

Abstract
Methylmercury (MeHg) is an environmental electrophile that covalently modifies cellular proteins with reactive thiols, resulting in the formation of protein adducts. While such protein modifications, referred to as S-mercuration, are thought to be associated with the enzyme dysfunction and cellular damage caused by MeHg exposure, the current consensus is that (1) there is a cellular response to MeHg through the activation of NF-E2-related factor 2 (Nrf2) coupled to S-mercuration of its negative regulator, Kelch-like ECH-associated protein 1 (Keap1), and (2) the Keap1/Nrf2 pathway protects against MeHg toxicity. In this review, we introduce our findings and discuss the observations of other workers concerning the S-mercuration of cellular proteins by MeHg and the importance of the Keap1/Nrf2 pathway in protection against MeHg toxicity in cultured cells and mice.
1. Introduction
Methylmercury (MeHg) is strongly accumulated by fish and marine mammals and is found at the highest concentrations in large predatory species at the top of the aquatic food chain. MeHg readily penetrates the blood-brain barrier and affects the central nervous system. It is transferred into cells by passive diffusion and/or transport by the L-type large neutral amino acid transporter (as its L-cysteine complex) and reacts covalently with protein thiols because of its high affinity for nucleophilic groups, having a dissociation constant of 10−15 [1]. This covalent modification by MeHg is referred to as “S-mercuration.” Some MeHg, however, binds to glutathione (GSH) that is present in a variety of cell types at relatively high concentrations (>1 mM) to form a MeHg-GSH adduct that is transported into the extracellular space by multidrug resistance-associated proteins (MRPs) [2–4] (Figure 1). While the MeHg-mediated S-mercuration of cellular proteins is believed to be involved in the toxicological significance of MeHg [5], oxidative stress from the presence of MeHg in the body is also a critical factor associated with its toxicity [6]. Interestingly, antioxidant proteins such as heme oxygenase-1 (HO-1), glutamate-cysteine ligase (GCL; a rate-limiting enzyme for GSH synthesis), and MRPs are regulated by transcription factor NF-E2-related factor 2 (Nrf2) [7–12].
Figure 1.
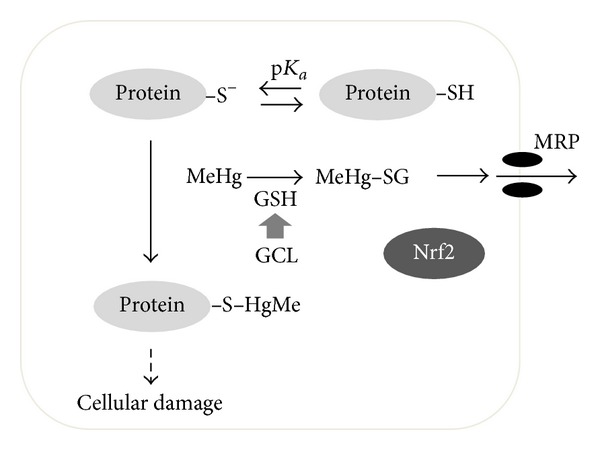
S-mercuration of cellular proteins by MeHg and the MeHg detoxification pathway.
2. S-Mercuration of Proteins by MeHg
Although there is little doubt that MeHg is able to covalently modify cellular proteins, and it is thought that a protein-MeHg complex is, at least partly, involved in MeHg toxicity (Figure 1), there have been only a limited number of studies in which the in vivo effects of MeHg have been investigated with regard to S-mercuration. This is probably because of the lack of appropriate methods for identifying S-mercuration. Here, we describe our efforts to investigate the effects of MeHg using chemical biology approaches (Table 1).
Table 1.
Molecular targets of MeHg.
| Target protein | In vivo effect | References |
|---|---|---|
| Mn-SOD | Reduction of catalytic activity in mouse brain | Shinyashiki et al. [13] |
| nNOS | Induction of NOS in rat brain | Shinyashiki et al. [15] |
| Arginase I | Reduction of activity in rat liver Decreased Mn levels in tissues |
Kanda et al. [18] |
| SDH | Not defined | Kanda et al. [19] |
We have found that the subcutaneous administration of MeHg (10 mg/kg) to mice resulted in a time-dependent decrease in brain manganese-superoxide dismutase (Mn-SOD) activity, whereas MeHg exposure had no effect on cuprozinc-SOD (Cu,Zn-SOD) activity [13]. Although levels of mRNA and protein synthesis of Mn-SOD were unaffected by MeHg administration, MeHg caused a facile reduction in the activity, and a drastic decrease in the native form, of Mn-SOD (but not of Cu,Zn-SOD) with purified enzyme preparations. It was subsequently shown that the S-mercuration of Mn-SOD, through Cys196, by MeHg results in a decrease in the enzyme activity in vivo [14]. Therefore, the selective inhibition of Mn-SOD activity caused by S-mercuration could play a critical role in MeHg-induced neurotoxicity because SOD protects the cell against oxidative stress by scavenging superoxide.
We also found that the subcutaneous administration of MeHg (10 mg kg−1 day−1 for 8 days) to rats caused significant increases in nitric oxide synthase (NOS) activities in the cerebrum and cerebellum with time [15]. The increase in NOS activity seems to be caused by an increase in neuronal NOS (nNOS) protein levels, but not an increase in inducible NOS. In contrast to the in vivo observations, however, the addition of MeHg to a cerebellar enzyme preparation in vitro caused a concentration-dependent decrease in nNOS activity, suggesting that MeHg modifies NOS through reactive thiol groups, thereby affecting its catalytic activity in vitro. Experiments with arylmercury column chromatography have confirmed that this is possible [15]. A reasonable explanation for our observations is that the increase in nNOS proteins observed from MeHg exposure in vivo may be one of the initial responses to counteract the metal-induced negative effects on the central nervous system.
In the body, MeHg accumulates to high concentrations in the liver [16, 17], but few molecular targets of MeHg have been identified in this tissue. We hypothesized that MeHg could interact with arginase I, an abundant Mn-binding protein in the liver, through covalent modification, resulting in alterations in not only its catalytic activity but also in hepatic Mn levels through in vivo MeHg exposure. After the subcutaneous administration of MeHg (10 mg kg−1 day−1 for 8 days) to rats, mercury levels in the liver were greater than those in the cerebrum or cerebellum [18]. A marked suppression of arginase I activity was also detected under these conditions. Using purified rat arginase I, we found that MeHg-induced S-mercuration of arginase I caused the protein to aggregate and substantial leakage of Mn ions from the arginase I active site. We speculate that the MeHg-mediated suppression of hepatic arginase I activity in vivo is, at least partly, attributable to the S-mercuration of this protein.
In the course of the study, we coincidentally detected another protein, with a 42 kDa subunit molecule, from the same hepatic preparation that readily underwent S-mercuration and subsequent aggregation. Two-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis was used to separate the protein, and it was identified by peptide mass fingerprinting using matrix-assisted laser desorption and ionization time-of-flight mass spectrometry (MALDI-TOF/MS). This 42 kDa protein was identified as sorbitol dehydrogenase (SDH) [19]. Using recombinant rat SDH possessing 10 cysteine residues in a subunit [20], we found that MeHg was covalently bound to SDH through Cys44, Cys119, Cys129, and Cys164, resulting in the inhibition of its catalytic activity and the release of zinc ions, facilitating protein aggregation [19]. Subsequent mutation analysis confirmed that Cys44, which ligates the active site zinc atom [21], and Cys129 play a crucial role in the MeHg-mediated aggregation of SDH [19].
3. Cellular Response to MeHg via the Keap1/Nrf2 Pathway
As mentioned above, MeHg reacts readily with cellular nucleophiles and also causes oxidative stress, implying that it may lead to decreased GSH levels in tissues. As a result, MeHg undergoes GSH conjugation and is excreted into the extracellular space through the action of MRPs [2–4]. However, several studies have indicated that MeHg exposure also upregulates GCL [22–28]. This suggests that there is an initial response to MeHg in the cells to compensate for decreased GSH levels and to repress oxidative cell damage. On the other hand, Dr. Yamamoto and his associates reported that transcription factor Nrf2 cooperatively regulates antioxidant proteins such as GCL and HO-1, phase-II xenobiotic detoxifying enzymes, and phase-III xenobiotic transporters such as MRPs [10, 29]. They subsequently found that Nrf2 is negatively regulated by Kelch-like ECH-associated protein 1 (Keap1) [30]. Interestingly, Keap1 has 27 and 25 cysteine residues in humans and mice, respectively [31, 32], and some thiols (e.g., Cys151, Cys273, and Cys288) that have been established as being highly reactive [31–34], so the “cysteine code,” which defines the preferential target cysteine(s) and distinct biological effects, was proposed [35–37]. These observations led us to assume that MeHg could modify Keap1 through S-mercuration and so activate the Nrf2-regulated gene expression of GCL, HO-1, and MRPs.
Figure 2 shows a summary of the Keap1 modification sites, consisting of NTR (N-terminal region), BTB (Board complex, Tramtrack, and Bric-à-brac), IVR (intervening region), and DC (DGR/CTR: double glycine repeat/C-terminal region) domains that can be modified by a variety of electrophiles [31, 33, 38, 39]. Among them, Cys151, Cys273, and Cys288 are well established as being essential for regulating Nrf2 function [31, 32, 40, 41], although other cysteine residues (e.g., Cys257, Cys297, and Cys613) in Keap1 can also be modified by dexamethasone 21-mesylate, biotinylated iodoacetamide, and 1,2-napthoquinone [31, 33, 38]. Our MALDI-TOF/MS analysis revealed that Keap1 undergoes S-mercuration by MeHg at three cysteine residues, Cys151, Cys368, and Cys489 (see Figure 3 and Table 2), suggesting that the S-mercuration of Cys151 in Keap1 potentially causes a structural alteration, activating Nrf2, while Cys368 and Cys489 are also targets for 1,2-naphthoquinone and tert-butyl-1,4-benzoquinone [38, 39].
Figure 2.
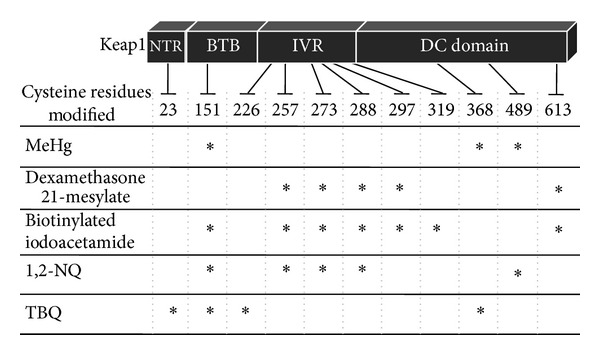
Covalent modification of Keap1 cysteine residues by electrophiles. The BTB, IVR, and DC domains are essential for the formation of homodimers, associated with cullin 3 and the binding of Nrf2. 1,2-NQ: 1,2-naphthoquinone; TBQ: tert-butyl-1,4-benzoquinone.
Figure 3.
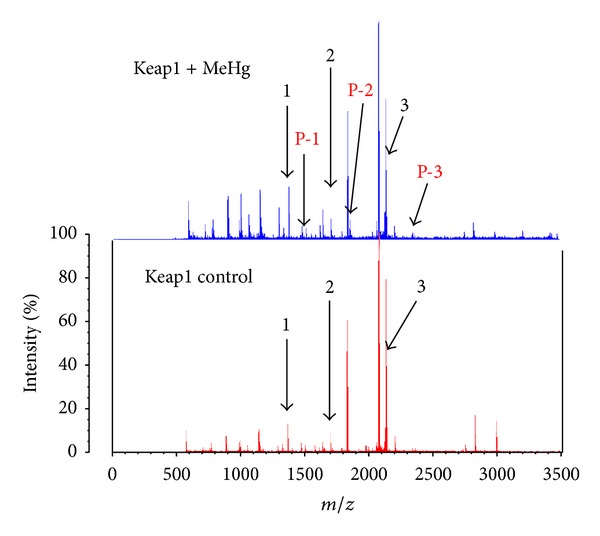
MALDI-TOF/MS analysis of Keap1 reacted with MeHg. Recombinant Keap1 protein (0.25 μg/μL) was incubated for 30 min at 37°C with or without MeHg (10 μM). After the reaction, the Keap1 proteins were trypsinized and subjected to MALDI-TOF/MS analysis. Compared with the calculated mass of the unmodified peptides, the modified peptides P-1 to P-3 showed increased masses of 214 or 216 Da, corresponding to the addition of a single equivalent of MeHg.
Table 2.
MeHg modification sites in Keap1.
| Peak no. | Position | Peptide sequence | Calculated mass | Observed mass |
|---|---|---|---|---|
| 1 | 484–494 | LNSAECYYPER | 1345.5 | 1345.1 |
| 2 | 363–380 | SGLAGCVVGGLLYAVGGR | 1649.9 | 1649.6 |
| 3 | 151–169 | CVLHVMNGAVMYQIDSVVR | 2135.6 | 2135.1 |
| P-1 | 484–494 +MeHg |
LNSAECYYPER +MeHg (214) |
1560.5 | 1559.6 |
| P-2 | 363–380 +MeHg |
SGLAGCVVGGLLYAVGGR +MeHg (216) |
1863.8 | 1864.8 |
| P-3 | 151–169 +MeHg |
CVLHVMNGAVMYQIDSVVR +MeHg (214) |
2350.6 | 2349.5 |
As expected, MeHg activated Nrf2 in SH-SY5Y cells in a concentration- and time-dependent manner and upregulated the downstream proteins such as the GCL catalytic subunit (GCLC) and the GCL modifier subunit (GCLM) (Figure 4) [42]. The same observation was seen in primary mouse hepatocytes and HepG2 cells (data not shown). Other researchers also reported that MeHg led to Nrf2 activation, its nuclear accumulation, and upregulation of Nrf2 downstream antioxidant genes such as HO-1 in a variety of cell types [43–45]. Interestingly, Ni et al. demonstrated different response kinetics in astrocytes and microglia upon MeHg treatment [46]. They concluded that these unique sensitivities appear to be dependent on the cellular thiol status of the particular cell type. This viewpoint is consistent with the evidence that reactive thiols of Keap1 undergo S-mercuration, resulting in Nrf2 activation. Pretreatment with Trolox, an antioxidant, blocked MeHg-mediated oxidative stress, determined by intracellular reactive oxygen species levels, to a significant degree but did not affect Nrf2 activation during the exposure of SH-SY5Y cells to MeHg (Toyama et al., unpublished data). We therefore speculate that S-mercuration, rather than oxidative stress, is involved in the MeHg-dependent activation of Nrf2.
Figure 4.
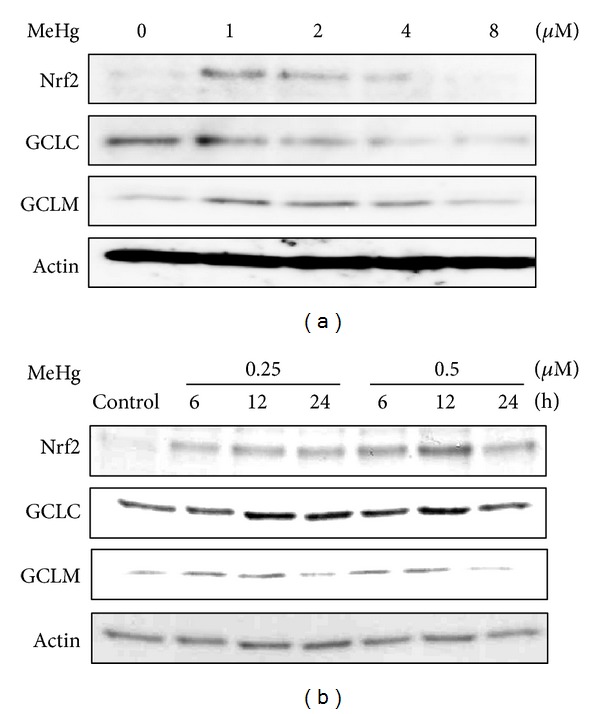
MeHg activates Nrf2 in SH-SY5Y cells. (a) Cells were exposed to MeHg (1, 2, 4, or 8 μM) for 6 h, and the total cell lysates (20 μg) were subjected to western blotting with the antibodies indicated. (b) Cells were exposed to MeHg (0.25 or 0.5 μM) for 6, 12, or 24 h, and the total cell lysates (20 μg) were subjected to western blotting with the antibodies indicated. Reprinted from Toyama et al. [42] with the permission of Biochemical Biophysical Research Communications.
4. Protection against MeHg Toxicity through the Keap1/Nrf2 Pathway
Several lines of evidence indicate that Nrf2-deficient mice are susceptible to the toxicity of a variety of chemicals, including acetaminophen, 7,12-dimethylbenz[a]anthracene, kainic acid, dextran sulfate sodium, and benzo[a]pyrene [47–52]. To examine the role of Nrf2 in protecting against MeHg, we used primary hepatocytes from Nrf2+/+ or Nrf2−/− mice. As shown in Figure 5(a), steady-state levels of Nrf2 downstream proteins, such as GCLC, GCLM, MRP1 [42], and MRP2 [42], were drastically decreased in Nrf2−/− cells. Under these condition, the Nrf2−/− cells were highly sensitive to MeHg compared with the Nrf2+/+ cells (Figure 5(b)) [42]. In agreement with these observations, Ni et al. demonstrated that Nrf2 knockdown by the small hairpin RNA approach reduced the upregulation of its downstream genes such as HO-1 in primary astrocytes and microglia and decreased viability for both cells [45, 46]. The accumulation of mercury in the cerebellum, cerebrum, and liver after oral MeHg administration was significantly higher in the Nrf2−/− mice than in the Nrf2+/+ mice (Figure 5(c)) [53] because detoxifying enzymes associated with MeHg excretion were relatively low in the Nrf2−/− mice. Consistent with this result, the Nrf2−/− mice were also highly susceptible to MeHg in vivo (Figure 5(d)). MeHg-induced neuropathological changes in the cerebellum were evaluated in the Nrf2+/+ and Nrf2−/− mice (Figure 6), and the degeneration of Purkinje cells (Figure 6(a)) and vacuolar degeneration of the medulla (Figure 6(b)) were observed in the cerebellum of MeHg-treated mice. Nrf2 deletion clearly increased these types of MeHg-induced degeneration. These results suggest that Nrf2 is a crucial transcription factor for protecting against MeHg toxicity in vitro and in vivo.
Figure 5.
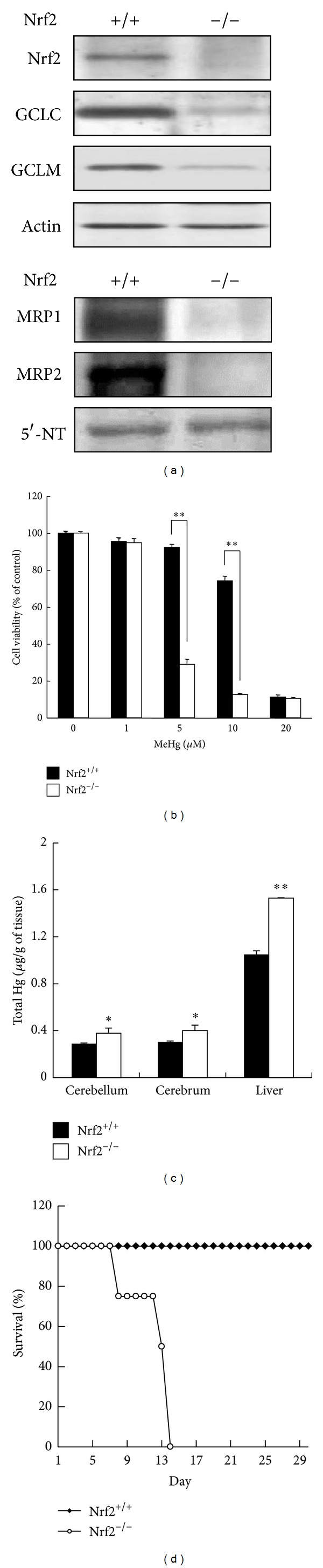
Nrf2 confers protection against MeHg toxicity. (a) Total cell lysates (upper) or crude membrane fractions (lower) from primary hepatocytes from Nrf2+/+ or Nrf2−/− mice were subjected to western blotting with the antibodies indicated. (b) Primary hepatocytes from Nrf2+/+ or Nrf2−/− mice were exposed to MeHg (1, 5, 10, and 20 μM) for 24 h, and then the MTT assay was performed. Each value represents the mean ± SE of three independent experiments. (c) Nrf2+/+ or Nrf2−/− mice were orally administrated MeHg (1 mg/kg). After 48 h, the total mercury contents of the cerebellum, cerebrum, and liver were determined by atomic absorption mercury detection. Each value represents the mean ± SE of five independent experiments. (d) Nrf2+/+ or Nrf2−/− mice were orally administered MeHg (5 mg kg−1 day−1) for 12 days (n = 4) and mortality was recorded. *P < 0.05 and **P < 0.01 for the Nrf2−/− mice compared to the Nrf2+/+ mice. Partially reprinted from Toyama et al. [42, 53] with the permission of Biochemical Biophysical Research Communications and Environmental Health Perspectives.
Figure 6.
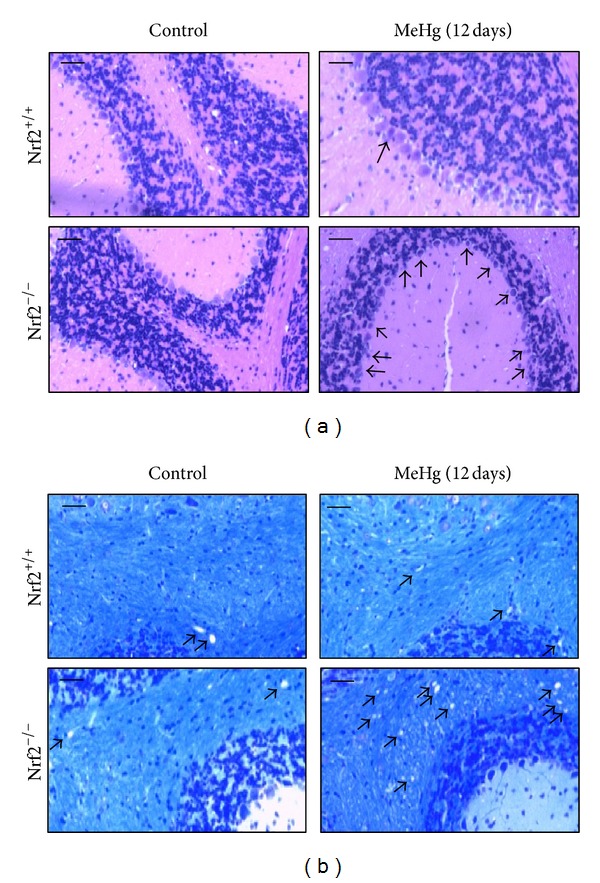
Pathological observations of brains from Nrf2+/+ or Nrf2−/− mice given MeHg. Nrf2+/+ or Nrf2−/− mice were orally administrated MeHg (5 mg kg−1 day−1) for 12 days, and pathophysiological changes in the cerebellum were observed. Bar = 50 μm. (a) Photographs of the cerebellar granule cell layer (HE stain). Arrows indicate the MeHg-induced degeneration of Purkinje cells. (b) Photographs of the cerebellum medulla (KB stain). Arrows indicate the MeHg-induced vacuolar degeneration.
5. Conclusions
We found that MeHg S-mercuration reactions on Mn-SOD and arginase I cause dysfunction in the activities of these catalysts and, therefore, possibly cause oxidative stress in mouse brain and the release of Mn from the active site, leading to a decrease in hepatic Mn levels (seen in rats). The evidence showed that MeHg also S-mercurates Keap1 at its reactive thiols, including Cys151, resulting in Nrf2 activation in a variety of cell types, and thereby upregulation of the downstream gene products such as antioxidant proteins, phase II xenobiotic-metabolizing enzymes, and phase III transporters (as shown in Figure 7). Because these Nrf2-regulated gene products contribute to the blocking of oxidative stress and the facilitation of the detoxification and excretion of MeHg, the findings indicate that the Keap1/Nrf2 system plays a critical role not only in the cellular response to MeHg, but also in the suppression of MeHg toxicity in vitro and in vivo.
Figure 7.
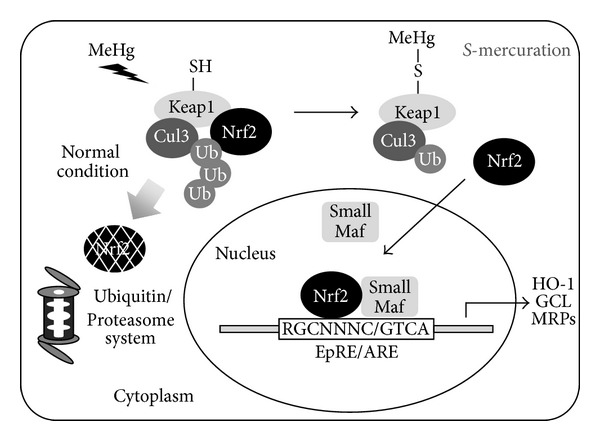
Cellular protective response to MeHg through the Keap1-Nrf2 system.
Conflicts of Interests
The authors declare that they have no conflict of interests.
Acknowledgments
The authors thank Dr. Masayuki Yamamoto, Department of Medical Biochemistry, Tohoku University Graduate School of Medicine, for his contributions to the studies. This work was supported by a grant-in-aid (no. 23117703 to Yoshito Kumagai) for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1.Simpson RB. Association constants of methylmercury with sulfhydryl and other bases. Journal of the American Chemical Society. 1961;83(23):4711–4717. [Google Scholar]
- 2.Ballatori N. Transport of toxic metals by molecular mimicry. Environmental Health Perspectives. 2002;110(supplement 5):689–694. doi: 10.1289/ehp.02110s5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Madejczyk MS, Aremu DA, Simmons-Willis TA, Clarkson TW, Ballatori N. Accelerated urinary excretion of methylmercury following administration of its antidote N-acetylcysteine requires Mrp2/Abcc2, the apical multidrug resistance-associated protein. The Journal of Pharmacology and Experimental Therapeutics. 2007;322(1):378–384. doi: 10.1124/jpet.107.122812. [DOI] [PubMed] [Google Scholar]
- 4.Bridges CC, Joshee L, Zalups RK. MRP2 and the handling of mercuric ions in rats exposed acutely to inorganic and organic species of mercury. Toxicology and Applied Pharmacology. 2011;251(1):50–58. doi: 10.1016/j.taap.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarkson TW. The pharmacology of mercury compounds. Annual Review of Pharmacology. 1972;12:375–406. doi: 10.1146/annurev.pa.12.040172.002111. [DOI] [PubMed] [Google Scholar]
- 6.Farina M, Aschner M, Rocha JB. Oxidative stress in MeHg-induced neurotoxicity. Toxicology and Applied Pharmacology. 2011;256(3):405–417. doi: 10.1016/j.taap.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. Journal of Biological Chemistry. 1999;274(37):26071–26078. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 8.Wild AC, Moinova HR, Mulcahy RT. Regulation of γ-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. The Journal of Biological Chemistry. 1999;274(47):33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- 9.Chan JY, Kwong M. Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochimica et Biophysica Acta. 2000;1517(1):19–26. doi: 10.1016/s0167-4781(00)00238-4. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi A, Suzuki H, Itoh K, Yamamoto M, Sugiyama Y. Transcription factor Nrf2 is required for the constitutive and inducible expression of multidrug resistance-associated protein 1 in mouse embryo fibroblasts. Biochemical and Biophysical Research Communications. 2003;310(3):824–829. doi: 10.1016/j.bbrc.2003.09.086. [DOI] [PubMed] [Google Scholar]
- 11.Vollrath V, Wielandt AM, Iruretagoyena M, Chianale J. Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochemical Journal. 2006;395(3):599–609. doi: 10.1042/BJ20051518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maher JM, Dieter MZ, Aleksunes LM, et al. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology. 2007;46(5):1597–1610. doi: 10.1002/hep.21831. [DOI] [PubMed] [Google Scholar]
- 13.Shinyashiki M, Kumagai Y, Homma-Takeda S, et al. Selective inhibition of the mouse brain Mn-SOD by methylmercury. Environmental Toxicology and Pharmacology. 1996;2(4):359–366. doi: 10.1016/s1382-6689(96)00070-1. [DOI] [PubMed] [Google Scholar]
- 14.Kumagai Y, Homma-Takeda S, Shinyashiki M, Shimojo N. Alterations in superoxide dismutase isozymes by methylmercury. Applied Organometallic Chemistry. 1997;11(8):635–643. [Google Scholar]
- 15.Shinyashiki M, Kumagai Y, Nakajima H, et al. Differential changes in rat brain nitric oxide synthase in vivo and in vitro by methylmercury. Brain Research. 1998;798(1-2):147–155. doi: 10.1016/s0006-8993(98)00400-4. [DOI] [PubMed] [Google Scholar]
- 16.Yasutake A, Nakano A, Miyamoto K, Eto K. Chronic effects of methylmercury in rats. I. biochemical aspects. The Tohoku Journal of Experimental Medicine. 1997;182(3):185–196. doi: 10.1620/tjem.182.185. [DOI] [PubMed] [Google Scholar]
- 17.Rodrigues JL, Serpeloni JM, Batista BL, Souza SS, Barbosa F., Jr. Identification and distribution of mercury species in rat tissues following administration of thimerosal or methylmercury. Archives of Toxicology. 2010;84(11):891–896. doi: 10.1007/s00204-010-0538-4. [DOI] [PubMed] [Google Scholar]
- 18.Kanda H, Sumi D, Endo A, et al. Reduction of arginase I activity and manganese levels in the liver during exposure of rats to methylmercury: a possible mechanism. Archives of Toxicology. 2008;82(11):803–808. doi: 10.1007/s00204-008-0307-9. [DOI] [PubMed] [Google Scholar]
- 19.Kanda H, Toyama T, Shinohara-Kanda A, et al. S-mercuration of rat sorbitol dehydrogenase by methylmercury causes its aggregation and the release of the zinc ion from the active site. Archives of Toxicology. 2012;86(11):1693–1702. doi: 10.1007/s00204-012-0893-4. [DOI] [PubMed] [Google Scholar]
- 20.Karlsson C, Jörnvall H, Höög JO. Sorbitol dehydrogenase: cDNA coding for the rat enzyme. variations within the alcohol dehydrogenase family independent of quaternary structure and metal content. European Journal of Biochemistry. 1991;198(3):761–765. doi: 10.1111/j.1432-1033.1991.tb16077.x. [DOI] [PubMed] [Google Scholar]
- 21.Johansson K, El-Ahmad M, Kaiser C, et al. Crystal structure of sorbitol dehydrogenase. Chemico-Biological Interactions. 2001;130–132(1–3):351–358. doi: 10.1016/s0009-2797(00)00260-x. [DOI] [PubMed] [Google Scholar]
- 22.Woods JS, Davis HA, Baer RP. Enhancement of γ-glutamylcysteine synthetase mRNA in rat kidney by methyl mercury. Archives of Biochemistry and Biophysics. 1992;296(1):350–353. doi: 10.1016/0003-9861(92)90583-i. [DOI] [PubMed] [Google Scholar]
- 23.Yasutake A, Hirayama K. Acute effects of methylmercury on hepatic and renal glutathione metabolisms in mice. Archives of Toxicology. 1994;68(8):512–516. doi: 10.1007/s002040050104. [DOI] [PubMed] [Google Scholar]
- 24.Li S, Thompson SA, Woods JS. Localization of γ-lutamylcysteine synthetase mRNA expression in mouse brain following methylmercury treatment using reverse transcription in situ PCR amplification. Toxicology and Applied Pharmacology. 1996;140(1):180–187. doi: 10.1006/taap.1996.0211. [DOI] [PubMed] [Google Scholar]
- 25.Thompson SA, White CC, Krejsa CM, et al. Induction of glutamate-cysteine ligase (γ-glutamylcysteine synthetase) in the brains of adult female mice subchronically exposed to methylmercury. Toxicology Letters. 1999;110(1-2):1–9. doi: 10.1016/s0378-4274(99)00133-2. [DOI] [PubMed] [Google Scholar]
- 26.Thompson SA, White CC, Krejsa CM, Eaton DL, Kavanagh TJ. Modulation of glutathione and glutamate-L-cysteine ligase by methylmercury during mouse development. Toxicological Sciences. 2000;57(1):141–146. doi: 10.1093/toxsci/57.1.141. [DOI] [PubMed] [Google Scholar]
- 27.Díaz D, Krejsa CM, White CC, Keener CL, Farin FM, Kavanagh TJ. Tissue specific changes in the expression of glutamate-cysteine ligase mRNAs in mice exposed to methylmercury. Toxicology Letters. 2001;122(2):119–129. doi: 10.1016/s0378-4274(01)00341-1. [DOI] [PubMed] [Google Scholar]
- 28.Díaz D, Krejsa CM, White CC, Charleston JS, Kavanagh TJ. Effect of methylmercury on glutamate-cysteine ligase expression in the placenta and yolk sac during mouse development. Reproductive Toxicology. 2004;19(1):117–129. doi: 10.1016/j.reprotox.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochemical and Biophysical Research Communications. 1997;236(2):313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 30.Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes & Development. 1999;13(1):76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, et al. Direct evidence that sulfhydryl groups of keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(18):11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang DD, Hannink M. Distinct cysteine residues in keap1 are required for keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Molecular and Cellular Biology. 2003;23(22):8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eggler AL, Liu G, Pezzuto JM, Van Breemen RB, Mesecar AD. Modifying specific cysteines of the electrophile-sensing human keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(29):10070–10075. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger keap1 ubiquitination and Nrf2 activation. Journal of Biological Chemistry. 2005;280(36):31768–31775. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto T, Suzuki T, Kobayashi A, et al. Physiological significance of reactive cysteine residues of keap1 in determining Nrf2 activity. Molecular and Cellular Biology. 2008;28(8):2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi M, Li L, Iwamoto N, et al. The antioxidant defense system keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Molecular and Cellular Biology. 2009;29(2):493–502. doi: 10.1128/MCB.01080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the keap1-Nrf2 pathway in stress response and cancer evolution. Genes to Cells. 2011;16(2):123–140. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- 38.Abiko Y, Miura T, Phuc BH, Shinkai Y, Kumagai Y. Participation of covalent modification of keap1 in the activation of Nrf2 by tert-butylbenzoquinone, an electrophilic metabolite of butylated hydroxyanisole. Toxicology and Applied Pharmacology. 2011;255(1):32–39. doi: 10.1016/j.taap.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 39.Miura T, Shinkai Y, Jiang HY, et al. Initial response and cellular protection through the keap1/Nrf2 system during the exposure of primary mouse hepatocytes to 1,2-naphthoquinone. Chemical Research in Toxicology. 2011;24(4):559–567. doi: 10.1021/tx100427p. [DOI] [PubMed] [Google Scholar]
- 40.Levonen AL, Landar A, Ramachandran A, et al. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochemical Journal. 2004;378(2):373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, et al. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the keap1 sensor modified by inducers. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(7):2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Toyama T, Sumi D, Shinkai Y, et al. Cytoprotective role of Nrf2/Keap1 system in methylmercury toxicity. Biochemical and Biophysical Research Communications. 2007;363(3):645–650. doi: 10.1016/j.bbrc.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 43.Wang L, Jiang H, Yin Z, Aschner M, Cai J. Methylmercury toxicity and Nrf2-dependent detoxification in astrocytes. Toxicological Sciences. 2009;107(1):135–143. doi: 10.1093/toxsci/kfn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lämsä V, Levonen AL, Leinonen H, Ylä-Herttuala S, Yamamoto M, Hakkola J. Cytochrome P450 2A5 constitutive expression and induction by heavy metals is dependent on redox-sensitive transcription factor nrf2 in liver. Chemical Research in Toxicology. 2010;23(5):977–985. doi: 10.1021/tx100084c. [DOI] [PubMed] [Google Scholar]
- 45.Ni M, Li X, Yin Z, et al. Methylmercury induces acute oxidative stress, altering Nrf2 protein level in primary microglial cells. Toxicological Sciences. 2010;116(2):590–603. doi: 10.1093/toxsci/kfq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ni M, Li X, Yin Z, et al. Comparative study on the response of rat primary astrocytes and microglia to methylmercury toxicity. Glia. 2011;59(5):810–820. doi: 10.1002/glia.21153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Enomoto A, Itoh K, Nagayoshi E, et al. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicological Sciences. 2001;59(1):169–177. doi: 10.1093/toxsci/59.1.169. [DOI] [PubMed] [Google Scholar]
- 48.Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicology and Applied Pharmacology. 2001;173(3):154–160. doi: 10.1006/taap.2001.9176. [DOI] [PubMed] [Google Scholar]
- 49.Xu C, Huang MT, Shen G, et al. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Research. 2006;66(16):8293–8296. doi: 10.1158/0008-5472.CAN-06-0300. [DOI] [PubMed] [Google Scholar]
- 50.Kraft AD, Lee JM, Johnson DA, Kan YW, Johnson JA. Neuronal sensitivity to kainic acid is dependent on the Nrf2-mediated actions of the antioxidant response element. Journal of Neurochemistry. 2006;98(6):1852–1865. doi: 10.1111/j.1471-4159.2006.04019.x. [DOI] [PubMed] [Google Scholar]
- 51.Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Research. 2006;66(24):11580–11584. doi: 10.1158/0008-5472.CAN-06-3562. [DOI] [PubMed] [Google Scholar]
- 52.Aoki Y, Hashimoto AH, Amanuma K, et al. Enhanced spontaneous and benzo(a)pyrene-induced mutations in the lung of Nrf2-deficient gpt delta mice. Cancer Research. 2007;67(12):5643–5648. doi: 10.1158/0008-5472.CAN-06-3355. [DOI] [PubMed] [Google Scholar]
- 53.Toyama T, Shinkai Y, Yasutake A, Uchida K, Yamamoto M, Kumagai Y. Isothiocyanates reduce mercury accumulation via an Nrf2-dependent mechanism during exposure of mice to methylmercury. Environmental Health Perspectives. 2011;119(8):1117–1122. doi: 10.1289/ehp.1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]