

Abstract
Neuroblastoma is the most common extracranial solid tumor of childhood and is responsible for over 15% of pediatric cancer deaths. Neuroblastoma tumorigenesis and malignant transformation is driven by overexpression and dominance of cell survival pathways and a lack of normal cellular senescence or apoptosis. Therefore, manipulation of cell survival pathways may decrease the malignant potential of these tumors and provide avenues for the development of novel therapeutics. This review focuses on several facets of cell survival pathways including protein kinases (PI3K, AKT, ALK, and FAK), transcription factors (NF-κB, MYCN and p53), and growth factors (IGF, EGF, PDGF, and VEGF). Modulation of each of these factors decreases the growth or otherwise hinders the malignant potential of neuroblastoma, and many therapeutics targeting these pathways are already in the clinical trial phase of development. Continued research and discovery of effective modulators of these pathways will revolutionize the treatment of neuroblastoma.
Neuroblastoma
Neuroblastoma is the most common extracranial solid tumor of childhood. This neuroendocrine tumor is most commonly located in the adrenal medulla, but may arise anywhere that sympathetic neural tissue is found. Over 50% of these tumors occur in children under 2 years of age1, and neuroblastoma is responsible for over 15% of pediatric cancer deaths.2 Despite recent advances in chemotherapy and surgical care, this tumor continues to carry a dismal prognosis for children presenting with advanced or metastatic disease, with a long term survival of only 18–30%.3 Neuroblastoma tumorigenesis and malignant transformation is driven by overexpression and dominance of cell survival pathways and a lack of normal cellular senescence or apoptosis. Therefore, manipulation of cell survival pathways may decrease the malignant potential of these tumors and provide avenues for the development of novel therapeutics. The current review will discuss some of the common cell survival signaling pathways in neuroblastoma.
Neuroblastoma and Kinases
One group of proteins involved in neuroblastoma cell survival pathways are kinases. Kinases serve to phosphorylate, and thereby activate, other factors in the cell signaling pathway. A well-known kinase pathway involved in cellular survival is the c-AKT kinase cascade. The AKT cascade is important in tumorigenesis as it controls inhibition of normal programmed cell death.4 Central to this cascade is AKT, a serine/threonine kinase that regulates many cellular functions including cell growth5, proliferation6, survival7, and angiogenesis.8 AKT is activated and recruited to the plasma membrane by phosphatidylcholine 38-OH (PI3K), a kinase stimulated by growth factors. Once at the plasma membrane, other kinases such as 3-phosphoinositide-dependent protein kinases (PDKs) phosphorylate AKT4, resulting in an active moiety.9,10 Many studies have shown the effects of AKT in reducing apoptosis in a variety of cell lines. Dudek et al showed that cerebellar neurons transfected with HA-AKT vector overexpressing AKT, had reduced apoptosis following withdrawal of growth factors.11 Khwaja et al showed decreased apoptosis following cell matrix detachment in kidney epithelial cells transfected with a constitutively active form of AKT.12
The PI3K-AKT cell survival pathway has been demonstrated to be important in many human cancers13 including neuroblastoma.14 AKT phosphorylation has been noted in a number of human neuroblastoma cell lines including SK-N-SH, SH-SY5Y, SK-N-BE, SH-EP, and IMR-32.15 In human tissue specimens, phosphorylation of AKT was more abundant in primary neuroblastoma samples than in benign ganglioneuromas or normal adrenal tissue.14 In addition, AKT phosphorylation correlated with advanced stage of disease, unfavorable histology and amplification of the MYCN oncogene in human neuroblastoma specimens.15 Upregulation of AKT has resulted in more aggressive neuroblastoma cell lines. One study treated human neuroblastoma cells with bilobalide, which increased phosphorylation of PI3K as well as AKT, prior to exposing them toxic stimuli. The cells with upregulated PI3K/AKT showed significantly less apoptosis when compared to cells with normal PI3K/AKT activity.16 In another example, SH-SY5Y neuroblastoma cells exposed to oxidative stress were protected from cell death when treated with Lonicera japonica, which enhanced AKT phosphorylation.17
A number of methods have been described to downregulate AKT in neuroblastoma cell lines, in attempts to increase apoptosis and cell death. In one study, long-term exposure of SH-SY5Y cells to interferon-β decreased activation of the PI3K-AKT pathway and increased apoptosis.18 Another method of AKT downregulation involved mTOR inhibition with rapamycin. mTOR is a serine/threonine kinase that is a downstream effector of AKT, regulating cell growth, proliferation, cytoskeleton organization, and energy metabolism.19,20,13 Johnsen and colleagues showed that rapamycin inhibition of mTOR in neuroblastoma cell lines had anti-proliferative effects.14 When rapamycin was administered to mice bearing SH-SY5Y xenografts, there was decreased tumor growth, increased apoptosis, decreased cell proliferation, and decreased small vessel density in the tumors. Finally, they showed that cell lines expressing high levels of MYCN were more sensitive to rapamycin treatment.14 In another study, small molecule inhibitors of the PI3K/AKT pathway, OSU03012 and PI103, were utilized to treat neuroblastoma cell lines and established subcutaneous xenografts in mice. These small molecules both downregulated the phosphorylation of AKT and resulted in decreased cell survival in vitro and decreased tumor growth in vivo. Again, the findings were more pronounced in cell lines with MYCN amplification.21 These observations were important as amplification of the MYCN oncogene is the most significant adverse prognostic indicator in neuroblastoma.22,23
All these findings lend evidence to the important role the PI3K/AKT pathway plays in neuroblastoma cell survival and tumorigenesis. Modulation of this pathway may be an important target for future chemotherapeutic agents in the treatment of neuroblastoma. In fact, current ongoing studies include several phase I clinical trials involving AKT inhibitors. The Children’s Oncology Group has an open protocol for a phase I study of temsirolimus (Table 1), a rapamycin analog, in combination with irinotecan and temozolomide for relapsed or refractory pediatric solid tumors including neuroblastoma. Perifosine (Table 1) is a synthetic oral alkylphospholipid that targets the lipid-binding PH domain of AKT, inhibiting the translocation of AKT to the plasma membrane.24 Recent studies utilizing perifosine in combination with temsirolimus have shown good results in some solid malignancies in adults25, and perifosine has been shown to increase the sensitivity of neuroblastoma cells to chemotherapeutic agents.26 Although the safety of perifosine has not yet been established in children, this drug may prove to be an adjunctive agent for neuroblastoma therapies.
Table 1.
| Inhibitor Name | Chemical Name | Structure | Company |
|---|---|---|---|
| Temsirolimus |

|
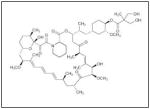
|
Wyeth |
| Perifosine |
|
|
Keryx |
| Crizotinib |

|
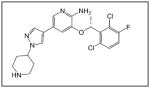
|
Pfizer |
| NVP-TAE684 |

|
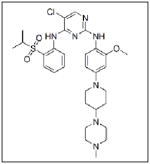
|
Novartis |
| NVP-TAE226 |

|
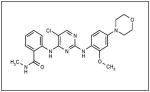
|
Novartis |
| Y15 |
|
|
Sigma |
| NVP-AEW541 |
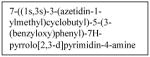
|
|
Novartis |
| BMS-754807 |

|
|
Bristol-Myers Squibb |
| MK0646 |
|
|
Merck |
| Gefitinib |

|
|
AstraZeneca |
| Erlotinib |
|
|
Genentech |
| Imatinib |

|
|
Novartis |
| SU11657 |

|

|
Sugen |
| SU101 |
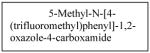
|
|
Sugen |
| AZD2171 |

|
|
AstraZeneca |
| Axitinib |
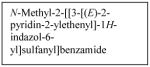
|
|
Pfizer |
| Vandetanib |

|
|
AstraZeneca |
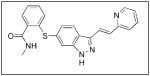
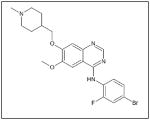
This table provides a list of the chemical names, structures and companies for the inhibitors discussed in the manuscript.
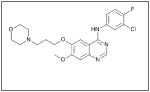
Anaplastic lymphoma kinase, ALK, is another kinase involved in neuroblastoma cell survival signaling. Originally discovered in anaplastic large cell lymphoma cells, ALK is part of the insulin receptor family of trans-membrane receptor tyrosine kinases27 involved in cell growth and development, particularly in the neuronal and central nervous system.28 It has been identified in several tumor types including inflammatory myofibroblastic tumors29, non-small cell lung cancer30,31, diffuse large B-cell lymphoma32, squamous cell cancer of the esophagus33,34, and neuroblastoma. In neuroblastoma, the ALK protein has been found to be expressed in over 90% of tumor samples examined35, and expression of the ALK protein was associated with mutations in the ALK gene.36 In fact, ALK gene mutations have been linked to familial as well as sporadic neuroblastoma. Mosse et al, using whole-genome scans of neuroblastoma pedigrees, showed that heritable mutations of ALK were the main cause of familial neuroblastoma. They also showed that somatically acquired mutations occurred in 12.4% of the tumors and cell lines tested, and that the presence of an aberrant ALK copy number was associated clinically with metastasis at diagnosis and death from disease.37 Furthermore, knockdown of ALK with siRNA resulted in significant inhibition of growth in neuroblastoma cell lines with mutant or amplified ALK.37 In a 2010 meta-analysis of neuroblastoma tumors, ALK gain-of-function mutations were found to be present in 6.9% of tumors, and the most common ALK mutation (F1174) was closely associated with MYCN amplification (58.8%) versus tumors with wild-type ALK.38 Recently, ALK has also been found to be an initiator of MYCN transcription, regulating the MYCN promoter in CLB-GA, CLB-GE, CLB-BAR, and IMR-32 neuroblastoma cell lines.39
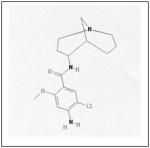
Recent studies have shown that manipulation of ALK can be used as a treatment target for neuroblastoma. One group found that neuroblastoma cell lines with mutated ALK and overactive ALK expression had greater cell death than those with non-mutated ALK when treated with the glycosylation inhibitor, tunicamycin. Downstream survival signaling products of ALK including AKT, ERK1/2, and STAT3 were decreased and apoptosis was activated after treatment with tunicamycin.40 Schonherr et al also found that the neuroblastoma cell lines overexpressing ALK were susceptible to treatment with ALK inhibitors. Treatment of cells with the small molecule ALK inhibitors crizotinib (PF-2341066, Table 1) or NVP-TAE684 (Table 1), and small interfering RNA against ALK, led to decreased cell proliferation and downregulation of MYCN transcription.39 These results demonstrate that ALK may be another kinase to target for innovative neuroblastoma therapies. To that end, patients are currently being accrued in a Phase 1/2 trial being conducted by the Children’s Oncology Group studying the oral small molecule ALK inhibitor, crizotinib, in children with relapsed or refractory neuroblastoma and other solid tumors.
A third kinase involved in neuroblastoma cell survival signaling is focal adhesion kinase (FAK). FAK is a non-receptor tyrosine kinase found on the cellular periphery in focal adhesions, and is a key regulator of pathways involved in proliferation, viability, and survival.41 Recently, it was shown that FAK was present in a number of human tumors including breast cancer and melanoma cells.42,43,44,45 FAK inhibition results in decreased cellular survival. For example, treatment of breast cancer cells (BT474 and MCF-7) with adenoviral gene transduction of a dominant-negative FAK protein (AdFAK-CD) resulted in loss of cell adhesion, degradation of native FAK, and induction of apoptosis while leaving normal mammary cells unaffected.42
FAK has been found to be overexpressed in neuroblastoma cell lines.46 It was also shown that FAK was present in human neuroblastoma tissues and the expression of FAK was related to tumor stage.47 In neuroblastoma, it was demonstrated with chromatin immunoprecipitation studies, electrophoretic mobility shift and dual luciferase assays, that MYCN bound to the FAK promoter and functioned as a transcription factor for FAK.46 Golubovskaya et al demonstrated that NF-κB is involved in the regulation of FAK transcription as well. Utilizing a luciferase assay, they showed that inhibition of NF-κB with a super-repressor of NF-κB resulted in decreased FAK transcriptional activation.48
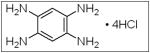
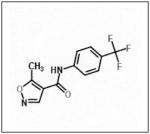
The inhibition of FAK in neuroblastoma using small interfering RNAs resulted in FAK dephosphorylation and cellular phenotypic changes including decreased cell attachment and rounding, and an increase in apoptosis. The effects of FAK inhibition with siRNA were more pronounced in the MYCN positive cells with greater FAK expression.46 When FAK expression was abrogated with a dominant-negative construct, AdFAK-CD, there was a significant decrease in cell survival, again more marked in MYCN+ cell lines.49 Other methods of FAK inhibition have been studied in neuroblastoma and they include small molecules targeting FAK or its phosphorylation, such as NVPTAE-226 (Table 1), or 1,2,4,5, benzenetetraamine tetrahydrochloride (Table 1). Treatment of neuroblastoma cell lines with NVPTAE-226 led to decreased neuroblastoma cell survival, increased apoptosis, and G2 cell cycle arrest.50 Another small molecule, 1,2,4,5, benzenetetraamine tetrahydrochloride (Y15), has also been shown to decrease neuroblastoma growth in vitro.51 In addition, this study also showed a significant decrease in tumor growth in a mouse xenograft model.51 There are currently two Phase 1 clinical trials investigating FAK inhibitors for the treatment of cancer.52 None involve children, but clearly targeting FAK may provide a novel treatment for this disease.
Neuroblastoma and Transcription Factors
Cell survival in neuroblastoma may also be regulated by activation of genes that promote survival or inhibit apoptosis. NF-κB is a transcription factor that is ubiquitously expressed, but is sequestered in the cytoplasm bound to inhibitor of κB (IκB) proteins.53 A variety of stimuli induce kinases that phosphorylate IκB, resulting in their degradation and freeing the NF-κB.54 Upon activation, NF-κB translocates into the nucleus where it binds to promoters of several survival genes including Bcl-2 and caspase inhibitors.4 Uninhibited activity of NF-κB was shown to lead to malignant transformation of NIH3T3 fibroblasts as measured by increased cell saturation density, increased growth in soft agar, and increased tumor growth in nude mice.55 NF-κB signaling has also been implicated in the inhibition of TNF-induced apoptosis in mouse fibroblasts. Fibroblasts with knockout of the p65 subunit of NF-κB were significantly more sensitive to apoptosis following TNF treatment, showing that active NF-κB had a protective effect on the cells.56 Finally, stimulation of multiple myeloma cells with insulin-like growth factor led to increased nuclear translocation of NF-κB and DNA binding activity, and increased resistance to TRAIL-induced apoptosis.57
NF-κB has been shown to be present and inducible in neuroblastoma cell lines.58 Russo et al showed that in glucose deprived neuroblastoma cells, insulin-like growth factor-1 led to increased transcription of NF-κB, which in turn promoted cell survival in the low glucose environment.59 NF-κB has also been implicated in neuroblastoma chemoresistance, as doxorubicin and VP16 have both been shown to trigger NF-κB activation in neuroblastoma cells, inhibiting apoptosis.60 In contrast, inhibition of NF-κB in neuroblastoma cell lines resulted in increased cell death. Karacay et al showed that inhibition of NF-κB in SK-N-SH neuroblastoma cells via adenoviral transfection of a dominant-negative mutant, Ad5-IKKβKA, resulted in significant cell death when the cells were exposed to an active TRAIL vector.61 Similarly, Ammann et al showed that inhibition of NF-κB activation by overexpression of a dominant-negative mutant IκBα-super-repressor resulted in sensitization of SH-EP neuroblastoma cells to TRAIL-induced apoptosis.60 Gao et al utilized synthetic oleanolic triterpenoid to inhibit cell growth and induce apoptosis in SK-N-MC neuroblastoma cells. The increased apoptosis seen was found to be the due partly to the inhibition of the NF-κB pathway.62 In a SCID mouse model, Orr et al showed that NF-κB inhibition with liposome-encapsulated curcumin resulted in a significant decrease in disseminated neuroblastoma burden, with decreased NF-κB activity, decreased cellular proliferation, and increased apoptosis.63
Another transcription factor involved in neuroblastoma cell survival is MYCN, which is encoded by the human MYCN proto-oncogene. MYCN is part of a regulatory network controlling many aspects of cell function including cell proliferation and apoptosis.64 As shown in a murine model, MYCN protein expression is usually restricted to early development and organogenesis. It is then severely downregulated within several days to weeks following birth and is usually only found in adult B cells.65 When MYCN expression does not cease following birth, cell overgrowth and tumors result.66 Mice genetically altered to express MYCN, had a significant incidence of developing neuroblastoma tumors. These tumors closely resembled those seen in humans.67 Brodeur et al showed in 1984 that amplification of MYCN was found in neuroblastoma cell lines, and was associated with more aggressive, advanced tumor types. About half of unfavorable neuroblastomas were MYCN amplified and expressed high levels of MYCN, and MYCN amplification was associated with rapid tumor progression and worse outcomes.23,68
As a significant number of patients have MYCN amplified neuroblastoma, it is logical that anti-tumor therapies may target MYCN itself. For example, Burkhart and others showed that antisense oligonucleotides directed to human MYCN resulted in decreased MYCN protein expression and decreased cell proliferation in IMR-32 neuroblastoma cells. They also treated transgenic MYCN mice with antisense oligonucleotides directed at MYCN and showed an in vivo decrease in tumor growth.69 Kang et al showed that MYCN protein expression could be significantly decreased using small interfering RNA against MYCN (siMYCN) in IMR-32 (MYCN amplified) neuroblastoma cells with no change in MYCN expression seen in SK-N-SH (MYCN non-amplified) cells. The silencing of MYCN inhibited cell growth and induced significant apoptosis after 48 hours of treatment in the MYCN amplified cell lines. Specifically, siMYCN was found to reduce expression of the anti-apoptotic protein Bcl-xL and increase caspase-3 mediated apoptosis. Other neuroblastoma cell lines (LAN-1, IMR-32, JF, SH-SY5Y, and SK-N-SH) were then treated with siMYCN, and significant apoptosis was found in each MYCN amplified cell line with no significant cell death in the non-amplified cell lines.70 Similar results were seen with selective inhibition of MYCN using an anti-gene peptide nucleic acid (PNA). When IMR-32 (MYCN amplified) and SJ-N-KP (MYCN non-amplified) cells were treated with PNA, MYCN mRNA abundance decreased significantly, resulting in decreased cell growth and increased apoptosis that was more pronounced in the MYCN-amplified, IMR-32, cell line.71 Since PNA form stable duplexes with DNA and RNA, and are resistant to nuclease and protease degradation, they show promise for potential antisense therapeutics against MYCN.72 In a study examining phenotypic changes of MYCN amplified neuroblastoma cells, Lynch et al showed microRNA (miRNA) treatment of MYCN amplified cell lines with miR-335 resulted in decreased cellular migration and invasion.73 Similarly, Buechner and colleagues utilized direct MYCN-targeting miRNAs, mir-101 and let-7e, in Kelly (MYCN amplified) neuroblastoma cells to block MYCN. They found a significant decrease in cellular proliferation when Kelly cells were treated with either of these two miRNAs.74 Although amplification of the MYCN oncogene is associated with more aggressive tumor types, expression of MYCN protein does not always correlate with disease outcome.75 For example, hyper-expression of MYCN protein in neuroblastoma cell lines with MCYN amplification, beyond that which is usually expressed, resulted in a significant reduction of neuroblastoma cell growth.76 Finally, analyzing MYCN as a target for further drug development, Lu et al developed a functional MYCN reporter gene assay using neuroblastoma cells stably transfected with a luciferase gene. This assay has been used to screen 2800 compounds from the Cancer Research-UK collection, identifying five compounds with significant reduction of MYCN-dependent luciferase activity (>50%). Further testing of these compounds is needed to determine the optimal method of MYCN inhibition.77
A third transcription factor found to play a role in cell survival in neuroblastoma is p53. This tumor suppressor gene is a regulator of the cell cycle that suppresses the growth of cancer cells, inhibits cell transformation, and induces apoptosis by inhibiting Bcl-2 protein and activating the pro-apoptotic protein BAX.78 Activation of p53 is triggered by DNA damage79 or hypoxia.80 Mutations in p53 have been implicated in a plethora of cancers, to the extent that a 1994 database maintained by Hollstein et al found over 2500 somatic mutations in the p53 gene in various human tumors and tumor cell lines.81
In neuroblastoma, p53 mutations have been found to occur infrequently, and are often seen only in relapsing tumors.82,83 Nonetheless, p53 plays an important role in these tumors through its influence upon other oncogenes. There are strong data to suggest that p53 and MYCN interact in neuroblastoma and may be mutually regulated in these tumors. Mutations of p53 and amplification of MYCN have been shown to occur simultaneously in the same neuroblastoma tumors.84 A study by Torres et al showed that co-transfection of the IMR5 neuroblastoma cell line with p53 and MYCN resulted in elevated p53 expression and reduced MYCN expression.76 Regan et al demonstrated that in MYCN-amplified neuroblastoma cell lines, an increase in p53 expression through inhibition of Hsp90, resulted in MYCN destabilization and suppression of cell growth.85 Finally, employing chromatin immunoprecipitation assays and site directed mutagenesis p53 has been shown to be a direct transcriptional target of MYCN in neuroblastoma.86
Suppression of p53-dependent apoptosis by other proteins, such as MDM287,88, is one process cancer cells employ to avoid normal cell regulation. MDM2 is the primary negative regulator of p53 and has been found in neuroblastoma. Corvi et al showed that the MDM2 gene was amplified in neuroblastoma cell lines, and also the MDM2 protein was present and bound to p53.89 High frequency of single nucleotide polymorphisms in the MDM2 gene promoter resulted in high levels of MDM2 expression that were associated with neuroblastoma disease aggressiveness.90 Studies have shown that MDM2 is regulated by MYCN thereby regulating p53. Slack and colleagues, using chromatin immunoprecipitation, oligonucleotide pulldown and luciferase reporter assays demonstrated that MYCN binds to the MDM2 promoter and induces MDM2 protein expression.91 Conversely, MDM2 regulates MYCN expression. It was recently reported that MDM2 enhanced the mRNA and protein expression of MYCN at a post-transcriptional level. MYCN protein was reduced in MYCN-amplified neuroblastoma cell lines with silencing of MDM2 with siRNA.92 In an in vitro study, He et al showed that in MYCN non-amplified neuroblastoma, MDM2 overexpression via gene transfection resulted in increased tumor growth and survival through suppression of p53. In MYCN amplified neuroblastoma, MDM2 served to induce MYCN expression, which was found to increase p53 expression as well. Subsequent inhibition of MDM2 via siRNA in these cells removed the p53 inhibition and resulted in decreased growth and induction of apoptosis due to the unrestricted p53 stimulation by MYCN itself.93 Similar findings have been demonstrated in vivo. Utilizing murine models of human neuroblastoma xenografts, MDM2 knockdown with siRNA in cell lines expressing wild-type p53 resulted in decreased tumor growth and increased animal survival.94
Golubovskaya and others have recently demonstrated that p53 and FAK interacted in breast cancer cell lines95 serving to sequester p53 in the cytoplasm and preventing its translocation to the nucleus.96 They have also shown an interaction between MDM2 and FAK, and that interruption of the MDM2-FAK interaction with a small molecule resulted in decreased breast and colon tumor cell growth in vitro and in vivo.97 Recent data in our laboratory indicate that p53 and FAK interact in neuroblastoma cell lines (Fig. 1) and that inhibition of the p53 and FAK interaction with interfering peptides results in decreased neuroblastoma tumor cell survival (Fig. 2). Therefore, targeting the inhibitors of p53 such as MDM2, or perhaps even better, the interactions between p53 and other proteins would be valid therapeutic strategies for neuroblastoma.
Figure 1.
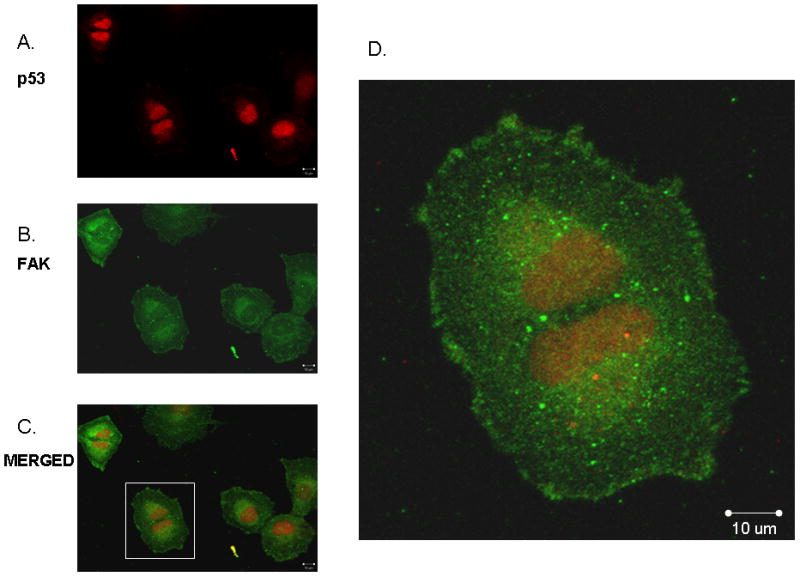
Immunofluorescence staining followed by confocal microscopy was employed to evaluate p53 and FAK colocalization. SK-N-AS neuroblastoma cells were stained for p53 (red, A.) and FAK (green, B.) and evaluated with confocal microscopy to determine colocalization. Merged image (C) shows colocalization of the two stains with a Manders coefficient, 153 MA and MB of 0.68 and 0.33, respectively. D. Enlarged area of (C) (white box) to demonstrate colocalization of the two stains in the nucleus and perinuclear areas.
Figure 2.
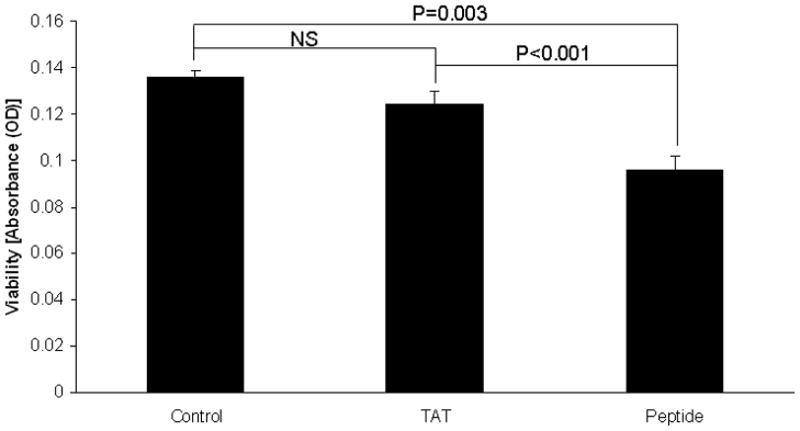
SH-EP neuroblastoma cells were treated with a 7-mer peptide to disrupt the p53-FAK interaction and compared to untreated cells and cells treated with the TAT sequence alone. Cell viability was measured with alamarBlue® assay. There was a significant decrease in cell viability following peptide treatment compared to either control cells or cells treated with TAT sequence alone. The TAT sequence did not significantly affect cell viability.
Neuroblastoma and Growth Factors
Growth factors also have an effect on cell survival. One of the better characterized growth factors functioning to promote cell survival is insulin-like growth factor (IGF), isoforms I (IGF-1) and II (IGF-II). Both type I and type II IGF receptors (IGF-IR, IGF-IIR) were present in neuroblastoma cells as shown by agonist binding studies, and IGF-1 and IGF-II stimulation both led to proliferation of the tumor cells, as Mattsson et al demonstrated in the SH-SY5Y cell line.98 IGF-II has been detected in a little less than half of the neuroblastoma tumor specimens analyzed99, and Tanno and colleagues found IGF-IR present in 86% of primary neuroblastoma tumor specimens studied.100 Evidence also exists to support the idea that MYCN regulates the transcription of IGF-IR. When MYCN non-amplified neuroblastoma cells were transfected with MYCN cDNA, there was a marked increase in IGF-IR mRNA.101 IGF stimulation caused neuroblastoma cells to initiate pro-survival pathways and avoid apoptosis. In 1996, Matthews and Feldman reported that administration of IGF-I to SH-SY5Y neuroblastoma cells during osmotic stress decreased apoptosis, and this effect was abrogated by a blocking antibody of IGF-IR.102 In another study of hyperosmolar-induced apoptosis in neuroblastoma cells, Kim and colleagues demonstrated that IGF-1 exerted a protective effect against hyperosmolarity, decreasing apoptosis and promoting cell survival through activation of the PI3K/AKT pathway.103 Another study showed IGF-1 protected SH-EP neuroblastoma cells from 1-methyl-4-phenylpyridinium (MPP+)-induced apoptosis, also via activation of the PI3K/AKT pathway.104
Insulin receptor substrate proteins 1 and 2 (IRS-1, IRS-2) are intracellular docking molecules that bind to phosphotyrosine residues on the IGF-1 receptor and mediate downstream transduction of the IGF signal.105 The importance of IRS signaling in neuroblastoma survival has been noted. Kim and colleagues demonstrated that SH-EP neuroblastoma cells transfected with IRS-2 were resistant to hyperglycemia-induced apoptosis.106 Similarly, it was shown that overexpression of IRS-1 or IRS-2 caused resistance to glucose-induced apoptosis via the PI3-kinase pathway.107
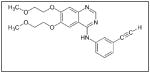
Some investigations evaluating IGF as a target for cancer therapies have been completed. NVP-AEW541 (Table 1), a small molecule inhibitor of IGF-IR activity, induced apoptosis in a number of neuroblastoma cell lines and also inhibited growth of HTLA-230 and SK-N-BE2(C) neuroblastoma xenografts in mice.100 BMS-754807 (Table 1), an oral, reversible, ATP-competitive small molecule inhibitor of IGF-1R, was shown to inhibit the growth of neuroblastoma both in vitro and in vivo; however, response to the drug was defined as progressive disease with growth delay.108 There are currently studies performing Phase I trials testing BMS-754807 in neuroblastoma, pending final results.109 A recent study evaluating another IGF-1R inhibitor, MK0646 (Table 1), in metastatic neuroendocrine tumors, not including neuroblastoma, showed no partial or complete responses.110 Therefore, single agent inhibition of the IGF pathway may not be beneficial, but its inhibition in combination with other chemotherapeutic agents may be a target for future neuroblastoma therapies.
Epidermal growth factor (EGF) and the epidermal growth factor receptor (EGFR) is another pathway of growth factor signaling in neuroblastoma. EGFR expression has been demonstrated in neuroblastoma tumor specimens111 and in a number of neuroblastoma cell lines.112 The expression of EGFR was also found to be significantly increased in multi-drug resistant neuroblastoma cell lines both at the mRNA and protein level.113 daMotta et al showed that EGFR ligand, EGF, treatment of SK-N-SH neuroblastoma cells resulted in cell proliferation.114 Ho and colleagues confirmed these findings, but found that the EGF-stimulated proliferation was the result of EGFR activation of the PI3K/AKT pathway.112 Tamura et al showed that gefitinib (Table 1), an EGFR tyrosine kinase inhibitor, induced apoptosis in neuroblastoma cells.111 Hatziagapiou also illustrated that gefitinib treatment of SH-SY5Y neuroblastoma cells led to decreased tyrosine phosphorylation of EGFR and decreased cell survival.115 Gefitinib was evaluated in a small study of children with relapsed neuroblastoma in combination with oral topotecan and cyclophosphamide, with encouraging results as measured as time to disease progression.116 Other investigators have not shown such encouraging results. Rossler and others found that inhibition of cellular proliferation in neuroblastoma cells in vitro required concentrations of gefitinib that were not achievable in the clinical setting and felt that clinical trials were not warranted.117 Another group looked at a small single-arm pilot study using gefitinib and irinotecan in 23 children with newly diagnosed neuroblastoma. They expected at least a 55% response rate, however, that level was not achieved in the 19 evaluable patients.118 Another EGFR inhibitor, erlotinib (Table 1), has already completed Phase I clinical trials in children with a variety of refractory solid tumors including neuroblastoma, with few dose limiting toxicities and some long term responders.119 Future research and clinical trials with EGFR inhibitors is warranted to develop a wider range of treatment options for neuroblastoma.
Platelet-derived growth factor (PDGF) is a platelet derived growth factor that influences neuroblastoma. PDGF is classically involved in the tissue healing processes as it stimulates cellular proliferation, chemotaxis, and matrix production120, but the mitogenic pathway of PDGF is also involved in cellular transformation and malignancy in human tumors.121 Pahlman et al demonstrated that a number of human neuroblastoma cell lines express PDGF receptor (PDGFR) but not PDGF. They also showed that PDGFR was functional in SH-SY5Y neuroblastoma cells in that PDGF stimulation resulted in a trophic and weakly mitogenic response.122 The presence of PDGF receptor in neuroblastoma was also supported by work from Matsui et al. They showed PDGFR mRNA and protein expression in a variety of neuroblastoma cell lines. They proved functionality by stimulating neuroblastoma cell lines with ligands PDGF-A or PDGF-B and finding an increase not only in DNA incorporation, but also in cellular migration.123 Eggert and others used RT-PCR to evaluate PDGF-A in human neuroblastoma specimens. They noted significantly more abundance of PDGF-A in advanced stage tumors (INSS Stage III and IV) compared to lower stage (I, II, IVS) tumors. In addition, they also found a positive correlation between PDGF-A mRNA and patient survival.124 Further investigations have shown that the AKT pathway was responsible for the PDGF-stimulated proliferation migration, and invasion in neuroblastoma cells.125
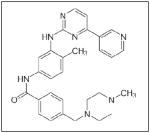
Abrogating the effects of PDGF by inhibiting the PDGF receptor diminishes the ability of tumor cells to survive and propagate. Several PDGF receptor inhibitors have been shown to be effective in neuroblastoma. For example, multiple studies showed that gangliosides modify the effects of PDGF. In SH-SY5Y cells, gangliosides GM1, GM2, GT1b and GD1a were shown to inhibit the PDGF-stimulated phosphorylation of PDGFR and subsequent cellular proliferation.126,127 Several drugs have been designed to inhibit the tyrosine kinase activity of PDGFR. Imatinib (Table 1), a tyrosine kinase inhibitor of PDGFR, was shown to inhibit the growth of a number of human neuroblastoma cell lines in vitro and neuroblastoma xenografts in vivo128, but a Phase II clinical trial of imatinib as a single agent failed to show a response in the treatment of recurrent or refractory neuroblastoma.129 However, in a study published in 2009, the researchers showed that metronomic dosing of imatinib, in combination with doxorubicin, a common chemotherapeutic agent for neuroblastoma, resulted in cell cycle arrest and apoptosis in neuroblastoma lines in vitro. In addition, they showed significant decreases in tumor growth in established neuroblastoma xenografts.130 A more recent study by Timeus et al investigated in vitro imatinib therapy in combination with the HIV protease inhibitor saquinavir. They discovered that the ability of saquinavir to inhibit cell proliferation and invasion as wells as induce apoptosis in neuroblastoma cell lines was significantly increased when combined with imatinib.131 Other small molecule inhibitors of the PDGF pathway have been investigated. Backman et al tested a multi-targeted tyrosine kinase inhibitor directed at PDGFR, SU11657 (Table 1), in mice with SK-N-AS, IMR-32, and SH-SY5Y subcutaneous neuroblastoma xenografts. Treatment resulted in significant inhibition of tumor growth and decreased overall expression of PDGFR by immunohistochemistry.132 A similar PDGFR pathway inhibitor, SU101 (Table 1), has been tested in children with refractory neuroblastoma and was well tolerated, although no clinical responses were documented.133 This promising research highlights that PDGFR inhibition may be a prudent target for drug development for neuroblastoma.
Lastly, vascular endothelial growth factor (VEGF) has been shown to promote tumorigenesis in many human cancers including breast134, non-small-cell lung135, and prostate.136 VEGF isoforms have been found in both neuroblastoma cell lines and in primary tumors137, and the mRNA abundance124,138 and protein expression were associated with higher stage tumors.138,139 Vascular endothelial growth factor receptors (VEFGR) have been identified on neuroblastoma cells140,141,142 as well as in primary tumor specimens.143,138 Both VEGF and VEGFR are involved in promoting neuroblastoma cell survival. Treating IMR-32 neuroblastoma cells with VEGF protected them from apoptosis secondary to serum starvation or TNF-α.144 In nude mouse neuroblastoma xenografts, inhibition of VEGF with anti-VEGF antibody resulted in significant decreases in xenograft growth.145 VEGFR-2 blockade with gene silencing RNAi resulted in significant increases in apoptosis in serum starved neuroblastoma cells.146 VEGF was especially significant in the subset of neuroblastomas with overexpression of MYCN. Kang et al found that MYCN inhibition with siMYCN significantly decreased VEGF secretion in MYCN-amplified neuroblastoma cells, and this effect was enhanced when combined with inhibition of PI3K/AKT. These same effects were not observed in cells with low MYCN.147 Inhibiting VEGF and VEGFR expression, therefore, should act on two fronts, reducing angiogenesis to the tumor as well as blocking cellular survival pathways.
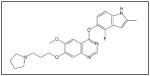
Multiple inhibitors of VEGF and VEGFR have been recently developed. Bevacizumab, a humanized anti-VEGF-A antibody, reduced SK-N-AS, IMR-32, and SH-SY5Y neuroblastoma xenograft growth148, and decreased tumor burden in animals with disseminated neuroblastoma.149 In addition, administration of bevacizumab to neuroblastoma tumor bearing mice, prior to chemotherapy, improved the penetration of the chemotherapeutic drugs by 81%.150 AZD2171 (Table 1), a relatively selective inhibitor of VEGFR, that is orally bioavailable, was shown to delay primary tumor growth in 5 of 6 neuroblastoma xenografts tested.151 A Phase 1 study evaluating the safety of VEGF Trap administration to children with refractory solid tumors including neuroblastoma has been concluded with results pending publication. In a study by Rossler et al, inhibition of VEGFR in neuroblastoma xenografts with the pan-VEGFR tyrosine kinase inhibitor axitinib (AG-013736, Table 1) resulted in a significant delay in time needed to increase tumor volume 5-fold and decreased microvessel density surrounding the tumors.152 Similar results were seen by Orr et al in studying curcumin. They discovered that curcumin treatment decreased VEGF levels and microvessel density in neuroblastoma xenografts.63 Another study utilized a small-molecule tyrosine kinase inhibitor, ZD6474 (vandetanib, Table 1), to block VEGFR-2. ZD6474 inhibited neuroblastoma cell viability in vitro and decreased neuroblastoma xenograft growth by 85%.153 These studies show that targeting both the tumor cells and the tumor vasculature may be a more effective strategy to treat neuroblastoma.
In conclusion, cellular survival pathways are important for tumorigenesis in neuroblastomas. Patients with neuroblastoma continue to have poor prognosis, especially children with more aggressive tumors, such as those that have MYCN amplification. It is encouraging that many of the treatments targeting cell survival pathways produce a more profound effect in these aggressive cell lines. Continued research and discovery of effective inhibitors of these pathways will revolutionize the treatment of neuroblastoma.
Acknowledgments
The authors wish to thank the UAB High Resolution Imaging Facility and Shawn Williams for his outstanding technical assistance with confocal microscopy. This work was supported, in part, by grants from the National Institutes of Health CA91078 (M.L. Megison, L.A. Gillory) and CA118178 (E.A. Beierle).
References
- 1.Spix C, Pastore G, Sankila R, Stiller CA, Steliarova-Foucher E. Neuroblastoma incidence and survival in European children (1978–1997): report from the Automated Childhood Cancer Information System project. Eur J Cancer. 2006;42(13):2081–91. doi: 10.1016/j.ejca.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 2.Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17(7):2264–79. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- 3.Cotterill SJ, Parker L, More L, Craft AW. Neuroblastoma: changing incidence, survival in young people aged 0–24 years A report from the North of England Young Persons’ Malignant Disease Registry. Med Pediatr Oncol. 2001;36(1):231–4. doi: 10.1002/1096-911X(20010101)36:1<231::AID-MPO1056>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 4.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13(22):2905–27. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 5.Scott PH, Brunn GJ, Kohn AD, Roth RA, Lawrence JC. Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1998;95(13):7772–7. doi: 10.1073/pnas.95.13.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E, Slingerland JM. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8(10):1153–60. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 7.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91(2):231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 8.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399(6736):601–5. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 9.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7(4):261–9. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 10.Bellacosa A, Testa JR, Staal SP, Tsichlis PN. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254(5029):274–7. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- 11.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275(5300):661–5. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 12.Khwaja A, Rodriguez-Viciana P, Wennström S, Warne PH, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 1997;16(10):2783–93. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 14.Johnsen JI, Segerström L, Orrego A, Elfman L, Henriksson M, Kågedal B, Eksborg S, Sveinbjörnsson B, Kogner P. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene. 2008;27(20):2910–22. doi: 10.1038/sj.onc.1210938. [DOI] [PubMed] [Google Scholar]
- 15.Opel D, Poremba C, Simon T, Debatin KM, Fulda S. Activation of Akt predicts poor outcome in neuroblastoma. Cancer Res. 2007;67(2):735–45. doi: 10.1158/0008-5472.CAN-06-2201. [DOI] [PubMed] [Google Scholar]
- 16.Shi C, Wu F, Yew DT, Xu J, Zhu Y. Bilobalide prevents apoptosis through activation of the PI3K/Akt pathway in SH-SY5Y cells. Apoptosis. 2010;15(6):715–27. doi: 10.1007/s10495-010-0492-x. [DOI] [PubMed] [Google Scholar]
- 17.Kwon SH, Hong SI, Kim JA, Jung YH, Kim SY, Kim HC, Lee SY, Jang CG. The neuroprotective effects of Lonicera japonica THUNB. against hydrogen peroxide-induced apoptosis via phosphorylation of MAPKs and PI3K/Akt in SH-SY5Y cells. Food Chem Toxicol. 2011;49(4):1011–9. doi: 10.1016/j.fct.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Dedoni S, Olianas MC, Onali P. Interferon-β induces apoptosis in human SH-SY5Y neuroblastoma cells through activation of JAK-STAT signaling and down-regulation of PI3K/Akt pathway. J Neurochem. 2010;115(6):1421–33. doi: 10.1111/j.1471-4159.2010.07046.x. [DOI] [PubMed] [Google Scholar]
- 19.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6(3):184–92. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 20.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8(3):179–83. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 21.Segerström L, Baryawno N, Sveinbjörnsson B, Wickström M, Elfman L, Kogner P, Johnsen JI. Effects of small molecule inhibitors of PI3K/Akt/mTOR signaling on neuroblastoma growth in vitro and in vivo. Int J Cancer. 2011;129(12):2958–65. doi: 10.1002/ijc.26268. [DOI] [PubMed] [Google Scholar]
- 22.Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3(3):203–16. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 23.Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224(4653):1121–4. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- 24.Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther. 2003;2(11):1093–103. [PubMed] [Google Scholar]
- 25.Sun W, Modak S. Emerging treatment options for the treatment of neuroblastoma: potential role of perifosine. Onco Targets Ther. 2012;5:21–9. doi: 10.2147/OTT.S14578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Oh DY, Nakamura K, Thiele CJ. Perifosine-induced inhibition of Akt attenuates brain-derived neurotrophic factor/TrkB-induced chemoresistance in neuroblastoma in vivo. Cancer. 2011;117(23):5412–22. doi: 10.1002/cncr.26133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–4. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 28.Palmer RH, Vernersson E, Grabbe C, Hallberg B. Anaplastic lymphoma kinase: signalling in development and disease. Biochem J. 2009;420(3):345–61. doi: 10.1042/BJ20090387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T, Perlman EJ. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res. 1999;59(12):2776–80. [PubMed] [Google Scholar]
- 30.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 31.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, Sullivan L, Mitchell J, Wetzel R, Macneill J, Ren JM, Yuan J, Bakalarski CE, Villen J, Kornhauser JM, Smith B, Li D, Zhou X, Gygi SP, Gu TL, Polakiewicz RD, Rush J, Comb MJ. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 32.Arber DA, Sun LH, Weiss LM. Detection of the t(2;5)(p23;q35) chromosomal translocation in large B-cell lymphomas other than anaplastic large cell lymphoma. Hum Pathol. 1996;27(6):590–4. doi: 10.1016/s0046-8177(96)90167-7. [DOI] [PubMed] [Google Scholar]
- 33.Du XL, Hu H, Lin DC, Xia SH, Shen XM, Zhang Y, Luo ML, Feng YB, Cai Y, Xu X, Han YL, Zhan QM, Wang MR. Proteomic profiling of proteins dysregulted in Chinese esophageal squamous cell carcinoma. J Mol Med (Berl) 2007;85(8):863–75. doi: 10.1007/s00109-007-0159-4. [DOI] [PubMed] [Google Scholar]
- 34.Jazii FR, Najafi Z, Malekzadeh R, Conrads TP, Ziaee AA, Abnet C, Yazdznbod M, Karkhane AA, Salekdeh GH. Identification of squamous cell carcinoma associated proteins by proteomics and loss of beta tropomyosin expression in esophageal cancer. World J Gastroenterol. 2006;12(44):7104–12. doi: 10.3748/wjg.v12.i44.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamant L, Pulford K, Bischof D, Morris SW, Mason DY, Delsol G, Mariamé B. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J Pathol. 2000;156(5):1711–21. doi: 10.1016/S0002-9440(10)65042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Passoni L, Longo L, Collini P, Coluccia AM, Bozzi F, Podda M, Gregorio A, Gambini C, Garaventa A, Pistoia V, Del Grosso F, Tonini GP, Cheng M, Gambacorti-Passerini C, Anichini A, Fossati-Bellani F, Di Nicola M, Luksch R. Mutation-independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients. Cancer Res. 2009;69(18):7338–46. doi: 10.1158/0008-5472.CAN-08-4419. [DOI] [PubMed] [Google Scholar]
- 37.Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, Hou C, Hakonarson H, Torkamani A, Schork NJ, Brodeur GM, Tonini GP, Rappaport E, Devoto M, Maris JM. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455(7215):930–5. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, Lakeman A, Vandesompele J, Hoebeeck J, Van Maerken T, De Paepe A, Laureys G, Schulte JH, Schramm A, Van Den Broecke C, Vermeulen J, Van Roy N, Beiske K, Renard M, Noguera R, Delattre O, Janoueix-Lerosey I, Kogner P, Martinsson T, Nakagawara A, Ohira M, Caron H, Eggert A, Cools J, Versteeg R, Speleman F. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res. 2010;16(17):4353–62. doi: 10.1158/1078-0432.CCR-09-2660. [DOI] [PubMed] [Google Scholar]
- 39.Schönherr C, Ruuth K, Kamaraj S, Wang CL, Yang HL, Combaret V, Djos A, Martinsson T, Christensen JG, Palmer RH, Hallberg B. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene. 2012 doi: 10.1038/onc.2012.12. [DOI] [PubMed] [Google Scholar]
- 40.Del Grosso F, De Mariano M, Passoni L, Luksch R, Tonini GP, Longo L. Inhibition of N-linked glycosylation impairs ALK phosphorylation and disrupts pro-survival signaling in neuroblastoma cell lines. BMC Cancer. 2011;11:525. doi: 10.1186/1471-2407-11-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gabarra-Niecko V, Schaller MD, Dunty JM. FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 2003;22(4):359–74. doi: 10.1023/a:1023725029589. [DOI] [PubMed] [Google Scholar]
- 42.Xu LH, Yang X, Bradham CA, Brenner DA, Baldwin AS, Craven RJ, Cance WG. The focal adhesion kinase suppresses transformation-associated anchorage-independent apoptosis in human breast cancer cells. Involvement of death receptor-related signaling pathways. J Biol Chem. 2000;275(39):30597–604. doi: 10.1074/jbc.M910027199. [DOI] [PubMed] [Google Scholar]
- 43.Xu LH, Yang X, Craven RJ, Cance WG. The COOH-terminal domain of the focal adhesion kinase induces loss of adhesion and cell death in human tumor cells. Cell Growth Differ. 1998;9(12):999–1005. [PubMed] [Google Scholar]
- 44.Xu LH, Owens LV, Sturge GC, Yang X, Liu ET, Craven RJ, Cance WG. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Differ. 1996;7(4):413–8. [PubMed] [Google Scholar]
- 45.Golubovskaya V, Beviglia L, Xu LH, Earp HS, Craven R, Cance W. Dual inhibition of focal adhesion kinase and epidermal growth factor receptor pathways cooperatively induces death receptor-mediated apoptosis in human breast cancer cells. J Biol Chem. 2002;277(41):38978–87. doi: 10.1074/jbc.M205002200. [DOI] [PubMed] [Google Scholar]
- 46.Beierle EA, Trujillo A, Nagaram A, Kurenova EV, Finch R, Ma X, Vella J, Cance WG, Golubovskaya VM. N-MYC regulates focal adhesion kinase expression in human neuroblastoma. J Biol Chem. 2007;282(17):12503–16. doi: 10.1074/jbc.M701450200. [DOI] [PubMed] [Google Scholar]
- 47.Beierle EA, Massoll NA, Hartwich J, Kurenova EV, Golubovskaya VM, Cance WG, McGrady P, London WB. Focal adhesion kinase expression in human neuroblastoma: immunohistochemical and real-time PCR analyses. Clin Cancer Res. 2008;14(11):3299–305. doi: 10.1158/1078-0432.CCR-07-1511. [DOI] [PubMed] [Google Scholar]
- 48.Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta. 2004;1678(2–3):111–25. doi: 10.1016/j.bbaexp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Beierle EA, Ma X, Trujillo A, Kurenova EV, Cance WG, Golubovskaya VM. Inhibition of focal adhesion kinase and src increases detachment and apoptosis in human neuroblastoma cell lines. Mol Carcinog. 2010;49(3):224–34. doi: 10.1002/mc.20592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beierle EA, Trujillo A, Nagaram A, Golubovskaya VM, Cance WG, Kurenova EV. TAE226 inhibits human neuroblastoma cell survival. Cancer Invest. 2008;26(2):145–51. doi: 10.1080/07357900701577475. [DOI] [PubMed] [Google Scholar]
- 51.Beierle EA, Ma X, Stewart J, Nyberg C, Trujillo A, Cance WG, Golubovskaya VM. Inhibition of focal adhesion kinase decreases tumor growth in human neuroblastoma. Cell Cycle. 2010;9(5):1005–15. doi: 10.4161/cc.9.5.10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schultze A, Fiedler W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin Investig Drugs. 2010;19(6):777–88. doi: 10.1517/13543784.2010.489548. [DOI] [PubMed] [Google Scholar]
- 53.Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242(4878):540–6. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 54.Lenardo MJ, Baltimore D. NF-kappa B: a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58(2):227–9. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- 55.Beauparlant P, Kwan I, Bitar R, Chou P, Koromilas AE, Sonenberg N, Hiscott J. Disruption of I kappa B alpha regulation by antisense RNA expression leads to malignant transformation. Oncogene. 1994;9(11):3189–97. [PubMed] [Google Scholar]
- 56.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274(5288):782–4. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 57.Mitsiades CS, Mitsiades N, Poulaki V, Schlossman R, Akiyama M, Chauhan D, Hideshima T, Treon SP, Munshi NC, Richardson PG, Anderson KC. Activation of NF-kappaB and upregulation of intracellular anti-apoptotic proteins via the IGF-1/Akt signaling in human multiple myeloma cells: therapeutic implications. Oncogene. 2002;21(37):5673–83. doi: 10.1038/sj.onc.1205664. [DOI] [PubMed] [Google Scholar]
- 58.Körner M, Tarantino N, Pleskoff O, Lee LM, Debré P. Activation of nuclear factor kappa B in human neuroblastoma cell lines. J Neurochem. 1994;62(5):1716–26. doi: 10.1046/j.1471-4159.1994.62051716.x. [DOI] [PubMed] [Google Scholar]
- 59.Russo VC, Kobayashi K, Najdovska S, Baker NL, Werther GA. Neuronal protection from glucose deprivation via modulation of glucose transport and inhibition of apoptosis: a role for the insulin-like growth factor system. Brain Res. 2004;1009(1–2):40–53. doi: 10.1016/j.brainres.2004.02.042. [DOI] [PubMed] [Google Scholar]
- 60.Ammann JU, Haag C, Kasperczyk H, Debatin KM, Fulda S. Sensitization of neuroblastoma cells for TRAIL-induced apoptosis by NF-kappaB inhibition. Int J Cancer. 2009;124(6):1301–11. doi: 10.1002/ijc.24068. [DOI] [PubMed] [Google Scholar]
- 61.Karacay B, Sanlioglu S, Griffith TS, Sandler A, Bonthius DJ. Inhibition of the NF-kappaB pathway enhances TRAIL-mediated apoptosis in neuroblastoma cells. Cancer Gene Ther. 2004;11(10):681–90. doi: 10.1038/sj.cgt.7700749. [DOI] [PubMed] [Google Scholar]
- 62.Gao X, Deeb D, Jiang H, Liu Y, Dulchavsky SA, Gautam SC. Synthetic triterpenoids inhibit growth and induce apoptosis in human glioblastoma and neuroblastoma cells through inhibition of prosurvival Akt, NF-kappaB and Notch1 signaling. J Neurooncol. 2007;84(2):147–57. doi: 10.1007/s11060-007-9364-9. [DOI] [PubMed] [Google Scholar]
- 63.Orr WS, Denbo JW, Saab KR, Myers AL, Ng CY, Zhou J, Morton CL, Pfeffer LM, Davidoff AM. Liposome-encapsulated curcumin suppresses neuroblastoma growth through nuclear factor-kappa B inhibition. Surgery. 2012 doi: 10.1016/j.surg.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol. 2000;16:653–99. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- 65.Zimmerman KA, Yancopoulos GD, Collum RG, Smith RK, Kohl NE, Denis KA, Nau MM, Witte ON, Toran-Allerand D, Gee CE. Differential expression of myc family genes during murine development. Nature. 1986;319(6056):780–3. doi: 10.1038/319780a0. [DOI] [PubMed] [Google Scholar]
- 66.Strieder V, Lutz W. Regulation of N-myc expression in development and disease. Cancer Lett. 2002;180(2):107–19. doi: 10.1016/s0304-3835(02)00020-4. [DOI] [PubMed] [Google Scholar]
- 67.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16(11):2985–95. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brodeur GM. Neuroblastoma: clinical significance of genetic abnormalities. Cancer Surv. 1990;9(4):673–88. [PubMed] [Google Scholar]
- 69.Burkhart CA, Cheng AJ, Madafiglio J, Kavallaris M, Mili M, Marshall GM, Weiss WA, Khachigian LM, Norris MD, Haber M. Effects of MYCN antisense oligonucleotide administration on tumorigenesis in a murine model of neuroblastoma. J Natl Cancer Inst. 2003;95(18):1394–403. doi: 10.1093/jnci/djg045. [DOI] [PubMed] [Google Scholar]
- 70.Kang JH, Rychahou PG, Ishola TA, Qiao J, Evers BM, Chung DH. MYCN silencing induces differentiation and apoptosis in human neuroblastoma cells. Biochem Biophys Res Commun. 2006;351(1):192–7. doi: 10.1016/j.bbrc.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tonelli R, Purgato S, Camerin C, Fronza R, Bologna F, Alboresi S, Franzoni M, Corradini R, Sforza S, Faccini A, Shohet JM, Marchelli R, Pession A. Anti-gene peptide nucleic acid specifically inhibits MYCN expression in human neuroblastoma cells leading to cell growth inhibition and apoptosis. Mol Cancer Ther. 2005;4(5):779–86. doi: 10.1158/1535-7163.MCT-04-0213. [DOI] [PubMed] [Google Scholar]
- 72.Pession A, Tonelli R. The MYCN oncogene as a specific and selective drug target for peripheral and central nervous system tumors. Curr Cancer Drug Targets. 2005;5(4):273–83. doi: 10.2174/1568009054064606. [DOI] [PubMed] [Google Scholar]
- 73.Lynch J, Fay J, Meehan M, Bryan K, Watters KM, Murphy DM, Stallings RL. MiRNA-335 suppresses neuroblastoma cell invasiveness by direct targeting of multiple genes from the non-canonical TGF-β signalling pathway. Carcinogenesis. 2012 doi: 10.1093/carcin/bgs114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Buechner J, Tømte E, Haug BH, Henriksen JR, Løkke C, Flægstad T, Einvik C. Tumour-suppressor microRNAs let-7 and mir-101 target the proto-oncogene MYCN and inhibit cell proliferation in MYCN-amplified neuroblastoma. Br J Cancer. 2011;105(2):296–303. doi: 10.1038/bjc.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cohn SL, London WB, Huang D, Katzenstein HM, Salwen HR, Reinhart T, Madafiglio J, Marshall GM, Norris MD, Haber M. MYCN expression is not prognostic of adverse outcome in advanced-stage neuroblastoma with nonamplified MYCN. J Clin Oncol. 2000;18(21):3604–13. doi: 10.1200/JCO.2000.18.21.3604. [DOI] [PubMed] [Google Scholar]
- 76.Torres J, Regan PL, Edo R, Leonhardt P, Jeng EI, Rappaport EF, Ikegaki N, Tang XX. Biological effects of induced MYCN hyper-expression in MYCN-amplified neuroblastomas. Int J Oncol. 2010;37(4):983–91. doi: 10.3892/ijo_00000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu X, Pearson A, Lunec J. The MYCN oncoprotein as a drug development target. Cancer Lett. 2003;197(1–2):125–30. doi: 10.1016/s0304-3835(03)00096-x. [DOI] [PubMed] [Google Scholar]
- 78.Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9(6):1799–805. [PubMed] [Google Scholar]
- 79.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51(23 Pt 1):6304–11. [PubMed] [Google Scholar]
- 80.Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379(6560):88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- 81.Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sørlie T, Hovig E, Smith-Sørensen B, Montesano R, Harris CC. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994;22(17):3551–5. [PMC free article] [PubMed] [Google Scholar]
- 82.Imamura J, Bartram CR, Berthold F, Harms D, Nakamura H, Koeffler HP. Mutation of the p53 gene in neuroblastoma and its relationship with N-myc amplification. Cancer Res. 1993;53(17):4053–8. [PubMed] [Google Scholar]
- 83.Vogan K, Bernstein M, Leclerc JM, Brisson L, Brossard J, Brodeur GM, Pelletier J, Gros P. Absence of p53 gene mutations in primary neuroblastomas. Cancer Res. 1993;53(21):5269–73. [PubMed] [Google Scholar]
- 84.Manhani R, Cristofani LM, Odone Filho V, Bendit I. Concomitant p53 mutation and MYCN amplification in neuroblastoma. Med Pediatr Oncol. 1997;29(3):206–7. doi: 10.1002/(sici)1096-911x(199709)29:3<206::aid-mpo7>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 85.Regan PL, Jacobs J, Wang G, Torres J, Edo R, Friedmann J, Tang XX. Hsp90 inhibition increases p53 expression and destabilizes MYCN and MYC in neuroblastoma. Int J Oncol. 2011;38(1):105–12. [PMC free article] [PubMed] [Google Scholar]
- 86.Chen L, Iraci N, Gherardi S, Gamble LD, Wood KM, Perini G, Lunec J, Tweddle DA. p53 is a direct transcriptional target of MYCN in neuroblastoma. Cancer Res. 2010;70(4):1377–88. doi: 10.1158/0008-5472.CAN-09-2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen J, Wu X, Lin J, Levine AJ. mdm-2 inhibits the G1 arrest and apoptosis functions of the p53 tumor suppressor protein. Mol Cell Biol. 1996;16(5):2445–52. doi: 10.1128/mcb.16.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378(6553):203–6. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 89.Corvi R, Savelyeva L, Breit S, Wenzel A, Handgretinger R, Barak J, Oren M, Amler L, Schwab M. Non-syntenic amplification of MDM2 and MYCN in human neuroblastoma. Oncogene. 1995;10(6):1081–6. [PubMed] [Google Scholar]
- 90.Cattelani S, Defferrari R, Marsilio S, Bussolari R, Candini O, Corradini F, Ferrari-Amorotti G, Guerzoni C, Pecorari L, Menin C, Bertorelle R, Altavista P, McDowell HP, Boldrini R, Dominici C, Tonini GP, Raschellà G, Calabretta B. Impact of a single nucleotide polymorphism in the MDM2 gene on neuroblastoma development and aggressiveness: results of a pilot study on 239 patients. Clin Cancer Res. 2008;14(11):3248–53. doi: 10.1158/1078-0432.CCR-07-4725. [DOI] [PubMed] [Google Scholar]
- 91.Slack A, Chen Z, Tonelli R, Pule M, Hunt L, Pession A, Shohet JM. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc Natl Acad Sci U S A. 2005;102(3):731–6. doi: 10.1073/pnas.0405495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gu L, Zhang H, He J, Li J, Huang M, Zhou M. MDM2 regulates MYCN mRNA stabilization and translation in human neuroblastoma cells. Oncogene. 2012;31(11):1342–53. doi: 10.1038/onc.2011.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.He J, Gu L, Zhang H, Zhou M. Crosstalk between MYCN and MDM2-p53 signal pathways regulates tumor cell growth and apoptosis in neuroblastoma. Cell Cycle. 2011;10(17):2994–3002. doi: 10.4161/cc.10.17.17118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen Z, Lin Y, Barbieri E, Burlingame S, Hicks J, Ludwig A, Shohet JM. Mdm2 deficiency suppresses MYCN-Driven neuroblastoma tumorigenesis in vivo. Neoplasia. 2009;11(8):753–62. doi: 10.1593/neo.09466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Golubovskaya VM, Finch R, Kweh F, Massoll NA, Campbell-Thompson M, Wallace MR, Cance WG. p53 regulates FAK expression in human tumor cells. Mol Carcinog. 2008;47(5):373–82. doi: 10.1002/mc.20395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Golubovskaya VM, Finch R, Zheng M, Kurenova EV, Cance WG. The 7-amino-acid site in the proline-rich region of the N-terminal domain of p53 is involved in the interaction with FAK and is critical for p53 functioning. Biochem J. 2008;411(1):151–60. doi: 10.1042/BJ20071657. [DOI] [PubMed] [Google Scholar]
- 97.Golubovskaya V, Palma NL, Zheng M, Ho B, Magis A, Ostrov D, Cance WG. A Small-Molecule Inhibitor, 5′-O-Tritylthymidine, Targets FAK And Mdm-2 Interaction, And Blocks Breast And Colon Tumorigenesis In Vivo. Anticancer Agents Med Chem. 2012 doi: 10.2174/1871520611313040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mattsson ME, Enberg G, Ruusala AI, Hall K, Påhlman S. Mitogenic response of human SH-SY5Y neuroblastoma cells to insulin-like growth factor I and II is dependent on the stage of differentiation. J Cell Biol. 1986;102(5):1949–54. doi: 10.1083/jcb.102.5.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sullivan KA, Castle VP, Hanash SM, Feldman EL. Insulin-like growth factor II in the pathogenesis of human neuroblastoma. Am J Pathol. 1995;147(6):1790–8. [PMC free article] [PubMed] [Google Scholar]
- 100.Tanno B, Mancini C, Vitali R, Mancuso M, McDowell HP, Dominici C, Raschellà G. Down-regulation of insulin-like growth factor I receptor activity by NVP-AEW541 has an antitumor effect on neuroblastoma cells in vitro and in vivo. Clin Cancer Res. 2006;12(22):6772–80. doi: 10.1158/1078-0432.CCR-06-1479. [DOI] [PubMed] [Google Scholar]
- 101.Chambéry D, Mohseni-Zadeh S, de Gallé B, Babajko S. N-myc regulation of type I insulin-like growth factor receptor in a human neuroblastoma cell line. Cancer Res. 1999;59(12):2898–902. [PubMed] [Google Scholar]
- 102.Matthews CC, Feldman EL. Insulin-like growth factor I rescues SH-SY5Y human neuroblastoma cells from hyperosmotic induced programmed cell death. J Cell Physiol. 1996;166(2):323–31. doi: 10.1002/(SICI)1097-4652(199602)166:2<323::AID-JCP10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 103.Kim B, Feldman EL. Insulin-like growth factor I prevents mannitol-induced degradation of focal adhesion kinase and Akt. J Biol Chem. 2002;277(30):27393–400. doi: 10.1074/jbc.M201963200. [DOI] [PubMed] [Google Scholar]
- 104.Wang L, Yang HJ, Xia YY, Feng ZW. Insulin-like growth factor 1 protects human neuroblastoma cells SH-EP1 against MPP+-induced apoptosis by AKT/GSK-3β/JNK signaling. Apoptosis. 2010;15(12):1470–9. doi: 10.1007/s10495-010-0547-z. [DOI] [PubMed] [Google Scholar]
- 105.Lee YH, White MF. Insulin receptor substrate proteins and diabetes. Arch Pharm Res. 2004;27(4):361–70. doi: 10.1007/BF02980074. [DOI] [PubMed] [Google Scholar]
- 106.Kim B, Feldman EL. Insulin receptor substrate (IRS)-2, not IRS-1, protects human neuroblastoma cells against apoptosis. Apoptosis. 2009;14(5):665–73. doi: 10.1007/s10495-009-0331-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stöhr O, Hahn J, Moll L, Leeser U, Freude S, Bernard C, Schilbach K, Markl A, Udelhoven M, Krone W, Schubert M. Insulin receptor substrate-1 and -2 mediate resistance to glucose-induced caspase-3 activation in human neuroblastoma cells. Biochim Biophys Acta. 2011;1812(5):573–80. doi: 10.1016/j.bbadis.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 108.Carboni JM, Wittman M, Yang Z, Lee F, Greer A, Hurlburt W, Hillerman S, Cao C, Cantor GH, Dell-John J, Chen C, Discenza L, Menard K, Li A, Trainor G, Vyas D, Kramer R, Attar RM, Gottardis MM. BMS-754807; a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol Cancer Ther. 2009;8(12):3341–9. doi: 10.1158/1535-7163.MCT-09-0499. [DOI] [PubMed] [Google Scholar]
- 109.Kolb EA, Gorlick R, Lock R, Carol H, Morton CL, Keir ST, Reynolds CP, Kang MH, Maris JM, Billups C, Smith MA, Houghton PJ. Initial testing (stage 1) of the IGF-1 receptor inhibitor BMS-754807 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2011;56(4):595–603. doi: 10.1002/pbc.22741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Reidy-Lagunes DL, Vakiani E, Segal MF, Hollywood EM, Tang LH, Solit DB, Pietanza MC, Capanu M, Saltz LB. A phase 2 study of the insulin-like growth factor-1 receptor inhibitor MK-0646 in patients with metastatic, well-differentiated neuroendocrine tumors. Cancer. 2012 doi: 10.1002/cncr.27459. [DOI] [PubMed] [Google Scholar]
- 111.Tamura S, Hosoi H, Kuwahara Y, Kikuchi K, Otabe O, Izumi M, Tsuchiya K, Iehara T, Gotoh T, Sugimoto T. Induction of apoptosis by an inhibitor of EGFR in neuroblastoma cells. Biochem Biophys Res Commun. 2007;358(1):226–32. doi: 10.1016/j.bbrc.2007.04.124. [DOI] [PubMed] [Google Scholar]
- 112.Ho R, Minturn JE, Hishiki T, Zhao H, Wang Q, Cnaan A, Maris J, Evans AE, Brodeur GM. Proliferation of human neuroblastomas mediated by the epidermal growth factor receptor. Cancer Res. 2005;65(21):9868–75. doi: 10.1158/0008-5472.CAN-04-2426. [DOI] [PubMed] [Google Scholar]
- 113.Meyers MB, Shen WP, Spengler BA, Ciccarone V, O’Brien JP, Donner DB, Furth ME, Biedler JL. Increased epidermal growth factor receptor in multidrug-resistant human neuroblastoma cells. J Cell Biochem. 1988;38(2):87–97. doi: 10.1002/jcb.240380203. [DOI] [PubMed] [Google Scholar]
- 114.da Motta LA, Galli P, Piva F, Maggi R. Effects of epidermal growth factor on the [3H]-thymidine uptake in the SK-N-SH and SH-SY5Y human neuroblastoma cell lines. Arq Neuropsiquiatr. 1997;55(3A):444–51. doi: 10.1590/s0004-282x1997000300016. [DOI] [PubMed] [Google Scholar]
- 115.Hatziagapiou K, Braoudaki M, Karpusas M, Tzortzatou-Stathopoulou F. Evaluation of antitumor activity of gefitinib in pediatric glioblastoma and neuroblastoma cells. Clin Lab. 2011;57(9–10):781–4. [PubMed] [Google Scholar]
- 116.Donfrancesco A, De Ioris MA, McDowell HP, De Pasquale MD, Ilari I, Jenkner A, Castellano A, Cialfi S, De Laurentis C, Dominici C. Gefitinib in combination with oral topotecan and cyclophosphamide in relapsed neuroblastoma: pharmacological rationale and clinical response. Pediatr Blood Cancer. 2010;54(1):55–61. doi: 10.1002/pbc.22219. [DOI] [PubMed] [Google Scholar]
- 117.Rössler J, Odenthal E, Geoerger B, Gerstenmeyer A, Lagodny J, Niemeyer CM, Vassal G. EGFR inhibition using gefitinib is not active in neuroblastoma cell lines. Anticancer Res. 2009;29(4):1327–33. [PubMed] [Google Scholar]
- 118.Furman WL, McGregor LM, McCarville MB, Onciu M, Davidoff AM, Kovach S, Hawkins D, McPherson V, Houghton PJ, Billups CA, Wu J, Stewart CF, Santana VM. A single-arm pilot phase II study of gefitinib and irinotecan in children with newly diagnosed high-risk neuroblastoma. Invest New Drugs. 2011 doi: 10.1007/s10637-011-9724-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jakacki RI, Hamilton M, Gilbertson RJ, Blaney SM, Tersak J, Krailo MD, Ingle AM, Voss SD, Dancey JE, Adamson PC. Pediatric phase I and pharmacokinetic study of erlotinib followed by the combination of erlotinib and temozolomide: a Children’s Oncology Group Phase I Consortium Study. J Clin Oncol. 2008;26(30):4921–7. doi: 10.1200/JCO.2007.15.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sprugel KH, McPherson JM, Clowes AW, Ross R. Effects of growth factors in vivoI Cell ingrowth into porous subcutaneous chambers. Am J Pathol. 1987;129(3):601–13. [PMC free article] [PubMed] [Google Scholar]
- 121.Nistér M, Libermann TA, Betsholtz C, Pettersson M, Claesson-Welsh L, Heldin CH, Schlessinger J, Westermark B. Expression of messenger RNAs for platelet-derived growth factor and transforming growth factor-alpha and their receptors in human malignant glioma cell lines. Cancer Res. 1988;48(14):3910–8. [PubMed] [Google Scholar]
- 122.Påhlman S, Johansson I, Westermark B, Nistér M. Platelet-derived growth factor potentiates phorbol ester-induced neuronal differentiation of human neuroblastoma cells. Cell Growth Differ. 1992;3(11):783–90. [PubMed] [Google Scholar]
- 123.Matsui T, Sano K, Tsukamoto T, Ito M, Takaishi T, Nakata H, Nakamura H, Chihara K. Human neuroblastoma cells express alpha and beta platelet-derived growth factor receptors coupling with neurotrophic and chemotactic signaling. J Clin Invest. 1993;92(3):1153–60. doi: 10.1172/JCI116684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Eggert A, Ikegaki N, Kwiatkowski J, Zhao H, Brodeur GM, Himelstein BP. High-level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin Cancer Res. 2000;6(5):1900–8. [PubMed] [Google Scholar]
- 125.Pola S, Cattaneo MG, Vicentini LM. Anti-migratory and anti-invasive effect of somatostatin in human neuroblastoma cells: involvement of Rac and MAP kinase activity. J Biol Chem. 2003;278(42):40601–6. doi: 10.1074/jbc.M306510200. [DOI] [PubMed] [Google Scholar]
- 126.Lee MC, Lee WS, Park CS, Juhng SW. The biologic role of ganglioside in neuronal differentiation--effects of GM1 ganglioside on human neuroblastoma SH-SY5Y cells. J Korean Med Sci. 1994;9(2):179–87. doi: 10.3346/jkms.1994.9.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hynds DL, Summers M, Van Brocklyn J, O’Dorisio MS, Yates AJ. Gangliosides inhibit platelet-derived growth factor-stimulated growth, receptor phosphorylation, and dimerization in neuroblastoma SH-SY5Y cells. J Neurochem. 1995;65(5):2251–8. doi: 10.1046/j.1471-4159.1995.65052251.x. [DOI] [PubMed] [Google Scholar]
- 128.Beppu K, Jaboine J, Merchant MS, Mackall CL, Thiele CJ. Effect of imatinib mesylate on neuroblastoma tumorigenesis and vascular endothelial growth factor expression. J Natl Cancer Inst. 2004;96(1):46–55. doi: 10.1093/jnci/djh004. [DOI] [PubMed] [Google Scholar]
- 129.Bond M, Bernstein ML, Pappo A, Schultz KR, Krailo M, Blaney SM, Adamson PC. A phase II study of imatinib mesylate in children with refractory or relapsed solid tumors: a Children’s Oncology Group study. Pediatr Blood Cancer. 2008;50(2):254–8. doi: 10.1002/pbc.21132. [DOI] [PubMed] [Google Scholar]
- 130.Palmberg E, Johnsen JI, Paulsson J, Gleissman H, Wickström M, Edgren M, Ostman A, Kogner P, Lindskog M. Metronomic scheduling of imatinib abrogates clonogenicity of neuroblastoma cells and enhances their susceptibility to selected chemotherapeutic drugs in vitro and in vivo. Int J Cancer. 2009;124(5):1227–34. doi: 10.1002/ijc.24069. [DOI] [PubMed] [Google Scholar]
- 131.Timeus F, Crescenzio N, Doria A, Foglia L, Pagliano S, Ricotti E, Fagioli F, Tovo PA, Cordero di Montezemolo L. In vitro anti-neuroblastoma activity of saquinavir and its association with imatinib. Oncol Rep. 2012;27(3):734–40. doi: 10.3892/or.2011.1582. [DOI] [PubMed] [Google Scholar]
- 132.Bäckman U, Christofferson R. The selective class III/V receptor tyrosine kinase inhibitor SU11657 inhibits tumor growth and angiogenesis in experimental neuroblastomas grown in mice. Pediatr Res. 2005;57(5 Pt 1):690–5. doi: 10.1203/01.PDR.0000156508.68065.AA. [DOI] [PubMed] [Google Scholar]
- 133.Adamson PC, Blaney SM, Widemann BC, Kitchen B, Murphy RF, Hannah AL, Cropp GF, Patel M, Gillespie AF, Whitcomb PG, Balis FM. Pediatric phase I trial and pharmacokinetic study of the platelet-derived growth factor (PDGF) receptor pathway inhibitor SU101. Cancer Chemother Pharmacol. 2004;53(6):482–8. doi: 10.1007/s00280-004-0769-2. [DOI] [PubMed] [Google Scholar]
- 134.van Iterson V, Leidenius M, von Smitten K, Bono P, Heikkilä P. VEGF-D in association with VEGFR-3 promotes nodal metastasis in human invasive lobular breast cancer. Am J Clin Pathol. 2007;128(5):759–66. doi: 10.1309/7FXVRMXF58PVRJUH. [DOI] [PubMed] [Google Scholar]
- 135.Saintigny P, Kambouchner M, Ly M, Gomes N, Sainte-Catherine O, Vassy R, Czernichow S, Letoumelin P, Breau JL, Bernaudin JF, Kraemer M. Vascular endothelial growth factor-C and its receptor VEGFR-3 in non-small-cell lung cancer: concurrent expression in cancer cells from primary tumour and metastatic lymph node. Lung Cancer. 2007;58(2):205–13. doi: 10.1016/j.lungcan.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 136.Jennbacken K, Vallbo C, Wang W, Damber JE. Expression of vascular endothelial growth factor C (VEGF-C) and VEGF receptor-3 in human prostate cancer is associated with regional lymph node metastasis. Prostate. 2005;65(2):110–6. doi: 10.1002/pros.20276. [DOI] [PubMed] [Google Scholar]
- 137.Meister B, Grünebach F, Bautz F, Brugger W, Fink FM, Kanz L, Möhle R. Expression of vascular endothelial growth factor (VEGF) and its receptors in human neuroblastoma. Eur J Cancer. 1999;35(3):445–9. doi: 10.1016/s0959-8049(98)00387-6. [DOI] [PubMed] [Google Scholar]
- 138.Fakhari M, Pullirsch D, Paya K, Abraham D, Hofbauer R, Aharinejad S. Upregulation of vascular endothelial growth factor receptors is associated with advanced neuroblastoma. J Pediatr Surg. 2002;37(4):582–7. doi: 10.1053/jpsu.2002.31614. [DOI] [PubMed] [Google Scholar]
- 139.Nowicki M, Konwerska A, Ostalska-Nowicka D, Derwich K, Miskowiak B, Kondraciuk B, Samulak D, Witt M. Vascular endothelial growth factor (VEGF)-C - a potent risk factor in children diagnosed with stadium 4 neuroblastoma. Folia Histochem Cytobiol. 2008;46(4):493–9. doi: 10.2478/v10042-008-0067-7. [DOI] [PubMed] [Google Scholar]
- 140.Lagodny J, Jüttner E, Kayser G, Niemeyer CM, Rössler J. Lymphangiogenesis and its regulation in human neuroblastoma. Biochem Biophys Res Commun. 2007;352(2):571–7. doi: 10.1016/j.bbrc.2006.11.062. [DOI] [PubMed] [Google Scholar]
- 141.Beierle EA, Dai W, Langham MR, Copeland EM, Chen MK. Expression of VEGF receptors in cocultured neuroblastoma cells. J Surg Res. 2004;119(1):56–65. doi: 10.1016/j.jss.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 142.Beierle EA, Dai W, Langham MR, Copeland EM, Chen MK. VEGF receptors are differentially expressed by neuroblastoma cells in culture. J Pediatr Surg. 2003;38(3):514–21. doi: 10.1053/jpsu.2003.50091. [DOI] [PubMed] [Google Scholar]
- 143.Langer I, Vertongen P, Perret J, Fontaine J, Atassi G, Robberecht P. Expression of vascular endothelial growth factor (VEGF) and VEGF receptors in human neuroblastomas. Med Pediatr Oncol. 2000;34(6):386–93. doi: 10.1002/(sici)1096-911x(200006)34:6<386::aid-mpo2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 144.Beierle EA, Strande LF, Chen MK. VEGF upregulates Bcl-2 expression and is associated with decreased apoptosis in neuroblastoma cells. J Pediatr Surg. 2002;37(3):467–71. doi: 10.1053/jpsu.2002.30868. [DOI] [PubMed] [Google Scholar]
- 145.Rowe DH, Huang J, Li J, Manley C, O’Toole KM, Stolar CJ, Yamashiro DJ, Kandel JJ. Suppression of primary tumor growth in a mouse model of human neuroblastoma. J Pediatr Surg. 2000;35(6):977–81. doi: 10.1053/jpsu.2000.6946. [DOI] [PubMed] [Google Scholar]
- 146.Gomes E, Rockwell P. p38 MAPK as a negative regulator of VEGF/VEGFR2 signaling pathway in serum deprived human SK-N-SH neuroblastoma cells. Neurosci Lett. 2008;431(2):95–100. doi: 10.1016/j.neulet.2007.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kang J, Rychahou PG, Ishola TA, Mourot JM, Evers BM, Chung DH. N-myc is a novel regulator of PI3K-mediated VEGF expression in neuroblastoma. Oncogene. 2008;27(28):3999–4007. doi: 10.1038/onc.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Segerström L, Fuchs D, Bäckman U, Holmquist K, Christofferson R, Azarbayjani F. The anti-VEGF antibody bevacizumab potently reduces the growth rate of high-risk neuroblastoma xenografts. Pediatr Res. 2006;60(5):576–81. doi: 10.1203/01.pdr.0000242494.94000.52. [DOI] [PubMed] [Google Scholar]
- 149.Sims TL, Williams RF, Ng CY, Rosati SF, Spence Y, Davidoff AM. Bevacizumab suppresses neuroblastoma progression in the setting of minimal disease. Surgery. 2008;144(2):269–75. doi: 10.1016/j.surg.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dickson PV, Hamner JB, Sims TL, Fraga CH, Ng CY, Rajasekeran S, Hagedorn NL, McCarville MB, Stewart CF, Davidoff AM. Bevacizumab-induced transient remodeling of the vasculature in neuroblastoma xenografts results in improved delivery and efficacy of systemically administered chemotherapy. Clin Cancer Res. 2007;13(13):3942–50. doi: 10.1158/1078-0432.CCR-07-0278. [DOI] [PubMed] [Google Scholar]
- 151.Maris JM, Courtright J, Houghton PJ, Morton CL, Gorlick R, Kolb EA, Lock R, Tajbakhsh M, Reynolds CP, Keir ST, Wu J, Smith MA. Initial testing of the VEGFR inhibitor AZD2171 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50(3):581–7. doi: 10.1002/pbc.21232. [DOI] [PubMed] [Google Scholar]
- 152.Rössler J, Monnet Y, Farace F, Opolon P, Daudigeos-Dubus E, Bourredjem A, Vassal G, Geoerger B. The selective VEGFR1-3 inhibitor axitinib (AG-013736) shows antitumor activity in human neuroblastoma xenografts. Int J Cancer. 2011;128(11):2748–58. doi: 10.1002/ijc.25611. [DOI] [PubMed] [Google Scholar]
- 153.Beaudry P, Nilsson M, Rioth M, Prox D, Poon D, Xu L, Zweidler-Mckay P, Ryan A, Folkman J, Ryeom S, Heymach J. Potent antitumor effects of ZD6474 on neuroblastoma via dual targeting of tumor cells and tumor endothelium. Mol Cancer Ther. 2008;7(2):418–24. doi: 10.1158/1535-7163.MCT-07-0568. [DOI] [PubMed] [Google Scholar]
- 154.Manders EMM, Verbeek FJ, Aten JA. Measurement of colocalization of objects in dual-color confocal images. J Microsc. 1993;169:375–82. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]