

Abstract
Four of the five members of the Toll-interleukin-1 receptor (TIR) domain-containing adaptor family are required for signaling downstream of Toll-like receptors, promoting innate immune responses against different pathogens. However, the role of the fifth member of this family, sterile alpha and TIR-domain containing 1 (SARM), is unclear. SARM is expressed primarily in the central nervous system where it is required for axonal death. Studies in C.elegans have also shown a role for SARM in innate immunity. To clarify the role of mammalian SARM in innate immunity, we infected SARM−/− mice with a number of bacterial and viral pathogens. SARM−/− mice show normal responses to Listeria monocytogenes, Mycobacterium tuberculosis (Mtb), and influenza virus, but show dramatic protection from death after CNS infection with vesicular stomatitis virus (VSV). Protection correlates with reduced CNS injury and cytokine production by non-hematopoietic cells, suggesting that SARM is a positive regulator of cytokine production. Neurons and microglia are the predominant source of cytokines in vivo, supporting a role for SARM as a link between neuronal injury and innate immunity.
Introduction
The innate immune system relies on TLRs as well as a number of other pattern recognition receptors to detect pathogen associated molecular patterns. The TLRs signal through the TIR-domain containing adaptor protein family, which includes MyD88, TRIF, MAL, and TRAM. Each of these family members play positive roles in innate immunity by inducing the expression of IFN-β or activation of NF-κB, and deficiency leads to increased susceptibility to infection. SARM is the fifth member of the family to be identified, and is comprised of 7 N-terminal HEAT/Armadillo motifs, 2 central sterile α motifs, and a C-terminal TIR domain (1), and is highly conserved between fly, worm, and mammals (2).
In C.elegans, TOL-1 is the sole TLR homolog and the SARM homolog TIR-1 is the sole cytoplasmic TIR-domain containing protein (3). TIR-1 knockdown worms display increased susceptibility to fungal infection and decreased anti-microbial peptide synthesis, supporting a positive role for SARM in innate immunity in worms. However, this susceptibility was not dependent on TOL-1 (4), suggesting that SARM may not only function as a TLR adaptor. TIR-1 is also expressed in C.elegans olfactory neurons where it regulates olfactory patterning. Genetic evidence suggests TIR-1 mediated patterning through a UNC-43(CaMKII)->TIR-1(SARM)->NSY(ASKI MAPKKK)->PMK-1(p38/JNK) pathway (5). Whether this pathway is also involved in the innate immune phenotype is unknown.
In contrast to results in C.elegans, human SARM has been suggested to function as a negative regulator of TRIF signaling in myeloid cells. Overexpression of SARM in 293T cells was shown to inhibit TRIF-dependent TLR signaling. In addition, LPS treatment of human PBMCs lead to increased expression of SARM (6). However, when SARM−/− mouse macrophage TLR responses were tested, they failed to show defects in cytokine production. This may be explained by the predominant expression of SARM at the RNA level in human and mouse brain, and relatively low expression in myeloid cells (7). Consistent with CNS expression, SARM has been reported to mediate stress-induced cell death, as neurons from SARM−/− mice are protected from glucose deprivation-induced death (7). In addition, SARM−/− mice infected with West Nile virus showed decreased TNF-α production and increased susceptibility to infection supportive of a positive role for SARM in innate immunity in mammals (8). Thus SARM expression can be either detrimental or protective depending on the context.
Recent evidence has identified SARM as a mediator of an active axonal destruction program termed Wallerian degeneration (9). Following injury to axons, neurons undergo degeneration distal to the injury site and a coordinated sequence of events leads to clearance of necrotic debris, degeneration, and subsequent axonal regeneration (10). SARM−/− axons were protected from Wallerian degeneration and the synaptic termini at the neuromuscular junctions were preserved following transection (9). Following trauma, cytokines and chemokines are produced locally by cells including microglia and oligodendrocytes, leading to the infiltration of glial cells and macrophages that remove axonal and myelin debris. This process is thought to aid in cytoskeletal rearrangements leading to growth cones and regeneration (11). The Wlds mouse, which displays a delayed Wallerian degeneration phenotype similar to SARM−/− mice, is also deficient in cytokine production suggesting that the processes are intricately linked (12). In addition to promoting repair of damaged brain tissue, glial activation and cytokine production in the CNS may injure bystander cells (13, 14). In other organs, this collateral damage is typically reversible, owing to the regenerative capacity of the tissue, but repopulation of cells is limited in the CNS (15). Therefore, establishing a balance between the protective and destructive effects of the neuroinflammatory response in the brain is critical (16, 17) and SARM may play a role in this balance.
In this study, we challenged SARM−/− mice with various pathogens to better understand the role of SARM in innate immune responses and disease. Consistent with a role of SARM in neurodegeneration, we found that VSV-infected SARM−/− mice were protected from neurodegeneration and neuropathology. In addition, SARM−/− mice showed dramatically reduced cytokine and chemokine production in the brain after VSV challenge supporting a positive role for SARM in cytokine production similar to its C.elegans homolog. In vivo and in vitro data suggest that cytokine production is mediated by CNS resident cells and relies on the cooperation between neurons and microglia. Collectively, these data support the role of SARM in neurodegeneration and suggest that SARM is a link between innate immune responses and neurodegeneration.
Materials and Methods
Mice
SARM−/− mice on the C57BL/6J background were generated previously (7) and compared to WT C57BL/6J mice purchased from Jackson. Animal studies were approved by the Institutional Animal Care and Use Committee of Weill Medical College of Cornell University and/or Icahn School of Medicine at Mount Sinai.
Bacterial infections
8-week old mice were intravenously infected with 5×103 L. monocytogenes. Livers were homogenized in a 0.3% collagenase solution (Collagenase Type IV, Worthington) and plated on BHI agar plates to determine bacterial burden. Using an Inhalation Exposure System (Glas-Col), 6 mL of M tuberculosis at OD = 0.15580 (approximately 7.5×106 bacteria) was nebulized for 40 min, which correlates with approximately 100 bacteria implanting. Lungs were homogenized and plated on 7H11 agar to determine bacterial burden.
Viral infections
6-8-week old animals were anesthetized with ketamine/xylazine and infected intranasally with 107 plaque forming units (pfu) of VSV-Indiana or 100 pfu of influenza A/PR/8/34 virus in 20 μl PBS. For intracranial infections, 5-week old mice were injected with 50 pfu in 30 μl in the parietal lobe, slightly in front of the bregma. Mice were monitored daily for weight and sacrificed when exhibiting severe paralysis or more than 25% weight loss. Organs were homogenized (MP Biomedical) in 0.2%BSA/PBS. VSV titers were determined on BHK cells with Avicel overlay (18); influenza titers were determined on MDCK cells with oxiod agar overlay and crystal violet staining.
Pathology
Mice were perfused with 4% PFA/PBS. Brains were fixed overnight (ON) in 4% PFA/PBS, paraffin embedded, and sectioned at the Histology Shared resource facility at Icahn Medical School at Mount Sinai. Serial sections were stained for H&E, VSV, or TUNEL. For VSV IHC sections were deparaffinized followed by antigen retrieval in Citra plus solution (Biogenex). Sections were blocked with 4% goat serum and incubated with rabbit polyclonal α-VSV-G (Abcam) followed by α-rabbit IgG(H+L)-bio, ABC kit, Nova red staining (Vector labs), and hematoxylin counterstain. TUNEL staining was performed using the DeadEnd Colorometric TUNEL System (Promega). Pathology was scored blindly by the Comparative Pathology Diagnostic Laboratory of the Icahn Medical School at Mount Sinai as follows: 1 = mild, few inflammatory cells, focal or multifocal, <5 cells thick; 2 = moderate, multifocally affected, inflammation is 5-10 cells thick; 3 = severe, multifocally affected, >10 cells thick, spreading into parenchyma. Points were given for necrosis or meningitis detected, and a total score was calculated for sagittal brain sections from each hemisphere. For Fluoro-Jade staining, brains were fixed for 2 hours in 4% PFA, ON in 4% PFA/20% sucrose, frozen in OCT, and sectioned. Slides were stained in Fluoro-Jade C (Chemicon) according to the manufacturers protocol. For MCP-1 IHC, mice were perfused with PBS and 4%PFA/PBS, and brains were fixed ON in 4%PFA/30%sucrose/PBS. Paraffin sections were blocked with 10% goat serum and stained with rabbit polyclonal α-MCP-1 (Millipore), α-rabbit-biotin (Jackson Immunoresearch), SA-HRP, and DAB substrate (Sigma). For MCP-1 IF, mice were perfused with PBS and formalin-free Zinc fixative (BD Biosciences) and 5 mm sections were incubated overnight in fixative. Samples were paraffin embedded, and sectioned at the Electron Microscopy & Histoloy core facility at Weill Cornell Medical College. Sections were deparaffinized and stained with rabbit polyclonal α-MCP-1 (Millipore), mouse α-CD11b (Serotec), mouse α-GFAP (Covance), and α-mouse Alexa488 and α-rabbit IgG Alexa 594 (Molecular Probes).
qRT-PCR and ELISA
Total RNA was isolated from perfused brain and lung with the RNeasy kit (Qiagen), and reverse transcribed with Oligo dT using MuLV reverse transcriptase (Perkin Elmer). cDNA was used for PCR with gene-specific primer and probes (BioSearch) using the ABI PRISM 7900HT sequence detection system (Perkin Elmer). MIP-1α, MCP-1 and RANTES ELISAs were from R&D systems.
Infiltration and flow cytometry analysis
Mice were perfused with PBS, and brains were mashed through a 70 μM filter in 0.06% BSA/300μM EDTA/Hanks. Leukocytes were isolated on a 30%/70% percoll gradient (GE Healthcare) after centrifugation at 2500 rpm for 30 min. Cells were washed an incubated with UV LIVE/DEAD stain (Invitrogen), α-CD11b-APC-Cy7, α-CD19-FITC, α-CD4-APC-Cy7, and α-CD8-PE-Cy7 (BD Pharmingen), and α-CD45-APC and α-NK1.1-APC (ebiosciences). Cell number was quantified by flow cytometry using AccuCount Particles (Spherotech) and plots were gated on live cells. Flow cytometry was performed on a BD LSRII.
Bone marrow chimeras
6-week old B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) and SARM−/− (CD45.2) mice were irradiated with two doses of 600 rads. Bone marrow cells from WT (CD45.2) or SARM−/− donor mice were prepared and 107 donor cells were intravenously injected into recipient mice 4 hours after irradiation. Reconstitution (95%) was confirmed by flow cytometry 6-weeks later and mice were infected 2-weeks later.
Neuronal cell culture
Primary hippocampal neurons were generated from E15-E17 embryos using previously described methods (19) and plated at 1.5×105 cells in 24-well PureCoat plates (BD Biosciences) in Neurobasal media supplemented with B27 and GlutaMAX (Invitrogen). Primary microglia were isolated from astrocyte monolayers from postnatal D1 mice (19) in the presence of 5 ng/ml M-CSF (R&D Systems) and removed by shaking at 125 rpm for 4 hrs. Astrocytes were isolated from the same cultures by trypsinization after microglia were removed. Bone-marrow derived macrophages were cultured for 6 days in DMEM supplemented with 20% L929 cell media. For mixed cultures, neurons were allowed to differentiate for 3 days in vitro, before microglia, astrocytes or bone marrow derived macrophages were directly added to neurons at a 10:1 ratio of neurons to other cells. 18 hours after mixing, cells were infected with VSV at MOI 1 for 30 minutes and supernatants were harvested at 8 hrs post-infection.
Microarray
Mice were infected intranasally with 107 pfu of VSV, at day 5 post-infection, mice were perfused with PBS, brains were harvested, and RNA was prepared by trizol extraction. Triplicate samples of 3 pooled mice were analyzed at the Biopolymers Facility at Harvard Medical School on the Mouse 430 2.0 chip (Affymetrix). Genes significantly different between WT and SARM−/− VSV-infected mice were determined using Genepattern and results were deposited in NCBI GEO (http://www.ncbi.nlm.nih.gov/geo/, accession GSE44331).
Results
SARM−/− mice show normal responses to Listeria, Mtb, and influenza virus, but are protected from VSV
In order to determine if SARM has a role in innate immunity similar to the other TIR-domain-containing adapters, we infected SARM−/− mice with various bacterial and viral pathogens. Bacterial burdens of SARM−/− mice in response to Mtb were similar to WT animals (Fig 1A) as were responses to Listeria (Fig 1B). SARM−/− mice also showed similar susceptibility to influenza virus and similar viral titers in the lung (Fig 1C).
Figure 1. SARM−/− mice are protected from VSV infection.
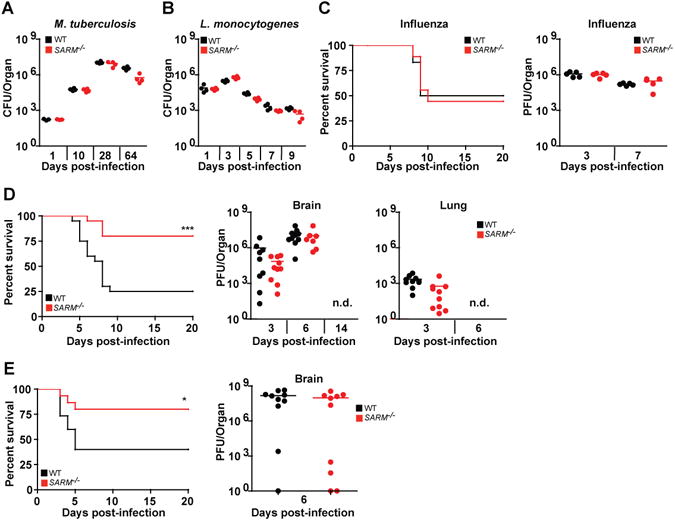
(A) WT and SARM−/− mice infected with nebulized M.tuberculosis have similar bacterial load in the lungs (n=4 per time point). (B) Similar bacterial burden in livers of WT and SARM−/− mice infected intravenously with 5×103 CFU L.monocytogenes (n=4 per time point). (C) WT and SARM−/− have similar survival rates (n=10) and viral titers in lungs (n=5) following intranasal infection with 100 PFU A/PR/8/34 influenza virus. (D) WT and SARM−/− mice were intranasally infected with 107 PFU VSV and monitored daily for mortality (n=20). Brains and lungs were harvested to determine viral burden by plaque assay (n=9−10). PFU data shown pooled from 2 independent experiments. (E) SARM−/− mice intracranially infected with 50 PFU VSV have decreased mortality as compared with WT animals, but viral burdens in the brains of SARM−/− mice were comparable to that of WT (n = 10). Bars represent mean bacterial or viral titers. For (D) and (E), *P<0.02 and ***P<0.0002 compared to WT mice, Students t-test. n.d. not detected.
To determine if SARM plays a unique role in the innate immune response in the brain, given its expression in the CNS (7) we next studied responses to vesicular stomatitis virus (VSV), a member of the family Rhabdoviridae, commonly used as a model to study neurotropic viral infection. Surprisingly, SARM−/− mice showed dramatic protection from intranasal VSV infection at a range of infectious doses (Fig 1D and S1). This protection was not due to differences in viral titers in the brain or lung (Fig 1D). To exclude possible differences in neuroinvasion similar to TLR3 deficiency (20), and to directly address whether the unique phenotype with VSV infection was related to CNS infection, mice were inoculated intracranially with VSV. Using this method, the same enhanced survival phenotype was observed in SARM−/− mice indicating that CNS infection with VSV results in increased survival in SARM−/− mice and suggesting neuroinvasion did not account for the observed difference in susceptibility. This phenotype was again independent of viral load in the brain (Fig 1E) suggesting differences in the immune response contributed to the enhanced survival of SARM−/− mice. Both intranasal and intracranial VSV infection lead to tail and hind limb paralysis and labored breathing that occurred with rapid onset between day 4 and 9 post-infection. Symptoms were similar in both WT and SARM−/− animals that succumbed to infection, however the occurrence of symptoms and lethality was less common in SARM−/− mice. Animals that survived infection either showed no symptoms, or signs of mild tail or hindlimb paralysis that did not persist beyond 14 days post-infection.
SARM−/− mice show reduced pathology in the brain
To compare the extent of neuronal damage, we next examined histological sections from WT and SARM−/− mice. Since WT mice succumb to disease with variable kinetics, brains were harvested between days 6 and 10 post-infection when animals showed signs of paralysis. SARM−/− animals with similar symptoms were harvested when available. WT animals showed multifocal necrosis and meningitis (11/11 and 10/11, Fig 2D), but SARM−/− mice showed reduced incidence of pathology (4/11 necrosis and 6/11 meningitis) and pathology was also less severe when present (Fig 2B and D). In serial sections from WT mice, we observed loss of cell architecture, eosinophilia of the cytoplasm, and appearance of pyknotic and karyorrhectic nuclei under H&E staining (Fig 2A top) as well as TUNEL staining indicative of necrosis in sections that stained positive for VSV antigen. However, while we did observe some necrosis in SARM−/− brain (Fig 2D), we also observed several areas that stained positive for VSV antigen, but showed no signs of necrosis under H&E staining or TUNEL staining (Fig 2A middle). In addition, WT mice had a higher incidence of more severe meningitis as indicated by expansion of the leptomeninges and accumulation of high numbers of inflammatory cells (Fig 2B top) as compared to milder meningitis observed in SARM−/− mice (Fig 2B bottom). We also observed less neurodegeneration in SARM−/− animals in the olfactory bulb, which is the site of initial VSV replication and spread to the brain (21) (Fig 2C and D), suggesting that neural tissue is protected from degeneration in SARM−/− mice during VSV infection as previously reported during axonal injury (9).
Figure 2. SARM−/− mice show reduced pathology in the brain.
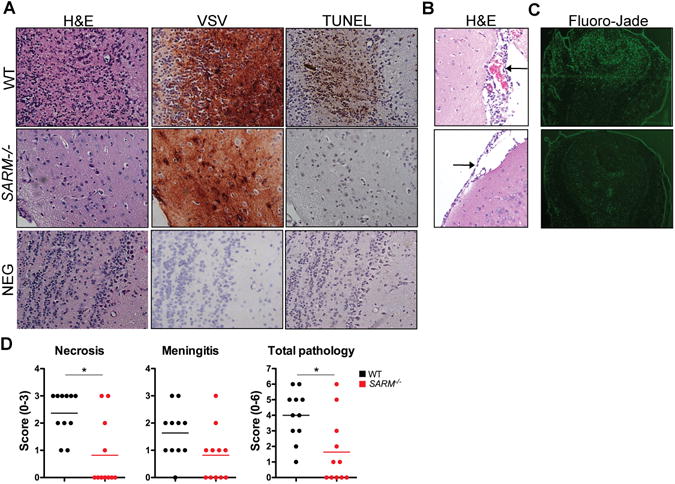
(A) Brain sections from WT (necrosis score 3) and SARM−/− (necrosis score 0) mice infected intranasally with VSV were stained for H&E, VSV, or apoptosis by TUNEL staining at day 5 post-infection showing the presence of VSV but absence of necrosis and TUNEL staining in SARM−/− brain. (B) H&E staining showing severe meningitis of WT (score 3) and mild meningitis of SARM−/− (score 1) brain. Images are shown at 40× and arrows indicate inflammation of the meninges. (C) Olfactory bulb sections were stained for Fluoro-Jade (n=4). Images are shown at 10× (D) Pathology for necrosis, meningitis, and total pathology (n=11) was scored as described in Materials and Methods. *P<0.02.
SARM−/− mice have reduced cytokines and infiltration in the brain
To determine whether there was a difference in the inflammatory response between WT and SARM−/− animals following VSV infection, RNA was isolated from brain and lung homogenates at day 6 post-infection and a number of chemokines and cytokines were tested by quantitative RT-PCR. Many inflammatory mediators are produced following intranasal infection with VSV including chemokines such as MCP-1 and RANTES, as well as cytokines such as Type I IFNs and TNF-α (22, 23). SARM−/− mice had severely blunted responses to all cytokines and chemokines examined in the brain (Fig 3A), but had similar levels to WT in the lung (Fig S2). In addition levels of MIP-1α, MCP-1, and RANTES protein were also significantly reduced in SARM−/− brains compared to WT. In contrast to the RNA data, we were unable to detect TNF-α protein in brain homogenates (Fig 3B). Microarray analysis on brain samples also corroborated these results. WT mice showed significantly higher upregulation of a number of chemokines and interferon-inducible genes as compared to SARM−/− animals (Table I, Fig 3C), confirming that SARM expression is important for the initiation of the innate immune response in the brain following VSV infection. Given the lack of type I interferon in the brains of SARM−/− mice, it was surprising that we did not observe differences in viral replication (Fig 1D). Analysis of the microarray data set showed that SARM−/− mice did produce levels of interferon-inducible genes above PBS controls, althought they failed to upregulate these genes to the same extent as WT mice (Fig 3C). This may indicate that low levels of type I interferon were present, and sufficient to control viral replication. XIAP associated factor, Xaf1 was highly upregulated in SARM−/− animals compared to WT, however western blot failed to show any change in Xaf1 levels (data not shown).
Figure 3. SARM−/− mice show reduced cytokines and infiltration in the brain.
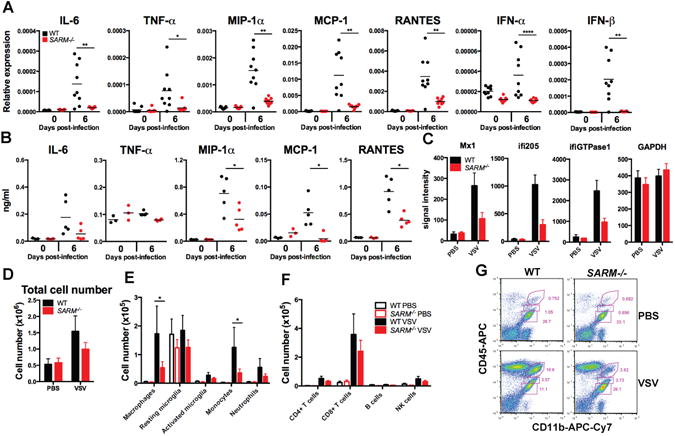
(A) qRT-PCR results from intranasally VSV-infected mouse brains expressed relative to GAPD (n=9, representative data from 2 independent experiments). (B) Whole brain homogenates were assayed for the production of chemokines by ELISA (n=5, representative data from 2 independent experiments). (C) Mas5.0 signal intensity for selected genes from microarray. (D) Leukocytes were isolated by Percoll gradient centrifugation at day 7 post-infection and quantified by flow cytometry. (E) Cells were stained CD11b, CD45 and Ly6C and populations were analyzed by flow cytometry (macrophages CD11bhiCD45hi, resting microglia CD11bloCD45lo, activated microglia CD11bintCD45int, monocytes CD11b+Ly6C+ and neutrophils CD11b+Ly6Cint). (F) Cells were stained with CD4, CD8, CD19 and NK1.1 to analyze CD4+ and CD8+ T cells, CD19+ B cells and NK1.1+ NK cells. (G) Representative FACS plot showing percentages of macrophages, resting microglia, and activated microglia. For (A-B) bars represent mean. For (D-F) data is from 6-7 mice per group and error bars show SEM. *P<0.05 **P<0.005 and ****P<0.00005 compared to WT mice, Students t-test.
Table I. Lack of upregulation of chemokines and interferon-indicible genes in VSV-infected SARM−/− mice.
WT and SARM−/− mice were infected with VSV and brains were harvested at day 5 post-infection for microarray analysis. Genes showing greater than 2-fold change between WT and SARM−/− mice are shown.
| Upregulated in WT compared to SARM−/− | ||||
|---|---|---|---|---|
| Score | Fold change | Gene symbol | Gene name | |
| 26.9 | 3.1 | Gbp2 | guanylate nucleotide binding protein 2 | |
| 15.0 | 4.4 | CXCL10 | chemokine (C-X-C motif) ligand 10 | |
| 13.8 | 3.1 | Gbp2 | guanylate nucleotide binding protein 2 | |
| 10.2 | 3.2 | Ifi205 | interferon activated gene 205 | |
| 10.2 | 8.1 | CDNA clone IMAGE:5719021 | ||
| 9.5 | 3.2 | Ifi203 | interferon activated gene 203 | |
| 9.2 | 6.0 | Erdr1 | erythroid differentiation regulator 1 | |
| 9.2 | 3.1 | Gdpd1 | glycerophosphodiester phosphodiesterase domain containing 1 | |
| 8.7 | 2.5 | Ifi203 | interferon activated gene 203, Ifi203 | |
| 8.2 | 2.5 | ISG15 like | ISG15 ubiquitin-like modifier | |
| 7.5 | 2.3 | CCL5 | chemokine (C-C motif) ligand 5 | |
| 7.4 | 7.2 | RIKEN cDNA G530011O06 gene | ||
| 7.1 | 3.4 | Rsad2 | radical S-adenosyl methionine domain containing 2 | |
| 7.1 | 6.8 | Mid1 | midline 1 | |
| 7.0 | 2.9 | Rsad2 | radical S-adenosyl methionine domain containing 2 | |
| 7.0 | 5.1 | Slfn4 | schlafen 4 | |
| 6.9 | 2.4 | CD274 | CD274 antigen | |
| 6.7 | 2.9 | Mx1 | myxovirus resistance 1 | |
| 6.7 | 2.6 | DNA segment, Chr 14, ERATO Doi 668 | ||
| 6.7 | 2.4 | Iigp1 | interferon inducible GTPase 1 | |
| 6.6 | 2.4 | Ms4a4c | membrane-spanning 4-domains, subfamily A, member 4C | |
| 6.0 | 3.0 | Ms4a4b | membrane-spanning 4-domains, subfamily A, member 4B | |
| 6.0 | 3.2 | predicted gene, EG633640 | ||
| 5.7 | 2.7 | CXCL9 | chemokine (C-X-C motif) ligand 9 | |
| 5.4 | 3.2 | CCL12 | chemokine (C-C motif) ligand 12 | |
| 5.4 | 3.8 | Plac8 | placenta-specific 8 | |
| 5.3 | 2.9 | Rsad2 | radical S-adenosyl methionine domain containing 2 | |
| 5.3 | 2.5 | Ifi44 | interferon-induced protein 44 | |
| 5.1 | 2.8 | Ms4a6d | membrane-spanning 4-domains, subfamily A, member 6D | |
| Upregulated in SARM−/− compared to WT | ||||
| 52.4 | 42.0 | Xaf1 | XIAP associated factor 1 | |
| 49.8 | 3.5 | Wdfy1 | WD repeat and FYVE domain containing 1 | |
| 27.6 | 3.6 | Wdfy1 | WD repeat and FYVE domain containing 1 | |
| 13.5 | 3.8 | Transcribed locus, moderately similar to XP_001477892.1 | ||
| 11.8 | 3.8 | Wdfy1 | WD repeat and FYVE domain containing 1 | |
Given the differences in chemokines, we next examined recruitment of inflammatory cells into the brain following infection. Consistent with published results and our histological examination (Fig 2A), we saw an increased number of infiltrating leukocytes in the brains of VSV-infected mice at day 7 post-infection, but there was a decrease in the total number of cells in the brains of infected SARM−/− mice (1×106) compared to WT mice (1.5×106) (Fig 3D). Using flow cytometry, we found significantly fewer macrophages and monocytes in SARM−/− brains than in WT brains (Fig 3E). In addition, SARM−/− mice showed a trend of decreased neutrophils, CD4+ T cells, and CD8+ T cells (Fig 3E-F), although the differences were not statistically significant at this time point. Although VSV-associated neuropathogenesis is reported to be T cell-independent, CD4+ and CD8+ T cells are required for viral clearance and host recovery (21). In addition to expression in the CNS, low levels of SARM were also detected in CD3+ splenocytes (7). SARM−/− mice also showed no differences in CD4+ or CD8+ T cell numbers in the thymus, spleen, or lymph nodes, and SARM−/− splenocytes proliferated normally in response to αCD3/αCD28 (Fig S3), indicating that SARM is dispensable for T cell development. Decreased numbers of activated microglia have been observed in SARM−/− mice during West Nile virus infection (8). We did observe a trend of less activated microglia (CD11bintCD45int) in SARM−/− mice (Fig 3E and G), however the differences were not statistically significant at this time point.
SARM expression in non-hematopoietic cells contributes to VSV susceptibility and cytokine production
The increased survival of SARM−/− mice compared to WT mice after VSV infection appeared to be correlated with a blunted inflammatory response in the brain. Therefore, we generated BM chimeras in order to dissect out the contribution of hematopoietic and non-hematopoietic SARM expressing cells to the inflammatory response in the brain. Strikingly, SARM deficiency in non-hematopoietic cells afforded protection from VSV (Fig 4A), as SARM−/− recipients of either WT or SARM−/− BM, were better able to survive VSV infection. In addition, cytokine and chemokine production was observed in WT→WT chimeras and SARM−/−→WT chimeras but not in SARM−/−→ SARM−/− or WT→SARM−/− chimeras, indicating that WT non-hematopoetic cells were responsible for increased cytokine production in the brain (Fig 4B). However, WT→WT chimeras showed higher levels for some cytokines compared to SARM−/−→WT chimeras, indicating that SARM expression in BM cells may make some contribution to cytokine production. The observed mortality of WT mice is likely related to the inflammatory response generated by the cells of the non-hematopoietic compartment, as mice depleted of macrophages displayed viral replication and encephalitis similar to WT (24).
Figure 4. SARM expression is critical in non-hematopoetic cells.
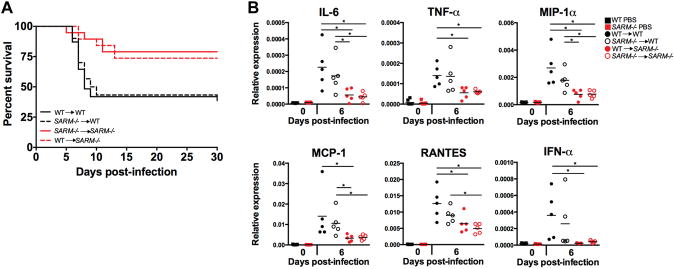
(A) WT recipients of bone marrow, irrespective of the genotype of bone marrow, were more susceptible to intranasal VSV infection. 13/30 SARM−/−→ WT mice survived VSV infection, as compared to 14/19 WT →SARM−/− mice, P<0.02. 13/31 WT → WT and 15/19 SARM−/− → SARM−/− mice survived VSV infection, P<0.02. (B) qRT-PCR results of VSV-infected BM chimeras. WT recipients of BM express higher levels of cytokines at day 6 following VSV infection than SARM−/− (n=5 per time point). Bars represent mean expression. WT and SARM−/− PBS controls from figure 3A are shown for comparison. *P<0.05 Students t-test.
Neurons and microglia cooperate to induce cytokine production
Because susceptibility to VSV correlated with cytokine and chemokine production in the CNS, we examined the cell types responsible for chemokine production. We chose MCP-1 because it is known to be induced following injury and prior to neurodegeneration and infilitration (25, 26). Olfactory bulb sections confirmed the presence of MCP-1 staining in VSV-infected WT but not SARM−/− mice (Fig 5A). Costaining with NeuN for neurons, CD11b for microglia, and GFAP for astrocytes indicated that both neurons and microglia from WT infected mice produced MCP-1 (Fig. 5B), with neurons being the predominant source.
Figure 5. Neurons are the predominant source of chemokines.
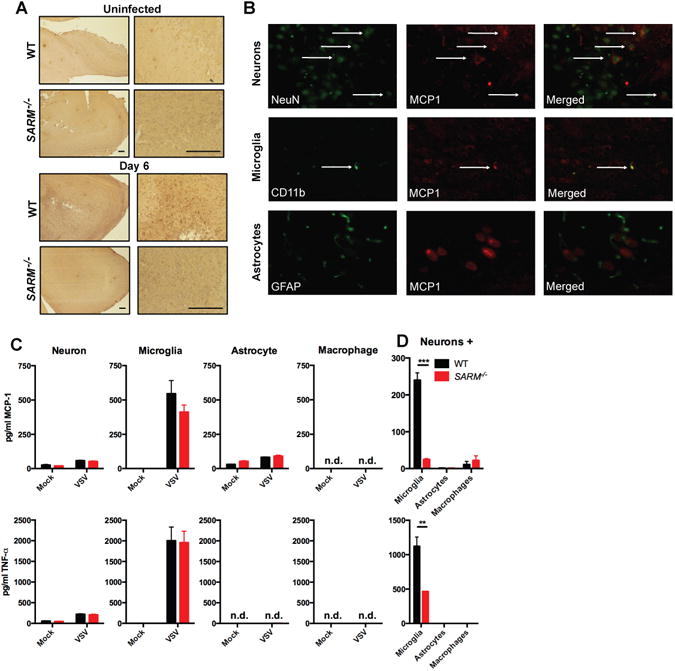
(A) Representative photomicrographs of olfactory bulb sections from mice infected intranasally with VSV at day 6 post-infection stained for MCP-1 are shown at 4× (left) and 63× (right) magnification, bar is equivalent to 200 μm. (B) Representative confocal microscopic images of MCP-1 (red) and NeuN (green, top), CD11b (green, middle) and GFAP (green, bottom) staining for neurons, microglia and astrocytes, respectively from mice infected intranasally with VSV at day 6 post-infection. (C) 1.5×105 neurons, microglia, astrocytes or macrophages were infected with VSV (MOI=1) for 8h and MCP-1 production measured by ELISA. (D) WT hippocampal neurons were co-cultured with microglia, astrocytes or macrophages at a 10:1 ratio (1.5×105 neurons to 1.5×104 glia), infected with VSV at an MOI of 1 for 8 hours and the supernatant was assayed for MCP-1 production. For (A-B), data is representative of 5 mice and for (C-D) bars represent SEM n=4, **P<0.005, ***P<0.0005, n.d. non-detectable.
In order to determine what cell types are important for cytokine production, we cultured neurons, microglia, and astrocytes in vitro. Neurons and astrocytes produced only low levels of MCP-1 and TNF-α (Fig 5C) in response to VSV infection. In contrast, microglia produced high levels of MCP-1 and TNF-α Surprisingly, no difference in cytokine production was observed between WT and SARM−/− cells suggesting that differences in cytokine production are not cell-intrinsic. Consistent with our in vivo results, we did not observe any differences in viral replication in isolated neuron or macrophage cultures (data not shown). Because neuroinflammatory responses often require cooperation between neurons and glia, we infected mixed cell cultures with VSV to assess the production of cytokines. WT neurons cultured at a 10:1 ratio with WT microglia produced high levels of MCP-1 and TNF-α (Fig 5D). In contrast, SARM−/− neurons cultured with SARM−/− microglia showed greatly diminished MCP-1 production (Fig 5D, note 10-fold more microglia are present in Fig 5C). To assess whether the interaction between neurons and microglia was unique, we co-cultured neurons and astrocytes or neurons and macrophages. Neither astrocytes nor macrophages were able to reproduce the cytokine production observed when neurons and microglia were co-cultured (Fig 5D), suggesting that a unique interaction between neurons and microglia was required for cytokine production. However at higher MOIs and later time points we did observe low levels of TNF-α production and high levels of IFN-α which were not affected by the absence of SARM. The low level of cytokine production is likely due to potent suppression of host protein translation by the M protein as infection with the M51R mutant enhanced cytokine production (Fig S4). Microglia are thought to be maintained in the healthy CNS in an active resting state and express a wide variety of receptors that allow them to very rapidly respond to changes in homeostasis (27), which may explain their ability to produce cytokines during VSV infection.
Discussion
We have infected SARM−/− mice with a number of bacterial and viral pathogens in order to investigate a possible role of SARM in innate immunity. We have found that SARM−/− mice display normal responses to most pathogens tested, but were dramatically protected from lethality and CNS damage from VSV infection. Normal responses to most infections support a role for SARM in specific CNS pathology consistent with its expression pattern. We also found that expression of SARM in non-hematopoetic cells was critical for lethality and cytokine production.
In contrast to our results, Szretter et al found that independently generated SARM−/− mice were more susceptible to West Nile virus infection (8). They similarly found reduced TNF-α production, but in contrast to our results observed more cell death in knockout animals. These results may reflect differences in the nature of the immune response required to clear specific pathogens (VSV versus West Nile virus) while limiting damage to the CNS. Differences in lethality may be a result of direct viral damage, or immune-mediated damage (28). Cytokine production and infiltration of the CNS may be beneficial for clearance of WNV infection, but may contribute to pathology during VSV infection. However, both studies support a positive role for SARM in cytokine production in the brain, with the outcome of infection differing depending on the viral infection. Mukherjee et al have also recently reported that SARM−/− mice are resistant to La Crosse virus infection and neuronal damage induced by the virus via a mechanism dependent on MAVS (29).
Osterloh et al have recently reported that both Drosophila and mouse SARM are required for injury-induced axonal cell death (9). Wallerian degeneration is an active cell death program akin to apoptosis with a well-established innate immune component that allows for clearance of debris from damaged neurons, and subsequent nerve regeneration (12). Consistent with this, we observed less overall pathology and less neurodegeneration in SARM−/− VSV-infected animals. Importantly, we also observed a dramatic reduction in cytokine production, with neurons being the predominant source of cytokines. This supports a role for SARM in linking neuronal damage with the innate immune response. It is unclear whether neurodegeneration or cytokine production and infiltration are more relevant for in vivo protection, and this is difficult to assess experimentally since they are likely to be linked. Future work will address whether a specific cytokine or chemokine is responsible for pathogensis, or a number of factors create a pro-inflammatory environment that leads to pathogenesis.
In addition to expression in neurons, low-level expression of SARM was observed in T cells from SARM-GFP BAC transgenic mice (7). Panneerselvam et al have recently reported that overexpression of SARM in 293T cells and CD8+ T cells causes increased apoptosis via the intrinsic mitochondrial pathway. In addition, knockdown of SARM in CD8 T cells lead to enhanced survival (30). Localization of SARM at the mitochondria and mitochondrial clustering has been reported when SARM is overexpressed (7). However, Osterloh et al report that endogenous dSARM-GFP is broadly localized to axons and not preferentially located at the mitochondria (9), and the poor quality of mouse antibodies make localization hard to access. We did not observe any defects in T cell proliferation in vitro (Fig S3) or increased T cell accumulation in vivo (Fig 5E), although differences may be apparent at later time points post-infection.
In vivo, we observed that neurons were the predominant source of cytokines, but we also observed cytokine production by microglia. In addition, it is important to note that microglia are predominantly of host origin in radiation bone-marrow chimeras (31), so microglia may contribute to cytokine production. In isolated neuronal and microglial cultures we were unable to observe differences in cytokine production in response to VSV infection, but our in vitro mixed neuronal cultures revealed a novel interaction between neurons and microglia leading to cytokine production. Activated microglia have been implicated in the pathogenesis of various CNS diseases, including Alzheimer's disease (32) and Parkinson's disease (33), as well as variety of different viral infections (34-36). Microglia have been described as the resident macrophage of the CNS, because they share many similar characteristics and effector functions (37), but they are a distinct population arising from different precursors than macrophages. Recent lineage tracing has shown that mouse adult microglia are derived from primitive myeloid precursors that arise from the extra-embryonic yolk sac and seed in the mouse brain (38) in a process that occurs before definitive hematopoiesis (39). Unlike adult mouse macrophages, microglia are self-renewing and are resistant to high doses of γ-ray irradiation (40). Although astrocytes can also initiate and enhance inflammation in the CNS, our co-culture experiments did not reveal a role for astocytes in VSV-induced cytokine production.
The nature of the interaction between neurons and microglia leading to cytokine production requires further investigation. Microglia are thought to be the predominant source of cytokines in the brain, but recent evidence suggests that neurons are also capable of producing cytokines (41). Our in vivo cytokine staining suggests that both neurons and microglia produce cytokines during infection. Glial cells can contribute to neuroinflammatory responses through the generation of pro-inflammatory mediators, but they can also communicate with neurons bi-directionally via contact-mediated or secreted factors to regulate the level of inflammatory responses. Many microglia-derived molecules can promote neurodegeneration, including IL-1β (42), reactive oxygen species (ROS) (43), nitric oxide (NO) (44), glutamate (45) and ATP (46). In addition to soluble mediators, there could be a cell-to-cell contact requirement between neurons and microglia that is disrupted in SARM−/− neurons and microglia. The neuroimmunoregulatory CX3CL1-CX3CR1 interaction is a mechanism utilized by neurons to prevent aberrant microglia activation. Another neuroimmunoregulatory molecule is CD200, which is expressed on neurons. By interacting with its receptor, CD200R, on microglia, microglia are kept quiescent in the normal state and prevent phagocytosis (13). It is possible that loss of any of these cell-to-cell interactions can lead to microglia activation and an inflammatory response. Future experiments will address whether soluble factors or cell-to-cell contact are required for cytokine production and whether viral PAMPs or endogenous danger signals are upstream of SARM.
In summary, our data demonstrate that mammalian SARM plays a role in neurodegeneration during viral CNS infection. In addition SARM plays a positive role in cytokine production similar to its C.elegans homolog. This is likely limited to infections of the CNS consistent with its expression pattern and its importance on non-hematopoeitic cells. The data suggest that SARM is crucial for CNS injury and cytokine production in the CNS and may provide a link between neurodegeneration and the innate immune response. Therapeutic targeting of SARM may be beneficial in neurodegenerative diseases as well as CNS viral infection.
Supplementary Material
Acknowledgments
We would like to thank Dr. Virginia Guillespie at the Comparative Pathology Diagnostic Laboratory at the Icahn Medical School at Mount Sinai for reading pathology slides. We also would like to thank Richard Cadagan, Osman Lizardo, Huihong Li, and Xiuju Jiang for excellent technical assistance.
This work was supported by NIH grant NS060885 to BT and NIAID grant U19AI083025 to AGS.
References
- 1.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 2.Mink M, Fogelgren B, Olszewski K, Maroy P, Csiszar K. A novel human gene (SARM) at chromosome 17q11 encodes a protein with a SAM motif and structural similarity to Armadillo/beta-catenin that is conserved in mouse, Drosophila, and Caenorhabditis elegans. Genomics. 2001;74:234–244. doi: 10.1006/geno.2001.6548. [DOI] [PubMed] [Google Scholar]
- 3.Harris TW, Antoshechkin I, Bieri T, Blasiar D, Chan J, Chen WJ, De La Cruz N, Davis P, Duesbury M, Fang R, Fernandes J, Han M, Kishore R, Lee R, Muller HM, Nakamura C, Ozersky P, Petcherski A, Rangarajan A, Rogers A, Schindelman G, Schwarz EM, Tuli MA, Van Auken K, Wang D, Wang X, Williams G, Yook K, Durbin R, Stein LD, Spieth J, Sternberg PW. WormBase: a comprehensive resource for nematode research. Nucleic Acids Res. 2010;38:D463–467. doi: 10.1093/nar/gkp952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Couillault C, Pujol N, Reboul J, Sabatier L, Guichou JF, Kohara Y, Ewbank JJ. TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat Immunol. 2004;5:488–494. doi: 10.1038/ni1060. [DOI] [PubMed] [Google Scholar]
- 5.Chuang CF, Bargmann CI. A Toll-interleukin 1 repeat protein at the synapse specifies asymmetric odorant receptor expression via ASK1 MAPKKK signaling. Genes Dev. 2005;19:270–281. doi: 10.1101/gad.1276505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carty M, Goodbody R, Schroder M, Stack J, Moynagh PN, Bowie AG. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat Immunol. 2006;7:1074–1081. doi: 10.1038/ni1382. [DOI] [PubMed] [Google Scholar]
- 7.Kim Y, Zhou P, Qian L, Chuang JZ, Lee J, Li C, Iadecola C, Nathan C, Ding A. MyD88-5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. J Exp Med. 2007;204:2063–2074. doi: 10.1084/jem.20070868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szretter KJ, Samuel MA, Gilfillan S, Fuchs A, Colonna M, Diamond MS. The immune adaptor molecule SARM modulates tumor necrosis factor alpha production and microglia activation in the brainstem and restricts West Nile Virus pathogenesis. J Virol. 2009;83:9329–9338. doi: 10.1128/JVI.00836-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH, Jr, Conforti L, Coleman M, Tessier-Lavigne M, Zuchner S, Freeman MR. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friede RL. The relationship of body size, nerve cell size, axon length, and glial density in the cerebellum. Proc Natl Acad Sci U S A. 1963;49:187–193. doi: 10.1073/pnas.49.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang JT, Medress ZA, Barres BA. Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol. 2012;196:7–18. doi: 10.1083/jcb.201108111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shamash S, Reichert F, Rotshenker S. The cytokine network of Wallerian degeneration: tumor necrosis factor-alpha, interleukin-1alpha, and interleukin-1beta. J Neurosci. 2002;22:3052–3060. doi: 10.1523/JNEUROSCI.22-08-03052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffiths MR, Gasque P, Neal JW. The regulation of the CNS innate immune response is vital for the restoration of tissue homeostasis (repair) after acute brain injury: a brief review. Int J Inflam. 2010;2010:151097. doi: 10.4061/2010/151097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butchi NB, Woods T, Du M, Morgan TW, Peterson KE. TLR7 and TLR9 trigger distinct neuroinflammatory responses in the CNS. Am J Pathol. 2011;179:783–794. doi: 10.1016/j.ajpath.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease--a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 16.Bjorklund A, Lindvall O. Self-repair in the brain. Nature. 2000;405:892–893. 895. doi: 10.1038/35016175. [DOI] [PubMed] [Google Scholar]
- 17.Noda M, Doi Y, Liang J, Kawanokuchi J, Sonobe Y, Takeuchi H, Mizuno T, Suzumura A. Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J Biol Chem. 2011;286:2308–2319. doi: 10.1074/jbc.M110.169839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matrosovich M, Matrosovich T, Garten W, Klenk HD. New low-viscosity overlay medium for viral plaque assays. Virol J. 2006;3:63. doi: 10.1186/1743-422X-3-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- 20.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 21.Huneycutt BS, I, Plakhov V, Shusterman Z, Bartido SM, Huang A, Reiss CS, Aoki C. Distribution of vesicular stomatitis virus proteins in the brains of BALB/c mice following intranasal inoculation: an immunohistochemical analysis. Brain Res. 1994;635:81–95. doi: 10.1016/0006-8993(94)91426-5. [DOI] [PubMed] [Google Scholar]
- 22.Ireland DD, Reiss CS. Gene expression contributing to recruitment of circulating cells in response to vesicular stomatitis virus infection of the CNS. Viral Immunol. 2006;19:536–545. doi: 10.1089/vim.2006.19.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Detje CN, Meyer T, Schmidt H, Kreuz D, Rose JK, Bechmann I, Prinz M, Kalinke U. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J Immunol. 2009;182:2297–2304. doi: 10.4049/jimmunol.0800596. [DOI] [PubMed] [Google Scholar]
- 24.Steel CD, Kim WK, Sanford LD, Wellman LL, Burnett S, Van Rooijen N, Ciavarra RP. Distinct macrophage subpopulations regulate viral encephalitis but not viral clearance in the CNS. J Neuroimmunol. 2010;226:81–92. doi: 10.1016/j.jneuroim.2010.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carroll SL, Frohnert PW. Expression of JE (monocyte chemoattractant protein-1) is induced by sciatic axotomy in wild type rodents but not in C57BL/Wld(s) mice. J Neuropathol Exp Neurol. 1998;57:915–930. doi: 10.1097/00005072-199810000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Perrin FE, Lacroix S, Aviles-Trigueros M, David S. Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in Wallerian degeneration. Brain. 2005;128:854–866. doi: 10.1093/brain/awh407. [DOI] [PubMed] [Google Scholar]
- 27.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 28.Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9:429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- 29.Mukherjee P, Woods TA, Moore RA, Peterson KE. Activation of the Innate Signaling Molecule MAVS by Bunyavirus Infection Upregulates the Adaptor Protein SARM1, Leading to Neuronal Death. Immunity. 2013;38:705–716. doi: 10.1016/j.immuni.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panneerselvam P, Singh LP, Selvarajan V, Chng WJ, Ng SB, Tan NS, Ho B, Chen J, Ding JL. T-cell death following immune activation is mediated by mitochondria-localized SARM. Cell Death Differ. 2013;20:478–489. doi: 10.1038/cdd.2012.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468:253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- 32.Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer's disease. Neurobiol Dis. 2010;37:503–509. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orr CF, Rowe DB, Halliday GM. An inflammatory review of Parkinson's disease. Prog Neurobiol. 2002;68:325–340. doi: 10.1016/s0301-0082(02)00127-2. [DOI] [PubMed] [Google Scholar]
- 34.Ovanesov MV, Ayhan Y, Wolbert C, Moldovan K, Sauder C, Pletnikov MV. Astrocytes play a key role in activation of microglia by persistent Borna disease virus infection. J Neuroinflammation. 2008;5:50. doi: 10.1186/1742-2094-5-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen CJ, Ou YC, Lin SY, Raung SL, Liao SL, Lai CY, Chen SY, Chen JH. Glial activation involvement in neuronal death by Japanese encephalitis virus infection. J Gen Virol. 2010;91:1028–1037. doi: 10.1099/vir.0.013565-0. [DOI] [PubMed] [Google Scholar]
- 36.Chauhan VS, Furr SR, Sterka DG, Jr, Nelson DA, Moerdyk-Schauwecker M, Marriott I, Grdzelishvili VZ. Vesicular stomatitis virus infects resident cells of the central nervous system and induces replication-dependent inflammatory responses. Virology. 2010;400:187–196. doi: 10.1016/j.virol.2010.01.025. [DOI] [PubMed] [Google Scholar]
- 37.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14:1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 41.Yang G, Meng Y, Li W, Yong Y, Fan Z, Ding H, Wei Y, Luo J, Ke ZJ. Neuronal MCP-1 mediates microglia recruitment and neurodegeneration induced by the mild impairment of oxidative metabolism. Brain Pathol. 2011;21:279–297. doi: 10.1111/j.1750-3639.2010.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tolosa L, Caraballo-Miralles V, Olmos G, Llado J. TNF-alpha potentiates glutamate-induced spinal cord motoneuron death via NF-kappaB. Mol Cell Neurosci. 2011;46:176–186. doi: 10.1016/j.mcn.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 43.Chao CC, Hu S, Ehrlich L, Peterson PK. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-D-aspartate receptors. Brain Behav Immun. 1995;9:355–365. doi: 10.1006/brbi.1995.1033. [DOI] [PubMed] [Google Scholar]
- 44.Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21:6480–6491. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen CJ, Ou YC, Chang CY, Pan HC, Liao SL, Chen SY, Raung SL, Lai CY. Glutamate released by Japanese encephalitis virus-infected microglia involves TNF-alpha signaling and contributes to neuronal death. Glia. 2012;60:487–501. doi: 10.1002/glia.22282. [DOI] [PubMed] [Google Scholar]
- 46.Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1111098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.