

Cellular senescence is an important defense against uncontrolled cellular replication and is a critical tumor suppressor mechanism. Although initially described as an in vitro phenomenon, in recent years, several lines of evidence have indicated that senescence also occurs in vivo. Disparate cellular stresses can provoke senescence including telomere shortening, activation of certain oncogenes, oxidative stress and stalled replication forks. These stresses are best known to engage the p16INK4a-RB and ARF-p53 pathways to effect growth arrest and prevent neoplasia. Among these, ‘oncogene-induced senescence’ (OIS) is particularly known for the prevention of melanoma, resulting from the expression of mutant B-RAF or N-RAS in this disease.
While a significant fraction of melanomas are believed to arise from progression within precursor nevi, the vast majority of nevi never undergo malignant transformation, despite the presence of activating mutations of B-RAF or other oncogenes. Recent work has suggested that many nevi comprise senescent melanocytes that are arrested in response to oncogenic mutations (Michaloglou et al., 2005). The low transformation rate and long-term persistence of nevi prove that senescence is a highly effective tumor suppressor mechanism, and vividly illustrates how senescent cells can endure in vivo for decades, accumulating with aging. Understanding why senescence fails to restrain malignant transformation in a rare subset of melanocytes harboring oncogenic events is a key question in cancer biology.
Against this background, Vredeveld et al. have recently reported on the role of PTEN/PI3K signaling in melanocyte senescence. In their study, the authors demonstrate that activation of the PI3K pathway (e.g. by reducing PTEN expression) was sufficient to abrogate OIS in melanocytes expressing activated B-RAF. To supplement these in vitro findings, the authors showed that delivery of short hairpin RNAs targeting Pten into established B-Raf-mutant nevi of mice was also sufficient to facilitate melanoma progression. Correspondingly, human melanoma specimens arising in the setting of contiguous nevi were also analyzed, showing conservation of B-RAF or N-RAS mutations during progression from nevus to melanoma, as well as activation of the PI3K pathway in the melanoma relative to the associated nevi. Finally, the authors showed that concomitant inhibition of PI3K signaling in B-RAF mutant cells increased the cytotoxicity of B-RAF inhibitors, suggesting a straightforward clinical application of these findings. In aggregate, this work suggests that melanocyte escape from senescence and tumor progression in B-RAF mutant melanocytes can result from PTEN inactivation and/or PI3K activation, providing a satisfying explanation for how some nevi progress to melanoma.
Aspects of this work fit nicely with prior studies. For example, Courtois-Cox et al. showed in other cell types that aberrant activation of RAS signaling promotes OIS by inducing a set of global negative feedback responses that potently suppress MAPK and PI3K signaling, and that persistent activation of PI3K signaling is sufficient to counter OIS resulting from RAS activation (Courtois- Cox et al., 2006). Moreover, the present work provides an explanation for the common co-occurrence of PTEN loss with B-RAF mutation in human melanoma (Tsao et al., 2004), as well as the observed decrease in PTEN expression from nevus to melanoma to metastasis (Whiteman et al., 2002). Because loss of CDKN2a (encoding p16INK4a and ARF) has been long known to be one of the most common events associated with melanoma progression, it appears RAF activation in melanocytes engages both an early p16INK4a(and/or ARF)-dependent form of senescence and a later CDKN2a-independent senescence associated with repressed PI3K signaling (Figure 1).
Figure 1.
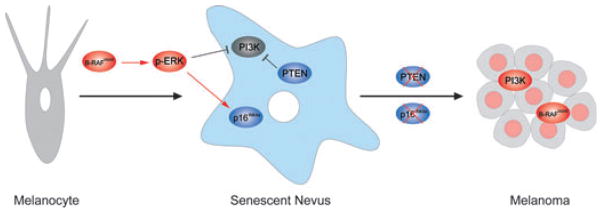
Senescence-Escape in Melanoma. Oncogenic activation of B-RAF leads to repressed PI3K signaling and elevated p16INK4a expression. The loss of PTEN and/or p16INK4a results in Senescence-Escape and melanoma progression.
This work does raise some new questions. First, is compromise of senescence resulting from PTEN loss only the result of PI3K activation? Although PI3K signaling may be the most familiar target of PTEN, several studies have suggested effects of PTEN that are independent of AKT and lipid phosphatase activity (Salmena et al., 2008). This issue is crucial, because it is becoming evermore dubious that the principal tumor-promoting effects of PTEN loss can be countered solely through the use of potent PIK3CA kinase inhibitors. Second, can cellular senescence – usually thought of as a deep, irreversible growth arrest – really be so fragile that it can be completely reverted by reducing the expression of a single gene? While some forms of senescence (e.g. associated with telomere-dysfunction and high p16INK4a expression in fibroblasts) appear to be very stable, resisting even the introduction of highly transforming viral oncoproteins, other reports have described senescent cells that require certain persistent tumor suppressor activities, e.g. p53 activation. It could be that OIS of melanocytes in nevi represents one of these less final forms of senescence, but an inevitable concern of these types of experiments is that populations of senescent cells are contaminated at low frequency by slowly cycling, non-senescent cells. Resolution of this issue will require a rigorous, in vivo cell tracking strategy to determine the durability of senescence.
These new data showing PI3K pathway activation facilitates melanomagenesis by abrogating B-RAF-induced OIS suggest an interesting and testable new therapeutic approach for patients with advanced melanoma. Vemurafenib, a B-RAF inhibitor, has been recently FDA-approved for use in B-RAF mutant melanoma, but disappointingly, responses to this drug are usually short-lived (~4 months). Although most of the clinical focus to date has been on combining B-RAF inhibitors with MEK inhibitors in an effort to prevent the emergence of drug resistance, the present work provides rationale for combining B-RAF and PIK3CA inhibitors in advanced melanoma. Several PIK3CA inhibitors (as well as downstream mTOR inhibitors) are at advanced stages of clinical development, and testing of these agents in combination with B-RAF inhibitors should begin soon. Of note, several trials combining inhibitors of PIK3CA and MEK, which is downstream of B-RAF, in cancer-bearing mice and humans have been reported with promising initial results. The mechanism of tumor response in such clinical setting is presently unknown, but based on these new results from Vredeveld et al., it will be interesting to see whether combined inhibition of B-RAF and PIK3CA can produce the entity, long-sought but to date largely theoretical, of ‘therapy-induced senescence’ in melanoma and other established cancers.
References
- Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, Mcgillicuddy LT, Johannessen CM, Hollstein PE, Maccollin M, Cichowski K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006;10:459–472. doi: 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, Van Der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence- like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004;122:337–341. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman DC, Zhou XP, Cummings MC, Pavey S, Hayward NK, Eng C. Nuclear PTEN expression and clinicopathologic features in a population-based series of primary cutaneous melanoma. Int J Cancer. 2002;99:63–67. doi: 10.1002/ijc.10294. [DOI] [PubMed] [Google Scholar]