metabolism of glutamine in cell proliferation at physiological oxygen levels (Fig. 4e). Given the almost exclusive use of reductive carboxylation for lipogenesis under hypoxia, a redundant or contributing role of mitochondrial IDH2 in this pathway is probable; however our data provide evidence that the reductive pathway involves IDH1-mediated catalysis in the cytoplasm. While the carbon source that cells use for lipid synthesis appears to be determined, at least partially, by HIF-mediated regulation of PDK1, additional hypoxia-associated changes may also promote reductive glutamine metabolism. For example, HIF-2α enhances c-MYC activity29, which in turn drives glutamine catabolism through the regulation of numerous genes including glutaminase30. This metabolic reprogramming provides an effective, glucose-independent means of generating AcCoA for biosynthesis. Since glucose is also delivered to cells via the vasculature it may be limited in microenvironments with decreased oxygen availability31. Reductively metabolizing amino acids for lipid synthesis under these conditions would allow cells to conserve glucose for production of ribose and other biosynthetic precursors (e.g. one carbon pool, hexosamines) that are not typically generated from other nutrients. Thus, reductive metabolism may allow cells to more efficiently distribute available nutrients in poorly vascularized microenvironments. These results add a new dimension to our understanding of cell metabolism and suggest potential therapeutic targets along the reductive carboxylation and glutamine catabolic pathways that could mitigate hypoxic tumor growth.
Methods Summary
For determination of steady state labeling of polar metabolites, cells were cultured for approximately 24 hours in the presence of 13C-labeled glutamine or glucose before extraction. For experiments involving stable isotopic labeling of lipid biomass, cells were grown for approximately 3 – 4 days in the presence of tracer before extraction. Details of the extraction and derivatization methods are described in Supplementary Methods. Computational determination of metabolic fluxes, confidence intervals, de novo lipogenesis, and the contribution of tracers to fatty acid carbon was accomplished using an in-house software package, Metran19. Details of the metabolic networks and GC/MS measurements used for modeling and complete results are described as Supplementary Information. The generation of cells stably expressing control shRNAs or those targeted IDH1 or IDH2 is described in Supplementary Methods; all experiments were conducted within 4 passages of initial selection. Hypoxic microenvironments were generated by feeding incubators with a pre-mixed gas composed of 1% O2, 5% CO2, and 94% N2, and O2 levels were confirmed to range between 1 and 3% using a Fyrite combustion analyzer. For details regarding recombinant IDH1 production and enzyme assays, T cell activation, medium analysis, [5-14C]glutamine experiment, and western blotting please see Supplementary Methods.
Supplementary Material
Acknowledgements
The authors would like to thank Natalie Vokes and Patrick Ward for helpful discussions. We also thank Stefan Gross and Agios Pharmaceuticals for providing the IDH1 construct. We acknowledge support from NIH grant RO1 DK075850-01. CMM is supported by a postdoctoral fellowship from the American Cancer Society. MVH is supported by the Burrough’s Wellcome Fund, the Smith Family, the Damon Runyon Cancer Research Foundation, and the National Cancer Institute. DJI is an investigator of the Howard Hughes Medical Institute. OI is supported by RO1 CA122591 and the Dana Farber/Harvard Cancer Center Kidney SPORE Grant Developmental Award.
Footnotes
Author Contributions C.M.M., P.A.G., E.L.B., J.Y., K.H., and C.M.J. performed cellular experiments and isotope tracing. C.M.M. and P.A.G. performed metabolite profiling and analyzed data. K.R.M. and M.G.V.H. performed enzyme assays and 14C experiments. E.L.B., J.Y., and Z.R.J. generated Western blots. D.J.I. and L.G. provided support and reagents. J.K.K., M.G.V.H., O.I., and G.S. provided conceptual advice. C.M.M., J.K.K., M.G.V.H., O.I., and G.S. wrote and edited the paper.
References
- 1.Hatzivassiliou G, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–321. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Wellen KE, et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Migita T, et al. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008;68:8547–8554. doi: 10.1158/0008-5472.CAN-08-1235. [DOI] [PubMed] [Google Scholar]
- 4.Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeBerardinis RJ, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinberg F, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Des Rosiers C, et al. Isotopomer analysis of citric acid cycle and gluconeogenesis in rat liver. Reversibility of isocitrate dehydrogenase and involvement of ATP-citrate lyase in gluconeogenesis. J Biol Chem. 1995;270:10027–10036. doi: 10.1074/jbc.270.17.10027. [DOI] [PubMed] [Google Scholar]
- 8.Yoo H, Antoniewicz MR, Stephanopoulos G, Kelleher JK. Quantifying reductive carboxylation flux of glutamine to lipid in a brown adipocyte cell line. J Biol Chem. 2008;283:20621–20627. doi: 10.1074/jbc.M706494200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metallo CM, Walther JL, Stephanopoulos G. Evaluation of 13C isotopic tracers for metabolic flux analysis in mammalian cells. J Biotechnol. 2009;144:167–174. doi: 10.1016/j.jbiotec.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lemons JM, et al. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol. 2010;8:e1000514. doi: 10.1371/journal.pbio.1000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward PS, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boros LG, et al. Defective RNA ribose synthesis in fibroblasts from patients with thiamine-responsive megaloblastic anemia (TRMA) Blood. 2003;102:3556–3561. doi: 10.1182/blood-2003-05-1537. [DOI] [PubMed] [Google Scholar]
- 13.Munger J, et al. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol. 2008 doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maier K, Hofmann U, Reuss M, Mauch K. Identification of metabolic fluxes in hepatic cells from transient 13C-labeling experiments: Part II. Flux estimation. Biotechnol Bioeng. 2008;100:355–370. doi: 10.1002/bit.21746. [DOI] [PubMed] [Google Scholar]
- 15.Kharroubi AT, Masterson TM, Aldaghlas TA, Kennedy KA, Kelleher JK. Isotopomer spectral analysis of triglyceride fatty acid synthesis in 3T3-L1 cells. Am J Physiol. 1992;263:E667–675. doi: 10.1152/ajpendo.1992.263.4.E667. [DOI] [PubMed] [Google Scholar]
- 16.Sazanov LA, Jackson JB. Proton-translocating transhydrogenase and NAD- and NADP-linked isocitrate dehydrogenases operate in a substrate cycle which contributes to fine regulation of the tricarboxylic acid cycle activity in mitochondria. FEBS Lett. 1994;344:109–116. doi: 10.1016/0014-5793(94)00370-x. [DOI] [PubMed] [Google Scholar]
- 17.Siebert G, Carsiotis M, Plaut GW. The enzymatic properties of isocitric dehydrogenase. J Biol Chem. 1957;226:977–991. doi: 10.1016/S0021-9258(18)70883-1. [DOI] [PubMed] [Google Scholar]
- 18.Sauer U. Metabolic networks in motion: 13C-based flux analysis. Mol Syst Biol. 2006;2:62. doi: 10.1038/msb4100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young JD, Walther JL, Antoniewicz MR, Yoo H, Stephanopoulos G. An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnol Bioeng. 2008;99:686–699. doi: 10.1002/bit.21632. [DOI] [PubMed] [Google Scholar]
- 20.Hartong DT, et al. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat Genet. 2008;40:1230–1234. doi: 10.1038/ng.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng T, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonnet S, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 23.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 24.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2:673–682. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 26.Ivan M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 27.Jaakkola P, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 28.Zimmer M, et al. Small-molecule inhibitors of HIF-2a translation link its 5′UTR iron-responsive element to oxygen sensing. Mol Cell. 2008;32:838–848. doi: 10.1016/j.molcel.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–347. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao P, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sutherland RM. Cell and environment interactions in tumor microregions: the multicell spheroid model. Science. 1988;240:177–184. doi: 10.1126/science.2451290. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.