

Abstract
The mitochondrial transcription factor A (TFAM) is required for mitochondrial DNA (mtDNA) replication and transcription. Disruption of TFAM results in heart failure and premature aging in mice. But very little is known about the role of TFAM in cancer development. Here, we report the identification of frequent frameshift mutations in the coding mononucleotide repeat of TFAM in sporadic colorectal cancer (CRC) cell lines and in primary tumors with microsatellite instability (MSI), but not in microsatellite stable (MSS) CRC cell lines and tumors. The presence of the TFAM truncating mutation, in CRC cells with MSI, reduced the TFAM protein level in vivo and in vitro and correlated with mtDNA depletion. Furthermore, forced overexpression of wild-type TFAM in RKO cells carrying a TFAM truncating mutation suppressed cell proliferation and inhibited RKO cell-induced xenograft tumor growth. Moreover, these cells showed more susceptibility to cisplatin-induced apoptosis due to an increase of cytochrome b (Cyt b) expression and its release from mitochondria. An interaction assay between TFAM and the heavy-strand promoter (HSP) of mitochondria revealed that mutant TFAM exhibited reduced binding to HSP, leading to reduction in Cyt b transcription. Collectively, these data provide evidence that a high incidence of TFAM truncating mutations leads to mitochondrial copy number reduction and mitochondrial instability, distinguishing most CRC with MSI from MSS CRC. These mutations may play an important role in tumorigenesis and cisplatin-induced apoptotic resistance of most microsatellite-unstable CRCs.
Introduction
Colorectal cancers (CRC) display 2 major types of genomic instability: chromosomal instability (CIN) and microsatellite instability (MIN or MSI; 1). CRC exhibiting MSI is characterized by the inactivation of DNA mismatch repair (MMR) proteins (2). As a consequence, tumors with MSI accumulate genetic alterations in both coding and noncoding microsatellite repeats, which are widely distributed throughout the genome (1, 3). MSI occurs in ~90% of cancers in patients with hereditary nonpolyposis CRC and in 10% to 15% of sporadic CRC (4). Since the inactivation of an MMR gene by itself is unlikely to result in a transforming event, additional genetic changes are believed to be required for cells to become malignant. In addition to the large number of microsatellite repeat alterations in noncoding regions that have been reported in CRC with MSI, many genes with frameshift mutations in their coding repeats have also been documented. Examples include TGFBRII, BAX, AXIN2, and HDAC2 (5–9). Functional analyses of some of these mutants have demonstrated that frameshift mutations can lead to loss of tumor suppressor function and are probably responsible for driving tumorigenesis in colonic tissues (6, 9). Therefore, it is critical to identify the genes carrying frameshift mutations in their coding repeats and defining their roles in tumorigenesis for an improved understanding of the etiology of CRC with MSI. Furthermore, this will provide valuable information for prognostic evaluation and better therapeutic intervention.
On screening for potential mutation targets of deficient MMR, we identified a coding poly(A)10 repeat in the TFAM gene. TFAM (also known as mtTF1, mtTFA, or TCF6) is a mitochondrial transcription factor (10). It regulates mtDNA transcription and replication, and plays a critical role in maintaining mtDNA copy number and mitochondrial morphology (10–12). Tfam heterozygous knockout mice exhibited a 50% reduction in both Tfam transcription and Tfam protein levels, resulting in a marked reduction in the mtDNA copy number (11). TFAM and mtDNA copy number have been shown to be critical for the regulation of mitochondrial gene expression; their reduction leads to reduced heart function and aging (13, 14). Moreover, the depletion of mtDNA and reduction of TFAM-dependent mtDNA expression are involved in infantile mitochondrial myopathy, Alzheimer disease, and Parkinson disease (13, 15, 16).
In the present study, we explored the role of TFAM in colorectal carcinogenesis. We identified a high frequency of TFAM truncating mutations, showed low level of TFAM protein and reduced mtDNA copy number in both CRC cell lines and primary CRC tissues with MSI. We also examined the effect of the mutant TFAM on tumorigenesis, mitochondrial gene expression, and cisplatin-induced apoptotic resistance. Our results indicate that TFAM mutations induce mtDNA depletion and result in decreased tumor sensitivity to cisplatin in most CRC with MSI. The presence of these alterations suggests the potential importance of TFAM in tumorigenesis of CRC with MSI and implicates TFAM-dependent mitochondrial instability as a unique pathogenetic factor for CRC with MSI. TFAM mutation may be potentially useful in predicting outcomes and selecting chemotherapy for patients with microsatellite-unstable CRC.
Materials and Methods
Tumor tissues and cell lines
Eighty-nine CRC tissue specimens including 43 MSI and 46 MSS collected at Mayo Clinic were previously described (6). Seventeen CRC cell lines including 11 with MSI (LS174T, Co115, LoVo, TC7, HCT-15, HCT 116, TC71, SW48, RKO, HCT-8, and LS411) and 6 MSS (SW480, SW837, COLO205, SW620, HT-29, and Caco-2), as well as 4 endometrial cancer cell lines with MSI (HEC-1-A, HEC59, Ishika14, and RL95–2), 1 gastric cancer cell line with MSI (SNU-1), and 4 non-CRC cell lines (LNCaP, SKBR3, MDA-MB231, and MCF-7) were obtained from American Type Culture Collection. Cells were cultured in medium by the standard protocol. Genomic DNA from tissues or cell lines was isolated using Easy-DNA Kit (Invitrogen) following the manufacturer’s instructions.
Mutation analysis
PCR was performed using the forward primer in exon 4 of TFAM: 5′-ATAAAGAAGAGATAAGCAGATT-3′ and the reverse primer in intron 5: 5′-TGCCTATTAAGAGAAAACTAC-3′ following the condition previously described (17). Mutations were determined by direct sequencing analyses of the PCR products (18). All mutations were confirmed by repeated PCR amplification and bidirectional sequencing analysis.
Western blotting
The whole cell lysates, cytosol, and mitochondria fractions were isolated from cells as previously described (19). Western blot analysis was performed following the protocol described before (17) using rabbit polyclonal TFAM antibody (20) and antibodies against Cyt b, cleaved PARP-1, and cleaved Casp-3 (Cell Signaling), β-actin and β-tubulin, and the corresponding HRP-conjugated secondary antibodies (Sigma), respectively.
Immunofluorescence and immunohistochemistry analyses
For immunofluorescence analysis, cells grown on the 12-well chamber slides were incubated with 100 nmol/L MitoTracker Red CMVRos (Invitrogen) for 20 minutes. Cells were then washed, fixed, and blocked in 1% bovine serum albumin–PBS, incubated with primary antibodies and goat antirabbit secondary antibody conjugated to Alexa 488 (Invitrogen). Microscopy was performed on LSM510 Carl Ziess Confocal Laser Scanning Microscope. For immunohistochemical analysis, tissue sections were deparaffinized, rehydrated, and treated in 3% H2O2–methanol to remove endogenous peroxidase. The sections were then boiled in 10 mmol/L citric acid buffer (pH 6.0) for 20 minutes for antigen retrieval, followed by incubation with anti-TFAM antibody (20), biotinylated goat antisera, and streptavidin peroxidase (Jackson ImmunoResearch). Detection was done using 3,3′-diamino-benzidine (Vector Labs). Sections were scored as 0 (−, negative), 1 (+, weak positive), 2 (++, medium positive), and 3 (+++, strong positive), respectively.
Determination of mtDNA copy number and mitochondrial mass
The mtDNA copy number was determined by quantitative real time PCR (qPCR) using the Platinum SYBR Green qPCR SuperMix-UDG with ROX (Invitrogen) on ABI PRISM 7900 Sequence Detector (Applied Biosystems; 21). The primer sequences used were ND1-F1, 5′-CACCCAAGAACAGGG-TTTGT-3′ and ND1-R1, 5′-TGGCCATGGGATTGTTGTTAA-3′ for mitochondrial NADH dehydrogenase 1 (ND1), and β-actin-F, 5′-TCCCAGCACACTTAACTTAGC-3′ and β-actin-R, 5′-AGCCACAAGAAACACTCAGG-3′, for β-actin (nuclear DNA control), respectively. Relative copy number was calculated from the threshold cycle value (Ct value) using the delta delta Ct (ΔΔCT) method (22). Each reaction was optimized and confirmed to be linear within an appropriate concentration range using genomic DNA from a normal colon epithelial cell line CRL1807. The mitochondrial mass was determined using flow cytometry. Briefly, 1 × 106 cells were labeled with 100 nmol/L MitoTracker Red CMXRos (M7512, Invitrogen) in Dulbecco’s modified Eagle medium containing 10% FBS and incubated at 37°C for 30 minutes, washed and resuspended in 1 mL PBS with 1% FBS. Individual cellular fluorescence signals were analyzed compared to unstained cells as controls using FACSCalibur and Cell-Quest Pro software (BD Biosciences).
RNA extraction and qRT-PCR
Total RNA was extracted using Trizol reagent (Invitrogen) following the manufacturer’s instructions. The first strand cDNA was synthesized by Superscript III First-Strand Kit (Invitrogen). qRT-PCR was carried out with ABI Prism 7900 using primers: ND1-F2, 5′-TCGCCCTATTCTTCATAGCC-3′ and ND1-R2, 5′-AGAAGTAGGGTCTTGGTGAC-3′ for ND1; CYTB-F, 5′-CTATCCATCCTCATCCTAGC-3′ and CYTB-R, 5′-TGGTTGTCCTCCGATTCAGG-3′ for mitochondrial cyto-chrome b gene (CYTB), respectively. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control with primers GAPDH-F, 5′-TCCCAGCACACTTAACTTAGC-3′ and GAPDH-R, 5′-AGCCACAAGAAACACTCAGG-3′. Relative expression levels of ND1 and CYTB were calculated by the ΔΔCT method as described above.
Constructs, lentivirus preparation, and infection
The full-length wild-type TFAM (Wt-TFAM) cDNA was PCR-amplified and cloned into pCMV-tag5b expression vector (Strategene). The mutant TFAM (Mut-TFAM) was created using the pCMV-tag5b-Wt-TFAM as a template by deleting 1 A in the poly(A)10 region using QuickChange XL site-directed mutagenesis kit (Stratagene) following the supplier’s instructions. Lentiviral constructs expressing Wt- or Mut-TFAM were generated by inserting the PCR products of the full-length Wt- or Mut-TFAM-Myc gene into pCDH-CMV-MCS-EF1-copGFP vector (System Biosciences) with the BamH1/NotI restriction sites, respectively. To express the GST (glutathione S transferase) fusion proteins of mature Wt- or Mut-TFAM in Escherichia coli, the Wt- or Mut-TFAM without the mitochondrial target sequence (1–43 amino acid residues) was amplified by PCR using pCMV-tag5b-Wt- or Mut-TFAM-Myc as templates and cloned into pGEX-4T-1 vector (GE Healthcare) with BamH1/NotI restriction sites. All constructs were verified by sequencing and immunoblotting. Lentivirus production and cell infection were performed according to the manufacturer’s instructions.
Separation and analyses of mitochondrial NP-40-soluble and -insoluble fractions
Mitochondrial NP-40-soluble and -insoluble fractions were separated and analyzed as previously described (23) with modification. Briefly, mitochondria (1.0 mg protein/mL) isolated from RKO cells expressing Wt-TFAM-Myc, Mut-TFAM-Myc, or control vector were resuspended in TES buffer (10 mmol/L Tris–HCl, pH 7.4, 0.25 mol/L sucrose, 1 mmol/L EDTA) containing 1× protease inhibitor mix and 0.5% NP-40. After incubation (30 minutes on ice) and centrifugation (20,000 × g, 30 minutes, 4°C), samples were separated into a pellet (P1) and a supernatant (S1). The P1 fractions were resuspended in mitochondrial lysis buffer, centrifuged (20,000 × g, 30 minutes) and supernatants (S2) were collected. Equal volumes of S1 or S2 fractions were subjected to 15% SDS-PAGE and detected by immunoblotting using anti-TFAM antibody. In addition, the mtDNA in P1 and S1 fractions were extracted (23) and subjected to PCR amplification and gel electrophoresis analysis.
Interaction assay of TFAM protein with mitochondrial heavy-strand promoter
GST-Wt-TFAM and GST-Mut-TFAM fusion proteins were generated as previously described (20) and purified using BugBuster GST Bind Purification Kit following manufacturer’s instruction (Novagen). The mitochondrial heavy-strand promoter (HSP) was generated by annealing 2 complementary oligonucleotides HSP-F: 5′-CACACACCGCTGCTAACCCCATACCCCGAACCAACCAAACCCCAAAGACACCCCC-3′ and HSP-R: 5′-GGGGGTGTCTTTGGGGTTTGGTTGGTTCGGGG-TATGGGGTTAGCAGCGGTGTGTG-3′. The interaction assay was performed as previously described (24) with modification. Briefly, the reactions were performed in 50 μL binding buffer (20 mmol/L HEPES pH 7.6, 100 mmol/L NaCl, 1 mmol/L EDTA, 0.1% NP40) containing 500 nmol/L HSP and 0, 20, 50, 100, 200, or 400 nmol/L of GST, GST-Wt-TFAM or GST-Mut-TFAM proteins. After incubation with glutathione-sepharose beads and centrifugation, the supernatants containing the unbound HSP were subjected to qPCR analysis using primers corresponding to the mitochondrial HSP, hsp-rtF: 5′-CACACACCGCTGCTAAC-3′ and hsp-rtR: 5′-GGGTGTCTTTGGGGTTT-3′. The levels of protein-bound HSP were determined by subtracting the unbound HSP.
Cell proliferation and apoptosis assays
For cell proliferation assay, cells infected with Lenti-vector, -Wt-TFAM, or -Mut-TFAM were seeded in 6-well plates (0.3 × 105 cells/well in triplicate) and counted daily for 5 days using a hemacytometer with trypan blue staining. For apoptosis analysis, the infected cells were treated with cisplatin (50 μmol/L/mL, 12 hours), followed by propidium iodide staining (25) and analyzed by the FACScan flow cytometer (Coulter Epics XL-MSL, Beckman Coulter).
Xenograft tumor growth assay
Cells infected with various lentiviral constructs grown to 70% to 80% confluences in complete medium were harvested with trypsin–EDTA and washed with PBS. Cells were counted and suspended in sterile PBS at a concentration of 2.0 × 107 cells/mL. Five-week-old nude mice (NCI) were inoculated s.c. in the left and right hind flanks with 200 μL cell suspension. Tumor development was closely monitored and pictures of mice were taken 3 weeks after inoculation.
Statistical analysis
Data are presented as mean ± SE. Student’s t test was used to compare means of 2 independent groups. One-way ANOVA and post hoc Turkey’s test was applied to analyze the difference of means of more than 2 groups. A P < 0.05 was considered statistically significant using SigmaStat 3.5 (Systat Software).
Results
TFAM frameshift mutations in primary CRCs and cell lines
We analyzed the poly(A)10 mononucleotide repeat within exon 4 of the TFAM gene for mutations in 17 CRCs cell lines and 89 CRC tissue specimens. We detected a mutation frequency of 100% (11/11) in CRC cell lines and 74.4% (32/43) in CRC tissue specimens with MSI. All the mutations displayed a heterozygous deletion of 1 base in the mononucleotide A track, except 1 cell line and 4 tissue specimens that displayed a 2-base deletion (Fig. 1A and Table 1). The TFAM frameshift mutation was also detected in 3 of 4 endometrial and 1 gastric cancer cell lines with MSI but not in 6 CRC cell lines and 46 CRC tissue specimens without MSI, nor in DNA isolated from normal individuals and cancer cell lines without MSI (Table 1 and data not shown). These data clearly demonstrate that the frequent frameshift mutation of the TFAM gene is unique to microsatellite-unstable cancers, particularly to microsatellite-unstable CRC, distinguishing them from MSS CRC.
Figure 1.

Sequence analysis of TFAM frameshift mutation in human CRC. A, sequence chromatograms of TFAM poly(A)10 mononucleotide repeat in normal (left) and CRC tissue (right). Arrow indicates the mutation site. B, genomic and domain structures of human TFAM. Frameshift TFAM mutation in the poly(A)10 mononucleotide repeat region of exon 4 results in a truncated protein lacking the C-terminus of TFAM, including a small portion of the linker domain, the entire HMG box2 domain, and the tail domain.
Table 1.
TFAM mutations identified in cancer cell lines with or without MSI
| Tumor cell lines | MS status | Cell line | A10 allele status |
|---|---|---|---|
| Colorectal cancer | |||
| MSI | Co115 | A9/A10 | |
| MSI | LS174T | A9/A10 | |
| MSI | LoVo | A9/A10 | |
| MSI | TC71 | A9/A10 | |
| MSI | HCT15 | A9/A10 | |
| MSI | HCT116 | A9/A10 | |
| MSI | TC7 | A9/A10 | |
| MSI | SW48 | A9/A10 | |
| MSI | RKO | A8/A10 | |
| MSI | HCT8 | A9/A10 | |
| MSI | LS411 | A9/A10 | |
| MSS | SW480 | A10 | |
| MSS | SW837 | A10 | |
| MSS | Colo205 | A10 | |
| MSS | SW620 | A10 | |
| MSS | HT-29 | A10 | |
| MSS | Caco-2 | A10 | |
| Endometrial cancer | |||
| MSI | HEC-1A | A9/A10 | |
| MSI | HEC59 | A9/A10 | |
| MSI | Ishika14 | A9/A10 | |
| MSI | RL95-2 | A10 | |
| Gastric cancer | |||
| MSI | SNU-1 | A9/A10 | |
TFAM mutation results in reduction in TFAM protein level
The TFAM frameshift mutation is predicted to result in a protein truncation at codon 149 (Leu149Stop; Δ149), leading to the deletion of the entire region of the second HMG domain and the tail region (Fig. 1B). To assess the putative effect of the mutation, we analyzed TFAM levels in 10 CRC cell lines (5 with MSI and 5 without MSI) by Western blotting using the TFAM polyclonal antibody. All 5 CRC cell lines with MSI showed reduced TFAM protein levels as compared to those in the 5 MSS CRC cell lines (Fig. 2A). Similar results were obtained by another TFAM antibody (ABCOM, Ab 47517, data not shown). In agreement with the Western blot data, immunofluorescence analysis showed relatively lower levels of TFAM protein in CRC cells with MSI than in MSS CRC cells (Fig. 2B). In addition, cells with reduced TFAM expression exhibited reduced mitochondrial mass compared to that in MSS CRC cell lines shown by decreased MitoTracker labeling (Fig. 2B and C). To examine the above observations in vivo, we performed immunohistochemical analysis of TFAM in 20 primary CRC specimens. Nine of the 10 CRC with MSI carrying TFAM mutations showed low levels of TFAM, while all 7 CRC with MSI carrying no TFAM mutations and 3 MSS CRC specimens displayed strong levels of TFAM (Fig. 2D). Taken together, these in vitro and in vivo data suggest that the TFAM truncating mutation correlates with the reduction in TFAM protein in most CRC with MSI.
Figure 2.

TFAM frameshift mutation results in reduction of TFAM protein and mtDNA in CRC with MSI. A, relatively low levels of TFAM protein in CRC cell lines with MSI and TFAM mutations compared to those in MSS CRC cell lines. B, immunofluorescent analysis of TFAM (green) and mitochondria (red) in MSS CRC cells (Caco2 and SW837) and CRC cells with MSI (HCT116 and TC71). Scale bar indicates 10 μm. C, mitochondrial mass of CRC cells. Shown here are flow cytometry curves for cells as in (B; top), and average values of the mean fluorescent intensity (MFI) of MitoTracker Red (bottom, P < 0.05). Results represent mean ± SE of 3 independent experiments. D, representative images of TFAM immunohistochemical analysis in 2 CRC specimens with MSI carrying TFAM mutations and 2 MSS CRC specimens. Positive TFAM immunoreactivity appeared brown in cytoplasm. Scale bar indicates 50 μm.
TFAM mutation leads to mtDNA depletion in CRC
Reduction in TFAM protein has been shown to induce mtDNA depletion in Tfam knockout mice and in cultured mammalian cells (11, 26). To determine whether this is also the case in CRC with MSI, we performed qPCR of mtDNA from 17 CRC cell lines, using the ND1 gene as the mtDNA marker and β-actin as a nuclear control. We first determined the efficiency and reproducibility of the method using a normal colon epithelial cell line (CRL-1807) as a control and generated a standard curve (Supplementary Fig. S1). The results indicated that the mtDNA copy number in 11 CRC cell lines with MSI was significantly lower than that in the 6 MSS CRC cell lines (1:2.63, P < 0.01; Fig. 3A, left). Similar results were obtained on analysis of 4 non-CRC cell lines with MSI (Table 1) and 5 non-CRC cell lines (LNCaP, SKBR3, RL95-2, MDA-MB231, and MCF-7), which do not carry TFAM mutations (Fig. 3A, right). These data demonstrate that the TFAM truncating mutation resulting in TFAM protein reduction led to the decrease in mtDNA copy number in CRC with MSI.
Figure 3.

Effects of mutant TFAM on mtDNA depletion and tumor cell growth. A, mtDNA copy number of CRC (left) and non-CRC cells (right) by qPCR analysis. Each dot represents the mean value of 3 independent measurements of each genomic DNA sample. Solid horizontal lines represent the mean value of each dataset. B, cell proliferation curves of RKO (left) and HCT116 cells (right). Data represent mean ± SE of 3 independent experiments in triplicates, P < 0.05 for all. C, representative photograph showing the difference of tumor size in nude mice injected with Wt-TFAM- or vector-expressing RKO cells (left, scale bar indicates 5 mm). The averaged tumor weights were significantly different (right, P < 0.01). Values represent mean ± SE of tumors from 5 pairs of mice.
Mut-TFAM promotes tumorigenesis in CRC
To assess the potential role of the mut-TFAM in CRC tumorigenesis, we performed a cell proliferation assay using the RKO CRC cell line, which harbors a TFAM truncating mutation. The growth rates of RKO cells infected with Lenti-Mut-TFAM or Lenti-Wt-TFAM were compared with the growth rates of RKO cells infected with Lenti-vector. The expression level of exogenous TFAM (Wt- or Mut-TFAM) was about 10% of the level of endogenous TFAM and appeared no apparent toxic effect on cells compared to the cells infected with Lenti-vector (Supplementary Fig. S2A and B). We found that the cells expressing Mut-TFAM grew faster, while the cells expressing Wt-TFAM grew slower than the control cells (P < 0.05, Fig. 3B, left). Similar results were observed in the HCT116 CRC cell line, which also harbors a TFAM truncating mutation (Fig. 3B, right). To determine the cell growth suppression potential of Wt-TFAM in vivo, we implanted athymic nude mice with RKO cells expressing the Lenti-vector or Lenti-Wt-TFAM on the right or left hind flank, respectively. Consistent with the in vitro cell proliferation data, RKO cells infected with Wt-TFAM induced much smaller tumors than those induced by the control RKO cells (Fig. 3C, left). The weight of the tumors induced by Wt-TFAM was reduced by more than 70% compared to those induced by control RKO cells (P < 0.01, Fig. 3C, right). These results suggest that the TFAM truncating mutation promotes, while Wt-TFAM inhibits cell growth in CRC with MSI.
Mut-TFAM renders CRC cells resistant to cisplatin-induced apoptosis
CRC with MSI exhibits low level of resistance to cisplatin (27, 28). To explore the role of the TFAM truncating mutation in cisplatin resistance of CRC with MSI, we treated Lenti-Wt-TFAM- or Lenti-vector-infected RKO cells with cisplatin and followed by apoptosis analysis using flow cytometry. The RKO cells expressing Wt-TFAM were more susceptible to cisplatin-induced apoptosis, as indicated by an up to 8-fold increase in sub-G1 stage cells (Fig. 4A) and an up to 4-fold increase in cytochrome c (Cyt c) levels before or after cisplatin treatment (Fig. 4B). In agreement with the Cyt c data, the levels of cleaved Parp-1 and Casp 3 were also increased (Fig. 4B). Since Cyt b is a mitochondrial gene encoded protein and plays an important role in apoptosis (29), we further examined Cyt b levels in those RKO cells. We found that the cytosolic levels of Cyt b and cleaved Cyt b were higher, while the mitochondrial level of Cyt b was lower, in RKO cells infected with Wt-TFAM than in those infected with the control vector (Fig. 4C). These results suggest that CRC cells harboring the TFAM frame-shift mutation are more resistant to cisplatin through a Cyt b-dependent apoptotic mechanism and that the resistance to cisplatin-induced apoptosis in CRC cells with MSI is probably due to the presence of a TFAM truncating mutation.
Figure 4.
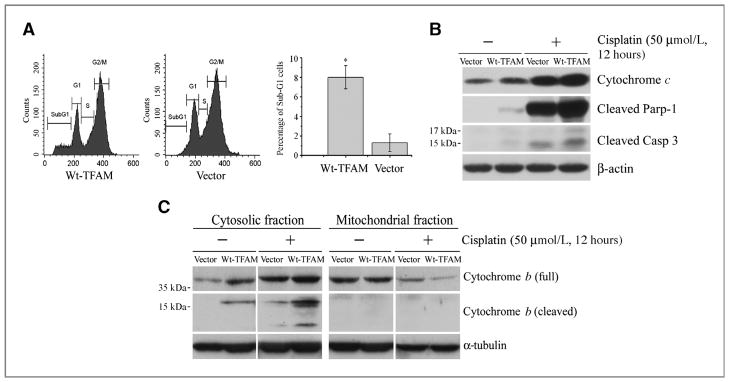
Mutant TFAM confers resistance to cisplatin-induced apoptosis in microsatellite-unstable RKO cells. A, representative flow cytometry images for apoptosis analysis. Cisplatin induced more apoptosis (sub-G1 population) in RKO cells expressing Wt-TFAM (left) than vector control (middle). The proportion of sub-G1 cells was increased by almost 8-fold (right, P < 0.05). Values represent mean ± SE of 3 independent experiments in triplicate. B, Western blots of RKO cell homogenates. The levels of Cyt c, cleaved Parp-1 and Casp-3 were higher in Wt-TFAM-expressing RKO cells and were enhanced by cisplatin treatment compared with vector-expressing RKO cells. C, Western blots showing Wt-TFAM overexpression promoted Cyt b release from mitochondria into cytoplasm of RKO cells under the same conditions as in (B). The release was enhanced by cisplatin treatment.
Mut-TFAM downregulates Cyt b transcription in CRC with MSI
To understand the mechanism of Mut-TFAM in the regulation of Cyt b, we assessed the expression levels of CYTB and ND1 in RKO cells expressing Wt-TFAM. The transcriptional levels of both ND1 and CYTB were elevated significantly (Fig. 5A, P < 0.05). The protein levels of both ND1 and Cyt b were also elevated (Fig. 5B). Since CYTB is a mitochondrial gene and TFAM regulates transcription of mitochondrial genes by binding to the HSP (30), we further tested our hypothesis that Mut-TFAM might impair the transcriptional regulation of CYTB by aberrant interaction with HSP in CRC with MSI. The interaction assay of equivalent amounts of GST-Wt-TFAM or GST-Mut-TFAM synthesized in E. coli with the HSP revealed that the relative binding ability of Wt-TFAM was significantly higher than that of Mut-TFAM (Fig. 5C and D, P < 0.01). Moreover, we found that more Mut-TFAM was present in the NP40-soluble fraction than Wt-TFAM, while more Wt-TFAM was in the pellet fraction, where most of the mtDNA was contained (Fig. 5E). Together, these data suggest that CRC carrying TFAM truncating mutation may increase resistance to cisplatin-induced apoptosis through reduced interaction with HSP and downregulation of CYTB transcription.
Figure 5.

Aberrant regulation of CYTB transcription by Mut-TFAM in CRC with MSI. A, qRT-PCR analysis of ND1 and CYTB mRNA in RKO cells expressing Wt-TFAM or vector. Values represent mean ± SD of 3 analyses, P < 0.05 for both. B, Western blots showing ND1 and CYTB protein levels in RKO cells as in (A). C, purified GST-Wt-TFAM and GST-Mut-TFAM fusion proteins were visualized by Coomassie blue staining (left) and Western blot with anti-TFAM antibody (right). D, relative binding ability of Wt-TFAM or Mut-TFAM to mitochondrial HSP. Data represent mean ± SE of 3 independent experiments in triplicate, P < 0.01. E, distribution of Wt-TFAM and Mut-TFAM in the NP40-soluble or -insoluble fraction of mitochondrial extracts from RKO cells expressing control vector, Wt- or Mut-TFAM. GRP75 was a loading control for mitochondrial protein; ND1 was PCR-amplified to serve as a mtDNA loading control.
Discussion
The mtDNA depletion or mitochondrial instability is a common event in human cancer, including CRC (31, 32). However, the cause of mtDNA depletion in tumorigenesis is currently unknown. In this study, we provided evidence that mutation in TFAM, an important component of the mitochondrial transcription and replication machinery, plays an important role in maintaining mitochondrial stability in CRC. The mtDNA copy number difference between CRC with or without MSI is so striking that low mtDNA copy number might be a potential genetic marker of CRC with MSI, once the current data are validated in a future large cohort analysis. Besides CRC, we also observed mtDNA depletion in non-CRC cell lines with TFAM mutations. However, the range of copy numbers in non-CRC cell lines without TFAM mutations was wider. Two breast cancer cell lines (MCF-7 and MDA-MB231) without TFAM mutations seemed to have low mtDNA copy numbers, similar to those in MSI cell lines with TFAM mutations (Fig. 3A, right). But the exact reason remains unknown and needs to be determined. Recently, more genes responsible for maintaining mtDNA stability have been identified, including TIM17 and OXA1 (33). It is possible that loss function mutations or epigenetic regulation of TFAM, TIM17, and OXA1 may induce mtDNA instability in these non-CRC cells without MSI.
CRC cells with MSI are prone to mutation throughout the genome. It thus presents a significant challenge to determine whether a truncating mutation induced by DNA mismatch repair deficiency functions as a “Driver” to facilitate tumor-igenesis or just as a “Bystander”. By comparing our data to the 5-point criteria for a bona fide target gene from bystander mutations proposed by the NCI group, we found that the TFAM truncating mutations fulfill 4 of the 5 criteria: (i) the frequency of the TFAM mutation in CRC with MSI is over 75%, way above that of the background; (ii) mut-TFAM promotes cell proliferation, suggesting a functional importance in tumorigenesis; (iii) TFAM interacts with p53 and although the role of TFAM in the p53 pathway is not defined, the p53 pathway genes are commonly mutated in CRC (34, 35); and (iv) biallelic inactivation is not applicable since loss of both TFAM alleles has been reported to be embryonic lethal in mouse and results in loss of all mitochondria and apoptosis in vivo (11, 36). We noted the prevalence of a 1-base or 2-base deletion detected in these samples. But when 95 subclones of PCR products amplified from a CRC tumor DNA with MSI were sequenced, we did detect low frequencies of 1-base insertion mutations (3.2%; data not shown). The presence of the low frequency of 1-base insertion in the tumor specimen harboring 1-base deletion mutation is probably due to the nature of mitochondrial heteroplasmy (37). Moreover, we did not detect any meaningful TFAM mutations in CRC cell lines and tumor specimens without MSI, except for some function-unknown SNPs (data not shown). The absence of TFAM mutations in MSS CRC could be due to the small sample size of the current study or the higher frequency of p53 mutations in MSS CRC than in CRC with MSI (38). It is, therefore, very likely that during mutation selection, only TFAM haploinsufficient cells are selected for tumor growth and the haploinsufficient TFAM truncating mutation becomes a predominant one during cancer progression.
Overexpression of Mut-TFAM in promoting cell growth is evident in both RKO and HCT116 cells by our in vitro cell proliferation assays. In contrast, Wt-TFAM suppressed cell growth in these 2 cell lines and in vivo xenograft tumor growth assays. Intriguingly, overexpression of Mut-TFAM in HEK293 cells induced apoptosis instead of cell growth (data not shown). This is not surprising since Tfam knockout mouse displayed massive apoptosis at embryonic day 9.5 and increased apoptosis was observed in the heart of the tissue-specific Tfam knockouts (39). These data suggest that the involvement of TFAM in apoptosis is probably temporally regulated and/or may be tissue specific. It will be necessary to find CRC cell lines with MSI, which do not carry TFAM truncating mutations to test whether overexpression of TFAM truncating mutation or knockdown of TFAM in these cells induces apoptosis. Although the detailed mechanism by which Mut-TFAM promotes tumor cell growth remains to be elucidated, our data indicate that loss of the ability to induce Cyt b expression and thus the inability to induce Cyt b-dependent apoptosis is probably one of the mechanisms for Mut-TFAM to induce tumor progression in most microsatellite-unstable CRC.
Accumulating evidence has indicated that the MMR protein is critical for cisplatin-dependent apoptosis in many cancer cells, including CRC, but how loss of MMR protein increases resistance to cisplatin-dependent apoptosis remains unclear (27, 40, 41). The results in our study demonstrated that CRC cells carrying TFAM truncating mutations were more resistant to cisplatin-induced apoptosis than were the same cells over-expressing Wt-TFAM. These data thus provide evidence that TFAM is required for the induction of cisplatin-dependent apoptosis in CRC with MSI. It is critical to initiate an investigation of the responsiveness of CRC patients who carry or do not carry TFAM truncating mutations to cisplatin-based chemotherapy. Such data may help to improve therapeutic intervention protocols for patients with or without TFAM truncating mutations.
In summary, we have shown the presence of high frequency of TFAM truncating mutations in human microsatellite-unstable CRC cell lines and primary tumors. The CRC with MSI harboring the TFAM truncating mutation displayed impaired mitochondrial stability, dysregulated cell proliferation, mitigated Cyt b-dependent apoptosis, and enhanced cisplatin-dependent apoptotic resistance. Our preliminary data also indicate a potential mechanism by which TFAM truncating mutation is involved in apoptosis through down-regulation of Cyt b transcription due to reduced interaction with mitochondrial HSP. These findings support the role of TFAM and mitochondrial stability in CRC tumorigenesis. This may have potential relevance in the pharmacogenetic selection of CRC patients for treatment with cisplatin or other drugs.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Jay K. Kolls and Dr. Matthew K. Gilbert for their critical review and revising this manuscript. This project was supported by Cancer Research Fund from Louisiana Cancer Research Consortium (LCRC) to W. Liu.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Lengauer C, Kinzler KW, Vogelstein B. DNA methylation and genetic instability in colorectal cancer cells. Proc Natl Acad Sci U S A. 1997;94:2545–50. doi: 10.1073/pnas.94.6.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–61. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 3.Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816–9. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 4.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–57. [PubMed] [Google Scholar]
- 5.Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res. 2002;62:2447–54. [PubMed] [Google Scholar]
- 6.Liu W, Dong X, Mai M, Seelan RS, Taniguchi K, Krishnadath KK, et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat Genet. 2000;26:146–7. doi: 10.1038/79859. [DOI] [PubMed] [Google Scholar]
- 7.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–8. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 8.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275:967–9. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- 9.Ropero S, Fraga MF, Ballestar E, Hamelin R, Yamamoto H, Boix-Chornet M, et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat Genet. 2006;38:566–9. doi: 10.1038/ng1773. [DOI] [PubMed] [Google Scholar]
- 10.Kang D, Kim SH, Hamasaki N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion. 2007;7:39–44. doi: 10.1016/j.mito.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 11.Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18:231–6. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 12.Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, et al. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13:935–44. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- 13.Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–7. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 14.Hansson A, Hance N, Dufour E, Rantanen A, Hultenby K, Clayton DA, et al. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc Natl Acad Sci U S A. 2004;101:3136–41. doi: 10.1073/pnas.0308710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A. 2007;104:1325–30. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39:17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 17.Guo J, Cagatay T, Zhou G, Chan CC, Blythe S, Suyama K, et al. Mutations in the human naked cuticle homolog NKD1 found in colorectal cancer alter Wnt/Dvl/beta-catenin signaling. PLoS One. 2009;4:e7982. doi: 10.1371/journal.pone.0007982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu W, Smith DI, Rechtzigel KJ, Thibodeau SN, James CD. Denaturing high performance liquid chromatography (DHPLC) used in the detection of germline and somatic mutations. Nucleic Acids Res. 1998;26:1396–400. doi: 10.1093/nar/26.6.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–62. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- 20.Ohno T, Umeda S, Hamasaki N, Kang D. Binding of human mitochondrial transcription factor A, an HMG box protein, to a four-way DNA junction. Biochem Biophys Res Commun. 2000;271:492–8. doi: 10.1006/bbrc.2000.2656. [DOI] [PubMed] [Google Scholar]
- 21.Liu CS, Tsai CS, Kuo CL, Chen HW, Lii CK, Ma YS, et al. Oxidative stress-related alteration of the copy number of mitochondrial DNA in human leukocytes. Free Radic Res. 2003;37:1307–17. doi: 10.1080/10715760310001621342. [DOI] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Ohgaki K, Kanki T, Fukuoh A, Kurisaki H, Aoki Y, Ikeuchi M, et al. The C-terminal tail of mitochondrial transcription factor a markedly strengthens its general binding to DNA. J Biochem. 2007;141:201–11. doi: 10.1093/jb/mvm020. [DOI] [PubMed] [Google Scholar]
- 24.Fukuoh A, Kang D. Methods for assessing binding of mitochondrial transcription factor A (TFAM) to DNA. Methods Mol Biol. 2009;554:87–101. doi: 10.1007/978-1-59745-521-3_6. [DOI] [PubMed] [Google Scholar]
- 25.Williams CD, Linch DC, Watts MJ, Thomas NS. Characterization of cell cycle status and E2F complexes in mobilized CD34+ cells before and after cytokine stimulation. Blood. 1997;90:194–203. [PubMed] [Google Scholar]
- 26.Kanki T, Ohgaki K, Gaspari M, Gustafsson CM, Fukuoh A, Sasaki N, et al. Architectural role of mitochondrial transcription factor A in maintenance of human mitochondrial DNA. Mol Cell Biol. 2004;24:9823–34. doi: 10.1128/MCB.24.22.9823-9834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res. 1998;4:1–6. [PubMed] [Google Scholar]
- 28.Honecker F, Wermann H, Mayer F, Gillis AJ, Stoop H, van Gurp RJ, et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J Clin Oncol. 2009;27:2129–36. doi: 10.1200/JCO.2008.18.8623. [DOI] [PubMed] [Google Scholar]
- 29.Komarov AP, Rokhlin OW, Yu CA, Gudkov AV. Functional genetic screening reveals the role of mitochondrial cytochrome b as a mediator of FAS-induced apoptosis. Proc Natl Acad Sci U S A. 2008;105:14453–8. doi: 10.1073/pnas.0807549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikeda S, Sumiyoshi H, Oda T. DNA binding properties of recombinant human mitochondrial transcription factor 1. Cell Mol Biol (Noisy-le-grand) 1994;40:489–93. [PubMed] [Google Scholar]
- 31.Lee HC, Yin PH, Lin JC, Wu CC, Chen CY, Wu CW, et al. Mitochondrial genome instability and mtDNA depletion in human cancers. Ann N Y Acad Sci. 2005;1042:109–22. doi: 10.1196/annals.1338.011. [DOI] [PubMed] [Google Scholar]
- 32.Lu J, Sharma LK, Bai Y. Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Res. 2009;19:802–15. doi: 10.1038/cr.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iacovino M, Granycome C, Sembongi H, Bokori-Brown M, Butow RA, Holt IJ, et al. The conserved translocase Tim17 prevents mitochondrial DNA loss. Hum Mol Genet. 2009;18:65–74. doi: 10.1093/hmg/ddn313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshida Y, Izumi H, Torigoe T, Ishiguchi H, Itoh H, Kang D, et al. P53 physically interacts with mitochondrial transcription factor A and differentially regulates binding to damaged DNA. Cancer Res. 2003;63:3729–34. [PubMed] [Google Scholar]
- 35.Wong TS, Rajagopalan S, Freund SM, Rutherford TJ, Andreeva A, Townsley FM, et al. Biophysical characterizations of human mitochondrial transcription factor A and its binding to tumor suppressor p53. Nucleic Acids Res. 2009;37:6765–83. doi: 10.1093/nar/gkp750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J, Silva JP, Gustafsson CM, Rustin P, Larsson NG. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc Natl Acad Sci U S A. 2001;98:4038–43. doi: 10.1073/pnas.061038798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25:4663–74. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 38.Samowitz WS, Holden JA, Curtin K, Edwards SL, Walker AR, Lin HA, et al. Inverse relationship between microsatellite instability and K-ras and p53 gene alterations in colon cancer. Am J Pathol. 2001;158:1517–24. doi: 10.1016/S0002-9440(10)64102-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Wang J, Wilhelmsson H, Hansson A, Thoren P, Duffy J, et al. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proc Natl Acad Sci U S A. 2000;97:3467–72. doi: 10.1073/pnas.97.7.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: the role of DNA repair pathways. Clin Cancer Res. 2008;14:1291–5. doi: 10.1158/1078-0432.CCR-07-2238. [DOI] [PubMed] [Google Scholar]
- 41.Topping RP, Wilkinson JC, Scarpinato KD. Mismatch repair protein deficiency compromises cisplatin-induced apoptotic signaling. J Biol Chem. 2009;284:14029–39. doi: 10.1074/jbc.M809303200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.