

Abstract
The molecular and cellular basis of coronavirus neurovirulence is poorly understood. Since neurovirulence may be determined at the early stages of infection of the central nervous system (CNS), we hypothesize that it may depend on the ability of the virus to induce proinflammatory signals from brain cells for the recruitment of blood-derived inflammatory cells. To test this hypothesis, we studied the interaction between coronaviruses (mouse hepatitis virus) of different neurovirulences with primary cell cultures of brain immune cells (astrocytes and microglia) and mouse tissues. We found that the level of neurovirulence of the virus correlates with its differential ability to induce proinflammatory cytokines (interleukin 12 [IL-12] p40, tumor necrosis factor alpha, IL-6, IL-15, and IL-1β) in astrocytes and microglia and in mouse brains and spinal cords. These findings suggest that coronavirus neurovirulence may depend on a novel discriminatory ability of astrocytes and microglia to induce a proinflammatory response in the CNS.
The brain has a unique immune system, which becomes activated during pathological insults including viral infections and immune-mediated diseases (50). The brain lacks elements of lymphoid tissue but contains instead two unique resident cells, astrocytes and microglia, with the potential for mediating immune responses (2, 38). Understanding the nature of the immune responses in the brain is important for understanding the pathogenesis of infectious and immune-mediated diseases of the central nervous system (CNS) such as multiple sclerosis (MS) (1). Experimental viral infections of the CNS provide an opportunity to study the function of the brain immune system.
Most neurotropic viruses reach the CNS by hematogenous spread, with the exception of viruses such as herpes simplex and rabies viruses, which invade the CNS also by retrograde interneuronal transport (21, 36). The hematogenous invasion of viruses begins by interaction between virus and elements of the blood-brain barrier (BBB) (34). Major components of the BBB are the foot processes of astrocytes while microglial processes are also found in proximity to the BBB. We hypothesize that events immediately after the initial entry of virus into the brain and the interaction between the virus and brain cells determine neurovirulence and the degree of encephalitis. This requires sending proinflammatory signals for the recruitment of exogenous inflammatory cells into the brain, including T and B lymphocytes, NK cells, and macrophages. The main task of proinflammatory signaling presumably falls on the local brain immune system of astrocytes and microglia. Although endothelial cells, pericytes, and even neurons can produce some proinflammatory signaling, astrocytes and microglia are generally considered the main source of proinflammatory molecules in the brain. Since immune cells communicate by cytokine secretion (7), we hypothesized that the cytokine response of brain immune cells to viral infection may help to determine the subsequent disease phenotype. If this theory is true, then neurotropic viruses induce a proinflammatory response that will subsequently contribute to the development of encephalitis, whereas nonneurotropic viruses induce significantly reduced proinflammatory signals, thus avoiding the development of encephalitis.
To test this hypothesis, we used the experimental model system of coronavirus-induced CNS infection in mice, which has been studied in our laboratory (27, 30). In this model, mice infected with a neurotropic strain (MHV-A59) develop acute meningoencephalitis, followed by chronic demyelination (24, 25, 49, 58). Mice infected with the nonneurotropic strain (MHV-2) develop only acute meningitis, without encephalitis or demyelination (12). Both MHV-A59 and MHV-2 also produce acute hepatitis in infected mice. During acute infection with MHV-A59, both neurons and glial cells become productively infected. Acute encephalitis is characterized by perivascular inflammatory infiltrates of mononuclear cells, especially lymphocytes and macrophages, microglial and astrocytic proliferation, and microglial nodules. Viral antigen can be detected in specific locations in the brain, especially in areas associated with the olfactory and limbic systems (23, 26). Inflammatory infiltrates start 2 to 3 days after infection, peak at days 5 to 7 after infection, and gradually decline over the next week. Viral antigen peaks at day 5 postinfection (p.i.) and disappears approximately 10 days after infection (23, 26). Virus then clears from neurons, but low levels of viral RNA persist in glial cells and mice develop a chronic inflammatory demyelinating disease similar to MS in humans (14, 24). In MHV-2 infection, there are no inflammatory infiltrates in the brain and no signs of acute encephalitis. Viral antigen can be detected only in the meninges and to a limited extent in some subpial and subventricular astrocytes (13). Although the molecular and cellular basis of coronavirus neurovirulence is poorly understood, neurovirulence can be measured by the mortality rate of mice infected by the virus (50% lethal dose [LD50]). In coronaviruses with liver susceptibility, the LD50 can be attributed to both hepatitis and encephalitis; however, in strains like JHM that do not produce hepatitis, the LD50 is almost exclusively due to neurovirulence. By analyzing the correlation between LD50 and viral replication of different neurotropic viruses in the brain, it becomes evident that brain virus titers alone cannot explain the degree of neurovirulence. However, the neurovirulence correlates well with the amount of inflammatory reaction in the brain (45, 46), which in turn depends on the amount of proinflammatory signaling by cytokines and chemokines. Recent observations of the highly virulent severe acute respiratory syndrome coronavirus (43) implicate a clinical and pathological picture of acute respiratory distress syndrome and multiorgan failure. The increased virulence of the virus is consistent with the manifestations of a cytokine storm, which has been reported in association with a variety of viral infections (17, 56, 61).
Consistent with this hypothesis, mice infected with another neurotropic-demyelinating virus strain, mouse hepatitis virus (MHV) JHM, develop a significant proinflammatory cytokine response in the brain during acute encephalitis (41, 42, 52). Similar cytokine induction has been shown with another demyelinating virus, Theiler's virus (57). In all of these cases, the proinflammatory immune response could be attributed to either infiltrating blood-borne inflammatory cells, such as lymphocytes and macrophages, and/or local brain immune cells, such as astrocytes and microglia. To evaluate the role of local CNS immune cells in cytokine immune responses, we studied cytokine profiles of primary glial cultures enriched with either astrocytes or microglia following infection with different MHVs. These cultures are devoid of blood-derived inflammatory cells, thus allowing us to assess the unique contribution of local CNS immune cells to the process of MHV disease without the contribution of blood-borne cells. We chose to compare MHV-A59 with MHV-2 because the two viruses are genetically closely related, and although both can grow equally well in tissue culture brain cells (astrocytes and microglia), the two viruses are very different in neurovirulence and produce strikingly different pathological pictures in the brain following intracerebral injection. We hypothesized that part of the difference in neurovirulence of these two viruses may be in their differential abilities to interact with brain immune cells such as astrocytes and microglia.
(Parts of this work were presented at the annual meetings of the American Society for Virology, Madison, Wis., July 2001, and Lexington, Ky., July 2002; at the International Society of Neurovirology meeting, Dusseldorf, Germany, June 2002; and at the 9th International Symposium of Nidoviruses, Egmond aan zee, The Netherlands, May 2003.)
MATERIALS AND METHODS
Viruses and mice.
The following plaque-purified viruses were used in this study: wild-type MHV-A59 (9, 25), wild-type MHV-2 (13, 15, 19), and wild-type JHM (51). Viral stocks were 106 to 107 PFU/ml. Viruses were propagated and virus titers were analyzed on the L2 murine fibroblast cell line. Late-pregnancy C57BL/6 mice (for cell culture experiments) and 4-week-old male C57BL/6 mice (for in vivo experiments) were purchased from NCI Laboratories (Frederick, Md.). Four-week-old tumor necrosis factor alpha (TNF-α) knockout mice, B6;129S6-Tnftm1Gkl, and 4-week-old interleukin 12 (IL-12) p40 knockout mice, B6.129S1-Il12btm1Jm, were purchased from Jackson Laboratory (Bar Harbor, Maine), and the absence of the specific cytokine gene in mouse tissues was confirmed by PCR.
Preparation of mouse astrocytes and microglia cultures.
Mixed glial cultures were prepared from newborn mice; astrocytes and microglia were further purified at 9 to 14 days in culture by the differential adhesion property and the shake-off technique, as previously described (28, 29, 53, 54). The purity of the astrocytes and microglial cultures was confirmed by immunofluorescence analysis with antibodies for glial fibrillary acidic protein (astrocytes) and CD11b-Mac-1 (microglia), which consistently showed purity levels of 98 and 95%, respectively. The astrocytes and microglia culture supernatants were collected at 0, 8, 16, and 24 h p.i.
Immunofluorescence of cell cultures.
Astrocytes were placed on coverslips and pretreated with 0.01% poly-l-lysine solution (Sigma) in 12-well plates at a concentration of 100,000 cells per well. The astrocyte cultures were infected with MHVs at a multiplicity of infection of 2 and incubated for 24 h. Cells were rinsed with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde in PBS at 4°C for 1 h. After rinsing three times in PBS for 5-min intervals, cells were permeabilized for 10 min with 1% Triton X-100 in PBS and washed three times with PBS. Cells were then incubated for 10 min with a blocking solution (10% bovine serum albumin [BSA] in PBS) and washed three times in PBS. Cells were incubated with the primary antibody (anti-MHV monoclonal antibody or anti-glial fibrillary acidic protein polyclonal antibody [Novus Bio Inc.] at a 1:200 dilution) for 1 h at 37 or 4°C overnight and then washed with PBS. Cells were incubated with the secondary antibody (chicken anti-mouse immunoglobulin G 488 or goat anti-rabbit immunoglobulin G 555 [Molecular Probes] at a 1:500 dilution) for 1 h at room temperature and washed with PBS three times for 5-min intervals. Nuclei were stained with propidium iodide-4′,6′-diamidino-2-phenylindole (DAPI) (Vector Laboratories) at 1.5 μg/ml of mounting media. The coverslips were inverted onto slides containing 10 μl of mounting media (Vector Laboratories). Slides were viewed with an Olympus BX60 fluorescence microscope.
Viral infection.
Confluent astrocytes or microglial cultures were treated with trypsin and distributed into 100-mm-diameter dishes. Cells were counted when they reached confluence again and then incubated with MHV-A59, JHM, or MHV-2 at 2 PFU/cell for 1 h and then washed and incubated at 37°C and 5% CO2 for 24 h, at which point cell culture supernatants were collected for enzyme-linked immunosorbent assay (ELISA) and cells were collected for RNA extraction. For infections with nonreplicating virus, virus was UV treated for 1 h prior to incubation.
Four-week-old C57BL/6 mice were injected intracerebrally with 50 PFU of MHVs suspended in 25 μl of sterile PBS and 0.75% BSA. Control animals were injected with 25 μl of sterile PBS-BSA. Animals were sacrificed at 5 and 30 days p.i., at which time brains, livers, and spinal cords were removed for total RNA extraction.
Viral titration.
The astrocytes and microglia culture supernatants were collected at 0, 8, 16, and 24 h p.i. The virus titers were detected on L2 cells by plaque assay as previously described (28).
RNA isolation from cells and tissues.
Total RNA was isolated from cultured cells and mouse tissues by using the StrataPrep Total RNA miniprep kit (Stratagene, La Jolla, Calif.) according to the manufacturer's instructions. Briefly, cells were collected and tissues were homogenized in lysis buffer, subjected to 70% ethyl alcohol precipitation, centrifuged (Eppendorf 5415C) at >10,000 rpm for 30 s, washed with high- and low-salt buffers, centrifuged again, dissolved in RNase-free water, and stored at −70°C for future use. The concentration and purity of the RNA samples were determined by measuring the absorbance at 260 nm (A260) and A280 in a spectrophotometer. The desired A260/A280 ratio of pure RNA was between 1.8 and 2.0. The integrity of the RNA samples was further confirmed by electrophoresis on 1% agarose gels. All the RNA samples were treated with DNase to avoid interference of genomic DNA.
Cytokine mRNA detection.
The cytokine mRNA levels of the MHV-infected cell cultures and animal tissues were detected by using the mouse common cytokine GEArray kit (Super Array Inc., Bethesda, Md.). Each array consists of 56 coordinates containing specific cDNA fragments spotted in duplicate as well as control sequences (PUC18 as a negative control; β-actin and glyceraldehyde-3-phosphate dehydrogenase [GAPDH] as loading controls). We compared the expression of 23 pathway-specific genes by using this array in astrocytes, microglia cultures, mouse brains, and spinal cords according to the manufacturer's instructions. In brief, 5 to 10 μg of total RNA was used as a template to mix with the primer mixtures for the synthesis of gene-specific cDNA by reverse transcription, which was labeled with biotin-dUTP. The labeled cDNA probe was then hybridized with gene-specific cDNA fragments spotted onto a nylon membrane at 68°C overnight. On the following day, the membrane was washed several times to remove nonspecific combinations and chemiluminescent detection was performed, including blocking the membrane and incubating it with alkaline phosphatase-conjugated streptavidin and CDP-star chemiluminescent substrate. The membrane was then exposed to an X-ray film for 5 to 10 min according to the strength of the signal. Specific array signal spots were analyzed with ImageQuant quantitation software (Molecular Dynamics) by scanning densitometry. The signal from the expression of each cytokine gene was normalized to the signal derived from β-actin on the same membrane and expressed as cytokine mRNA arbitrary units. These were calculated with the following formula:
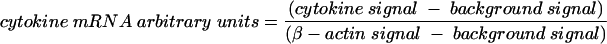 |
For cell cultures, the experiment with each group was repeated three times with different RNA from separate infections. For the in vivo study, equivalent amounts of total RNA from 3 mice were mixed together for cDNA synthesis and hybridization and each experiment was repeated twice with different membranes. The membranes were stripped and reused up to three times. In addition, control sham-infected mice and sham-infected cell cultures with uninfected L2 cell lysate (similar to the medium that was used for viral preparations) were analyzed in parallel for each experiment.
The statistical significances of the different groups were determined by using the Student t test. A twofold increase in the level of gene expression over that of the control is regarded as significant in this study, as referenced in a previous report in which the same technique was used (10, 47).
Protein analysis by ELISA.
The cell culture supernatants, which were collected at different time points, were used for the analysis of cytokine protein expression by sandwich ELISA (BD Pharmingen, San Diego, Calif.) according to the manufacturer's instructions. The lower detection limits for TNF-α, IL-6, and IL-12 p40 were all 15 pg/ml. Briefly, 96-well Nunc Maxisorb plates were coated with anti-cytokine capture antibodies overnight at 4°C, followed by blocking with assay diluent and washing with 0.05% Tween-PBS. The cytokine standard and samples diluted in assay buffer were added to coated wells and incubated at 4°C overnight and then washed with PBS. Biotin-labeled detection antibodies were then added, followed by washing and the addition of streptavidin-horseradish peroxidase conjugate. A yellow color in the reaction mixture was developed by a 3,3′,5,5′-tetramethylbenzidine substrate solution, and 2 N H2SO4 was used as a stop solution. The plates were then read by a microplate reader at 405 nM, and 570 nm was used as a correction λ. The cytokine protein concentration was obtained from a standard curve.
Statistical analysis.
Results presented as mean values ± standard deviations were analyzed by analysis of variance.
RESULTS
Viral replication in astrocytes and microglial cells.
We have previously studied the kinetics of MHV-A59 replication in primary glial cells. Virus titers reach a plateau at 24 h p.i. and may persist as long as the cultures survive (28). Thus, we studied by plaque assays the titers of both neurotropic (MHV-A59) and nonneurotropic (MHV-2) viruses in astrocytes and microglial cultures at 0, 8, 16, 24, and 48 h following infection. The growth curves of both viruses were very similar. The titers at the peak of viral replication are shown in Fig. 1, where MHV-A59 and MHV-2 had similar titers in both astrocytes and microglia cultures at 24 h p.i. (the time point used for assessment of cytokine levels). Astrocyte cultures were also analyzed for amounts of viral antigen by immunofluorescence by using monoclonal antibodies against the nucleocapsid protein of MHV, which cross-reacts with MHV-A59 and MHV-2. Cultures infected with MHV-A59 expressed viral antigen in about 20 to 30% of cells, consistent with previous results (28). Cultures infected with MHV-2 expressed viral antigen in up to 70 to 80% of cells in some cultures. The similar abilities of both viruses to replicate in astrocytes and microglia precludes the possibility that different viral loads may be responsible for the difference in cytokine response by the different viruses. The level of viral replication in microglia was 1 to 2 logs lower than in astrocytes, indicating that astrocytes are more susceptible to MHV infection than microglia.
FIG. 1.

Virus titers in astrocytes and microglial cultures at 24 h postinfection with MHV-A59 and MHV-2, detected by plaque assay.
Cytokine mRNA analysis in vitro.
Cytokine mRNA profiles in astrocytes infected with a neurotropic MHV-A59 and the nonneurotropic MHV-2 at 24 h p.i. were assessed by the gene array assay with a 23-cytokine profile (Fig. 2 and 3). Constitutive expression of cytokines in control uninfected cultures included IL-2, IL-7, IL-13, and lymphotoxin. Following infection of astrocytes with either MHV-A59 or MHV-2, there was a significant up-regulation of both pro- and anti-inflammatory cytokines. However, the mRNA levels of IL-12 p40, TNF-α, IL-15, and IL-6 were significantly higher in MHV-A59-infected cells than in MHV-2-infected cells (Fig. 4). The mRNA level of IL-1β was slightly more up-regulated following MHV-A59 infection than in MHV-2-infected astrocytes. The mRNA of cytokines IL-5, TNF-β, IL-10, IL-11, IL-13, and IL-1α was up-regulated in both neurotropic and nonneurotropic viral infections (data not shown). None of the cytokines were down-regulated compared to the control uninfected culture.
FIG. 2.

Analysis of mRNA levels of cytokines 24 h following infection of astrocytes with a neurotropic (MHV-A59) and a nonneurotropic (MHV-2) virus compared with an uninfected control culture. The blots of mouse cytokine array assays are shown. The cytokine key is as follows: A, colony-stimulating factor granulocyte; B, gamma interferon; C, IL-1α; D, IL-1β; E, IL-2; F, IL-3; G, IL-4; H, IL-5; I, IL-6; J, IL-7; K, IL-9; L, IL-10; M, IL-11; N, IL-12 p35; O, IL-12 p40; P, IL-13; Q, IL-15; R, IL-16; S, IL-17; T, IL-18; U, lymphotoxin B; V, TNF-α; W, TNF-β; X, GAPDH; Y, β-actin; Z, bacterial plasmid (pUC18).
FIG. 3.

The blot assays shown in Fig. 2 were scanned and analyzed for relative quantitative levels of mRNA. The results are shown in a graphical form as arbitrary units based on the cytokine levels relative to the level of the housekeeping genes as explained in the text. The results for two alpha interferon (IFN-α) genes from another kit were also included in this figure. LT-b, lymphotoxin B; G-CSF, colony-stimulating factor granulocyte.
FIG. 4.

Analysis of mRNA levels of 5 cytokines (TNF-α, IL-12 p40, IL-6, IL-15, and IL-1β) following infection with a neurotropic (MHV-A59) and a nonneurotropic (MHV-2) virus compared with an uninfected control. Cytokine levels were examined in astrocytes (24 h postinfection), microglia (24 h postinfection), brains during acute encephalitis (5 days postinfection), and spinal cords during chronic demyelinating disease (30 days postinfection). Cytokine mRNA was expressed in relative strength, as evaluated against the expression of a housekeeping gene, β-actin (arbitrarily designated as 1.0 U of strength).
We then examined the cytokine response to viral infection in microglia, another brain immune cell with the potential for mediating immune responses. Although infection of microglia by MHV viruses is less productive than astrocyte infections, the ability to induce cytokines may not require high levels of viral replication. We tested the cytokine mRNA profiles of microglia cultures by cDNA gene array at 24 h p.i. with either MHV-A59 or MHV-2. The mRNA of IL-1β, IL-7, IL-13, IL-2, and TNF-β was constitutively expressed in uninfected cells (data not shown). However, the levels of mRNA of IL-12 p40, TNF-α, IL-15, IL-6, and IL-1β were higher in MHV-A59-infected cells than in MHV-2-infected cells and uninfected control cultures (Fig. 4). None of the cytokine mRNA levels was up-regulated in MHV-2-infected cells more than in MHV-A59 infected cells. Thus, in both astrocytes and microglia, the same core of five proinflammatory cytokine mRNAs (IL-12 p40, TNF-α, IL-15, IL-6, and IL-1β) was up-regulated by the neurotropic virus more than by the nonneurotropic virus.
To test whether cytokine induction resulted from active viral replication, we incubated astrocytes and microglial cultures with UV-inactivated, nonreplicating viruses. Cytokine mRNA profiles following this treatment were not up-regulated above the background of uninfected control cultures, except for a slight up-regulation of TNF-α in both MHV-2- and A59-infected cells and a minimal up-regulation of IL-1β only in MHV-2-infected cells (data not shown).
Up-regulation of a specific set of cytokines may be a general phenomenon in all cell types susceptible to MHV infection. To test whether the change in cytokine gene expression profile found in CNS cells following MHV infection was specific for the CNS, we compared the cytokine mRNA profiles in astrocytes and microglia to the profile in the L2 murine fibroblast cell line, which is known to be susceptible to MHV infection. In L2 cells, IL-13 and TNF-α mRNA levels were up-regulated with both neurotropic (MHV-A59) and nonneurotropic (MHV-2) viral infections, whereas IL-1α mRNA was up-regulated in MHV-A59 infection more than in MHV-2 infection (data not shown). None of the cytokines were up-regulated in MHV-2-infected cells more than in MHV-A59-infected cells. Thus, the differential cytokine profile seen in CNS immune cells is not a universal phenomenon present in all cell types that are infected by MHVs.
Cytokine mRNA analysis in vivo.
Since the above results were based on in vitro studies, we wanted to know whether a similar picture is observed in mice in vivo. Pathologically, the inflammatory CNS disease shifts from acute encephalitis in the brain to chronic demyelinating disease, primarily in the spinal cord, following MHV-A59 infection. In comparison, there is no encephalitis or demyelination following MHV-2 infection. Thus, we wanted to see whether the up-regulation of the cytokine profile in vivo correlates with the disease activity in MHV-A59 compared with the lack of disease activity in MHV-2. Although both the brain and spinal cord were tested in both acute and chronic stages, we only show the relevant comparison (Fig. 4), i.e., acute disease in the brain and chronic disease in the spinal cord following infection with both viruses. We found that the same set of cytokines (IL-12 p40, TNF-α, IL-6, IL-15, and IL-1β) that was up-regulated in vitro was also up-regulated in the mouse brain at the peak of acute disease (day 5 p.i.) and in the spinal cord at 30 days p.i. during the chronic demyelinating disease following infection with MHV-A59 (Fig. 4). These five cytokines were not up-regulated in the brain during chronic disease or in the spinal cord during acute disease (data not shown). There was also minimal, if any, change in cytokine expression in MHV-2 brains or spinal cords during acute or chronic disease. Thus, the close correlation between cytokine induction and the pathology of inflammatory CNS disease strongly suggests a cause-and-effect relationship between cytokine up-regulation in specific brain cells (i.e., astrocytes and microglia) and the disease process in vivo.
Cytokine protein analysis.
To test whether protein profiles were consistent with the mRNA profile, we tested by ELISA the protein levels of the main three proinflammatory cytokines (IL-12 p40, TNF-α, and IL-6) that were up-regulated in the supernatant of astrocyte cultures infected with MHV-A59 and MHV-2 (Fig. 5). The levels of all three cytokine proteins were significantly higher in astrocytes infected with the neurotropic virus (MHV-A59) than in those infected with the nonneurotropic virus (MHV-2). These results showed that the cytokine mRNA and protein profiles in astrocytes infected with different neurotropic viruses correlated well with the degree of neurovirulence of the viruses.
FIG. 5.

Cytokine protein levels in astrocytes at 24 h postinfection with MHV-A59 and MHV-2 compared with an uninfected control group. The levels of cytokine proteins are expressed in picograms per milliliter.
Do all neurotropic viruses produce similar cytokine profiles?
Although MHV-A59 is the primary neurotropic virus studied in our lab, another neurotropic MHV (MHV-JHM) has been extensively investigated (45, 54). To assess whether MHV-A59 induces a profile of proinflammatory cytokines similar to that induced by MHV-JHM, we studied the cytokine profile of all three viruses (A59, JHM, and MHV-2) in astrocytes. The results are shown in Fig. 6 and indicate that both neurotropic viruses up-regulate the same set of cytokines in astrocytes as MHV-2.
FIG. 6.

Analysis of mRNA levels of 5 cytokines (TNF-α, IL-12 p40, IL-6, IL-15, and IL-1β) in astrocytes following infection with neurotropic viruses (MHV-A59 and MHV-JHM) and a nonneurotropic virus (MHV-2) compared with an uninfected control. Cytokine mRNA levels were examined by 24 h postinfection and were expressed in relative strength, as evaluated against the expression of a housekeeping gene, β-actin (arbitrarily designated as 1.0 U of strength).
Sequence of cytokine induction.
To understand the sequence of cytokine induction following a neurotropic viral infection, we studied cytokine profiles following infections of astrocytes derived from IL-12 p40 and TNF-α knockout mice (Table 1). The mRNA levels of IL-6 and IL-15 were completely dependent on the availability of TNF-α, whereas the level of IL-12 p40 mRNA was completely independent of TNF-α. TNF-α exhibited a high level of expression upon virus infection in astrocytes from IL-12 p40 knockout mice. IL-1β depended on both IL-12 and TNF-α. The levels of inducible nitric oxide synthase (iNOS) were also completely dependent on TNF-α (data not shown). Thus, based on these studies, we propose a sequence of cytokine induction in the CNS following neurotropic viral infection (Fig. 7). The two major regulatory cytokines in this diagram are IL-12 p40 and TNF-α.
TABLE 1.
Relative levels of cytokine mRNA compared with β-actin determined by cDNA gene array assaya
| Culture type and treatmentb | Amt of cytokine
|
||||
|---|---|---|---|---|---|
| IL-1β | IL-6 | IL-12 p40 | IL-15 | TNF-α | |
| B6 + medium | 0 | 0 | 0 | 0 | 0 |
| B6 + virus | 12 | 24 | 44 | 7 | 46 |
| B6 IL-12 p40−/− + medium | 0 | 6 | 0 | 0 | 16 |
| B6 IL-12 p40−/− + virus | 3 | 22 | 0 | 6 | 69 |
| B6 TNF-α−/− + medium | 0 | 0 | 2 | 0 | 0 |
| B6 TNF-α−/− + virus | 2 | 0 | 40 | 0 | 0 |
The level of β-actin was arbitrarily designated 100. The levels of all other cytokines are in arbitrary units relative to the level of β-actin.
Each culture was treated with either 1 PFU of MHV-A59 (virus) or uninfected medium/cell. Astrocyte cultures were prepared from newborn mice (normal B6, IL-12 p40−/− B6 knockout mice, and TNF-α−/− B6 knockout mice).
FIG. 7.

A schematic diagram of the proposed sequence of events of proinflammatory cytokine secretion from astrocytes following a neurotropic viral infection with MHV-A59 at 2 PFU/cell (multiplicity of infection = 2).
DISCUSSION
A number of exogenous and endogenous factors control the expression, transcription, and secretion of cytokines to maintain a dynamic balance within the cytokine network. Dysregulation at any level, particularly exaggerated cytokine production, can contribute to disease (33, 48). Previous reports showed that during infection of the brain with neurotropic MHV viruses there is an increase in the mRNA levels of certain cytokines, including some of those described here (41, 42). However, only neurotropic MHVs were analyzed in those reports. A recent report analyzed several cytokines and chemokines in the brains of JHM-infected mice lacking most components of the immune system (Rag−/− mice). In these mice, up-regulation of IL-12 p40, TNF-α, IL-6, and IL-1β was shown following MHV infection, demonstrating the contribution of brain immune cells to proinflammatory signaling during acute encephalitis (16).
When a neurotropic virus infects the brain, the brain immune system is the first line of defense before any hematogenous inflammatory cells are recruited. It has been previously shown that brain immune cells such as astrocytes and microglia respond to various stimulations, such as lipopolysaccharide, or to viral infection in vitro by the induction of cytokines (4, 32, 40). It therefore appears that the CNS immune cells may respond to the invading pathogen in a discriminatory way, which will determine the fate of the infection and the subsequent pathological process. Surprisingly, this intuitive hypothesis has never been directly proven. We therefore hypothesized that a neurotropic pathogen would elicit a proinflammatory response from brain immune cells while a nonneurotropic virus would produce a significantly lower level of that response. We describe here for the first time the ability of astrocytes and microglia, the innate immune cells of the brain, to produce discriminatory immune responses to different phenotypes of viral infections. This ability is parallel to the discriminatory immune response of lymphocytes to various stimulations. T-helper lymphocytes are composed of two distinct subpopulations that can be distinguished based on physical and functional properties (55). The characteristic feature of these subpopulations of lymphocytes is their differential cytokine immune response: the Th1 subset produces a proinflammatory cytokine response while the Th2 subset produces an anti-inflammatory cytokine response (35). We show that although the cytokine mRNA profiles are different between astrocytes and microglia cultures, a neurotropic virus (MHV-A59) elicits a profoundly more elaborate proinflammatory immune response in astrocytes and microglia than does the nonneurotropic virus MHV-2, even though both viruses infect astrocytes and microglia at the same titer levels. Since there are no lymphocytes in our cultures, the pattern of proinflammatory response in astrocytes and microglia cannot be labeled a Th1 response and should therefore be labeled based on the cell type it originated in. We provisionally labeled it the “Ast/Mic 1” response, which is parallel to the Th1 proinflammatory response of T-helper lymphocytes. The proinflammatory response of the neurotropic virus (A59) includes a set of five up-regulated proinflammatory cytokines. Thus, the cytokine profile described in this report reflects the neurovirulence of the strain of virus and is probably related to the neurotropic phenotype of the virus. However, neurotropism and neurovirulence depend on other factors in addition to cytokine secretion, including the affinity of the virus for neuronal cell infection and the ability of the virus to travel within the CNS by intra- and interneuronal transport.
This picture of the cytokine profile is entirely consistent with the pathological process of the inflammatory disease, which is manifested first as encephalitis, primarily in the brain, and then as an inflammatory demyelinating disease, primarily in the spinal cord. A very strong support for the idea that induction of the Ast/Mic 1 cytokine response is indeed a crucial step in the disease process of a neurotropic viral infection comes from the study in which antibody treatment against IL-12 significantly reduced the amount of inflammatory demyelination caused by Theiler's viral infection (18).
The set of cytokines that is up-regulated following a neurotropic viral infection consists of proinflammatory cytokines which would normally be associated with the recruitment of inflammatory cells, including lymphocytes and macrophages, to the site of infection. These in turn will contribute to the characteristic manifestations of encephalitis, which mainly consists of perivascular mononuclear inflammatory infiltrates. These cytokines may also contribute to the disruption of the BBB, which facilitates additional dissemination of the virus within the CNS. Previous publications on the involvement of these cytokines in autoimmune processes suggest the following points. (i) IL-12 induction involves innate immune response in viral and autoimmune conditions such as Theiler's virus encephalomyelitis, experimental autoimmune encephalomyelitis (EAE) and experimental colitis (11, 18, 20, 40). (ii) TNF-α is an important mediator of proinflammatory functions in autoimmune and viral diseases (5, 6, 59) and may also induce iNOS, which can also contribute to the development of the pathological process. Although demyelination can occur in the absence of nitric oxide synthase (60), iNOS may still contribute to the pathogenesis of MHV-induced disease (22). TNF-α is also known as an inducer of the secretion of IL-15, IL-6, and IL-1β. (iii) IL-15 mediates T-cell response and innate activity and is also known as an inducer of additional TNF-α (31, 33). (iv) IL-6 is a cytokine frequently induced by TNF-α and other proinflammatory regulator molecules and has been associated with neurotropic viral infections, EAE, and other autoimmune conditions of the CNS (3, 8, 37, 39, 40, 44).
The difference in cytokine response between the neurotropic and nonneurotropic virus is consistent with the idea that cytokine induction is a pivotal factor in the development of an inflammatory CNS disease. The relevance of the proinflammatory cytokine response to MHV-A59-induced demyelination in mice is important because this disease has many similarities with the human demyelinating disease MS. The increased expression of this set of cytokines has also been reported in MS and in another animal model of MS (EAE). Whether this cytokine up-regulation is absolutely necessary for MHV-induced demyelination awaits further investigation.
Acknowledgments
This work was supported in part by grants from the National Multiple Sclerosis Society (RG 2615) and the NIH (NS30606).
REFERENCES
- 1.Allen, I., and B. Brankin. 1993. Pathogenesis of multiple sclerosis-the immune diathesis and the role of viruses. J. Neuropathol. Exp. Neurol. 52:95-105. [DOI] [PubMed] [Google Scholar]
- 2.Aloisi, F. 1999. The role of microglia and astrocytes in CNS immune surveillance and immunopathology. Adv. Exp. Med. Biol. 468:123-133. [DOI] [PubMed] [Google Scholar]
- 3.Aloisi, F., A. Care, G. Borsellino, P. Gallo, S. Rosa, A. Bassani, A. Cabibbo, U. Testa, G. Levi, and C. Peschle. 1992. Production of hemolymphopoietic cytokines (IL-6, IL-8, colony-stimulating factors) by normal human astrocytes in response to IL-1 beta and tumor necrosis factor-alpha. J. Immunol. 149:2358-2366. [PubMed] [Google Scholar]
- 4.Aranguez, I., C. Torres, and N. Rubio. 1995. The receptor for tumor-necrosis-factor on murine astrocytes-characterization, intracellular degradation, and regulation by cytokines and Theiler's murine encephalomyelitis virus. Glia 13:185-194. [DOI] [PubMed] [Google Scholar]
- 5.Arend, W. P. 2001. Cytokine imbalance in the pathogenesis of rheumatoid arthritis: the role of interleukin-1 receptor antagonist. Semin. Arthritis Rheum. 30:1-6. [DOI] [PubMed] [Google Scholar]
- 6.Barbara, J. A. J., X. Van Ostade, and A. Flopez. 1996. Tumor necrosis factor-alpha (TNF-α): The good, the bad and potentially very effective. Immunol. Cell Biol. 74:434-443. [DOI] [PubMed] [Google Scholar]
- 7.Benveniste, E. N. 1992. Inflammatory cytokines within the central nervous system: sources, function and mechanism of action. Am. J. Physiol. Cell. Physiol. 263:C1-C16. [DOI] [PubMed] [Google Scholar]
- 8.Breen, E. C., A. R. Rezai, K. Nakajima, G. N. Beall, R. T. Mitsuyasu, T. Hirano, T. Kishimoto, and O. Martinez-Maza. 1990. Infection with HIV is associated with elevated IL-6 levels and production. J. Immunol. 144:480-484. [PubMed] [Google Scholar]
- 9.Budzilowicz, C. J., S. P. Wilczynski, and S. R. Weiss. 1985. Three intergenic regions of mouse hepatitis virus strain A59 genome RNA contain a common nucleotide sequence that is homologous to the 3′ end of the viral mRNA leader sequence. J. Virol. 53:834-840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chapoval, A. I., J. Ni, J. S. Lau, R. A. Wilcox, D. B. Flies, D. Liu, H. Dong, G. L. Sica, G. Zhu, K. Tamada, and L. Chen. 2001. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat. Immunol. 2:269-274. [DOI] [PubMed] [Google Scholar]
- 11.Constantinescu, C. S., B. Hilliard, E. Ventura, M. Wysocka, L. Showe, E. Lavi, T. Fujioka, P. Scott, G. Trinchieri, and A. Rostami. 2001. Modulation of susceptibility and resistance to an autoimmune model of multiple sclerosis in prototypically susceptible and resistant strains by neutralization of interleukin-12 and interleukin-4, respectively. Clin. Immunol. 98:23-30. [DOI] [PubMed] [Google Scholar]
- 12.Das Sarma, J., L. Fu, S. T. Hingley, M. M. Lai, and E. Lavi. 2001. Sequence analysis of the S gene of recombinant MHV-2/A59 coronaviruses reveals three candidate mutations associated with demyelination and hepatitis. J. Neurovirol. 7:432-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Das Sarma, J., L. Fu, S. T. Hingley, and E. Lavi. 2001. Mouse hepatitis virus type-2 infection in mice: an experimental model system of acute meningitis and hepatitis. Exp. Mol. Pathol. 71:1-12. [DOI] [PubMed] [Google Scholar]
- 14.Das Sarma, J., L. Fu, J. C. Tsai, S. R. Weiss, and E. Lavi. 2000. Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J. Virol. 74:9206-9213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu, L., J. Das Sarma, and E. Lavi. 2001. Differential expression of tumor necrosis factor in primary glial cell cultures infected with demyelinating and non-demyelinating MHVs. Adv. Exp. Med. Biol. 494:663-668. [DOI] [PubMed] [Google Scholar]
- 16.Haring, J. S., L. L. Pewe, and S. Perlman. 2002. Bystander CD8 T cell-mediated demyelination after viral infection of the central nervous system. J. Immunol. 169:1550-1555. [DOI] [PubMed] [Google Scholar]
- 17.Imashuku, S., Y. Tabata, T. Teramura, and S. Hibi. 2000. Treatment strategies for Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis (EBV-HLH). Leuk. Lymphoma 39:37-49. [DOI] [PubMed] [Google Scholar]
- 18.Inoue, A., C. S. Koh, M. Yamazaki, H. Yahikozawa, M. Ichikawa, H. Yagita, and B. S. Kim. 1998. Suppressive effect on Theiler's murine encephalomyelitis virus-induced demyelinating disease by the administration of anti-IL-12 antibody. J. Immunol. 161:5586-5593. [PubMed] [Google Scholar]
- 19.Keck, J. G., L. H. Soe, S. Makino, S. A. Stohlman, and M. M. C. Lai. 1988. RNA recombination of murine coronavirus: recombination between fusion-positive mouse hepatitis virus A59 and fusion-negative mouse hepatitis virus 2. J. Virol. 62:1989-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komastu, T., D. D. Ireland, and C. S. Reiss. 1998. IL-12 and viral infections. Cytokine Growth Factor Rev. 9:277-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kristensson, K., I. Nennesmo, L. Persson, and E. Lycke. 1982. Neuron to neuron transmission of herpes simplex virus. Transport of virus from skin to brainstem nuclei. J. Neurol. Sci. 54:149-156. [DOI] [PubMed] [Google Scholar]
- 22.Lane, T. E., A. D. Paoletti, and M. J. Buchmeier. 1997. Disassociation between the in vitro and in vivo effects of nitric oxide on neurotropic murine coronavirus. J. Virol. 71:2202-2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lavi, E., S. P. Fishman, M. K. Highkin, and S. R. Weiss. 1988. Limbic encephalitis following inhalation of murine coronavirus MHV-A59. Lab. Investig. 58:31-36. [PubMed] [Google Scholar]
- 24.Lavi, E., D. H. Gilden, M. K. Highkin, and S. R. Weiss. 1984. Persistence of MHV-A59 RNA in a slow virus demyelinating infection in mice as detected by in situ hybridization. J. Virol. 51:563-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavi, E., D. H. Gilden, Z. Wroblewska, L. B. Rorke, and S. R. Weiss. 1984. Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 34:597-603. [DOI] [PubMed] [Google Scholar]
- 26.Lavi, E., E. M. Murray, S. Makino, S. A. Stohlman, M. M. Lai, and S. R. Weiss. 1990. Determinants of coronavirus MHV pathogenesis are localized to 3′ portions of the genome as determined by ribonucleic acid-ribonucleic acid recombination. Lab. Investig. 62:570-578. [PubMed] [Google Scholar]
- 27.Lavi, E., T. Schwartz, Y. P. Jin, and L. Fu. 1999. Nidovirus infections: experimental model systems of human neurologic diseases. J. Neuropathol. Exp. Neurol. 58:1197-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lavi, E., A. Suzumura, M. Hirayama, M. K. Highkin, D. M. Dambach, D. H. Silberberg, and S. R. Weiss. 1987. Coronavirus MHV-A59 causes a persistent, productive infection in glial cells. Microb. Pathog. 3:79-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lavi, E., A. Suzumura, E. M. Murray, D. H. Silberberg, and S. R. Weiss. 1989. Induction of MHC class I antigens on glial cells is dependent on persistent mouse hepatitis virus infection. J. Neuroimmunol. 22:107-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lavi, E., and S. R. Weiss. 1989. Coronaviruses, p. 101-139. In D. H. Gilden and H. L. Lipton (ed.), Clinical and molecular aspects of neurotropic viral infections. Kluwer Academic Publishers, Boston, Mass.
- 31.Lee, Y. B., J. Satoh, D. G. Walker, and S. U. Kim. 1996. Interleukin-15 gene expression in human astrocytes and microglia in culture. Neuroreport 7:1062-1066. [DOI] [PubMed] [Google Scholar]
- 32.Lieberman, A. P., P. M. Pitha, H. S. Shin, and M. L. Shin. 1989. Production of tumor necrosis factor and other cytokines by astrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proc. Natl. Acad. Sci. USA 86:6348-6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McInnes, I. B., B. P. Leung, R. D. Sturrock, M. Field, and F. Y. Liew. 1997. Interleukin-15 mediates T cell-dependent regulation of tumor necrosis factor-alpha production in rheumatoid arthritis. Nat. Med. 3:189-195. [DOI] [PubMed] [Google Scholar]
- 34.Mims, C. A. 1982. The pathogenesis of infectious disease, 2nd ed., p. 82-108. Academic Press, London, United Kingdom.
- 35.Mosmann, T. R., H. Cherwinski, M. W. Bond, M. A. Giedlin, and R. L. Coffman. 1986. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136:2348-2357. [PubMed] [Google Scholar]
- 36.Murphy, F. A., S. P. Bauer, A. K. Harrison, and W. C. Winn. 1973. Comparative pathogenesis of rabies and rabies-like viruses. Viral infection and transit from inoculation site to the central nervous system. Lab. Investig. 28:361-376. [PubMed] [Google Scholar]
- 37.Nakajima, K., O. Martinez-Maza, T. Hirano, E. C. Breen, P. G. Nishanian, J. F. Salazar-Gonzalez, J. L. Fahey, and T. Kishimoto. 1989. Induction of IL-6 (B cell stimulatory factor-2/IFN-beta 2) production by HIV. J. Immunol. 142:531-536. [PubMed] [Google Scholar]
- 38.Nelson, P. T., L. A. Soma, and E. Lavi. 2002. Microglia in diseases of the central nervous system. Ann. Med. 34:491-500. [DOI] [PubMed] [Google Scholar]
- 39.Okuda, Y., S. Sakoda, C. C. Bernard, H. Fujimura, Y. Saeki, T. Kishimoto, and T. Yanagihara. 1998. IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int. Immunol. 10:703-708. [DOI] [PubMed] [Google Scholar]
- 40.Olson, J. K., A. M. Girvin, and S. D. Miller. 2001. Direct activation of innate and antigen-presenting functions of microglia following infection with Theiler's virus. J. Virol. 75:9780-9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parra, B., D. R. Hinton, M. T. Lin, D. J. Cua, and S. A. Stohlman. 1997. Kinetics of cytokine mRNA expression in the central nervous system following lethal and nonlethal coronavirus-induced acute encephalomyelitis. Virology 233:260-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pearce, B. D., M. V. Hobbs, T. S. McGraw, and M. J. Buchmeier. 1994. Cytokine induction during T-cell-mediated clearance of mouse hepatitis virus from neurons in vivo. J. Virol. 68:5483-5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peiris, J. S. M., S. T. Lai, L. L. M. Poon, Y. Guan, L. Y. C. Yam, J. Nicholls, W. K. S. Yee, W. W. Yan, M. T. Cheung, V. C. C. Cheng, K. H. Chan, D. N. C. Tsang, R. W. H. Yung, T. K. Ng, K. Y. Yuen, and SARS Study Group. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 1361:1319-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perrin, P. J., C. A. Rumbley, R. L. Beswick, E. Lavi, and S. M. Phillips. 2000. Differential cytokine and chemokine production characterizes experimental autoimmune meningitis and experimental autoimmune encephalomyelitis. Clin. Immunol. 94:114-124. [DOI] [PubMed] [Google Scholar]
- 45.Phillips, J. J., M. M. Chua, E. Lavi, and S. R. Weiss. 1999. Pathogenesis of chimeric MHV4/MHV-A59 recombinant viruses: the murine coronavirus spike protein is a major determinant of neurovirulence. J. Virol. 73:7752-7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phillips, J. J., M. M. Chua, G. F. Rall, and S. R. Weiss. 2002. Murine coronavirus spike glycoprotein mediates degree of viral spread, inflammation, and virus-induced immunopathology in the central nervous system. Virology 301:109-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharif, S., G. A. Arreaza, P. Zucker, Q. S. Mi, J. Sondhi, O. V. Naidenko, M. Kronenberg, Y. Koezuka, T. L. Delovitch, J. M. Gombert, M. Leite-De-Moraes, C. Gouarin, R. Zhu, A. Hameg, T. Nakayama, M. Taniguchi, F. Lepault, A. Lehuen, J. F. Bach, and A. Herbelin. 2001. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune type 1 diabetes. Nat. Med. 7:1057-1062. [DOI] [PubMed] [Google Scholar]
- 48.Siegmund, B., G. Fantuzzi, F. Rieder, F. Gamboni-Robertson, H. A. Lehr, G. Hartmann, C. A. Dinarello, S. Endres, and A. Eigler. 2001. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281:R1264-R1273. [DOI] [PubMed] [Google Scholar]
- 49.Stohlman, S. A., and L. P. Weiner. 1981. Chronic central nervous system demyelination in mice after JHM virus infection. Neurology 31:38-44. [DOI] [PubMed] [Google Scholar]
- 50.Streit, W. J., and C. A. Kincaid-Colton. 1995. The brain's immune system. Sci. Am. 273(5):54-55, 58-61. [DOI] [PubMed] [Google Scholar]
- 51.Sturman, L., and K. Holmes. 1985. The novel glycoproteins of coronaviruses. Trends Biochem. Sci. 10:17-20. [Google Scholar]
- 52.Sun, N., D. Grzybicki, R. F. Castro, S. Murphy, and S. Perlman. 1995. Activation of astrocytes in the spinal cord of mice chronically infected with a neurotropic coronavirus. Virology 213:482-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzumura, A., E. Lavi, S. Bhat, D. M. Murasko, S. R. Weiss, and D. H. Silberberg. 1988. Induction of glial cell MHC antigen expression in neurotropic coronavirus infection: characterization of the H-2 inducing soluble factor elaborated by infected brain cells. J. Immunol. 140:2068-2072. [PubMed] [Google Scholar]
- 54.Suzumura, A., E. Lavi, S. R. Weiss, and D. H. Silberberg. 1986. Coronavirus infection induces H-2 antigen expression on oligodendrocytes and astrocytes. Science 232:991-993. [DOI] [PubMed] [Google Scholar]
- 55.Tada, T., T. Takemori, K. Okumura, M. Nonaka, and T. Tokuhisa. 1978. Two distinct types of helper T cells involved in the secondary antibody response: independent and synergistic effects of Ia− and Ia+ helper T cells. J. Exp. Med. 147:446-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsuda, H., and K. Shirono. 1996. Successful treatment of virus-associated haemophagocytic syndrome in adults by cyclosporin A supported by granulocyte colony-stimulating factor. Br. J. Haematol. 93:572-575. [DOI] [PubMed] [Google Scholar]
- 57.Tsunoda, I., and R. S. Fujinami. 1996. Two models for multiple sclerosis: experimental allergic encephalomyelitis and Theiler's murine encephalomyelitis virus. J. Neuropathol. Exp. Neurol. 55:673-686. [DOI] [PubMed] [Google Scholar]
- 58.Weiner, L. P. 1973. Pathogenesis of demyelination induced by a mouse hepatitis virus (JHM virus). Arch. Neurol. 28:298-303. [DOI] [PubMed] [Google Scholar]
- 59.Wesselingh, S. L., J. Glass, J. C. McArthur, J. W. Griffin, and D. E. Griffin. 1994. Cytokine dysregulation in HIV-associated neurological disease. Adv. Neuroimmunol. 4:199-206. [DOI] [PubMed] [Google Scholar]
- 60.Wu, G. F., L. Pewe, and S. Perlman. 2000. Coronavirus-induced demyelination occurs in the absence of inducible nitric oxide synthase. J. Virol. 74:7683-7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yokota, S., T. Imagawa, T. Miyamae, S. Ito, S. Nakajima, A. Nezu, and M. Mori. 2000. Hypothetical pathophysiology of acute encephalopathy and encephalitis related to influenza virus infection and hypothermia therapy. Pediatr. Int. 42:197-203. [DOI] [PubMed] [Google Scholar]