

Abstract
Through their functional diversification, distinct lineages of CD4+ T cells play key roles in either driving or constraining immune-mediated pathology. Transcription factors are critical in the generation of cellular diversity, and negative regulators antagonistic to alternate fates often act in conjunction with positive regulators to stabilize lineage commitment1. Genetic polymorphisms within a single locus encoding the transcription factor BACH2 are associated with numerous autoimmune and allergic diseases including asthma2, Crohn’s disease3–4, coeliac disease5, vitiligo6, multiple sclerosis7 and type 1 diabetes8. While these associations point to a shared mechanism underlying susceptibility to diverse immune-mediated diseases, a function for Bach2 in the maintenance of immune homeostasis has not been established. Here, we define Bach2 as a broad regulator of immune activation that stabilizes immunoregulatory capacity while repressing the differentiation programmes of multiple effector lineages in CD4+ T cells. Bach2 was required for efficient formation of regulatory (Treg) cells and consequently for suppression of lethal inflammation in a manner that was Treg cell dependent. Assessment of the genome-wide function of Bach2, however, revealed that it represses genes associated with effector cell differentiation. Consequently, its absence during Treg polarization resulted in inappropriate diversion to effector lineages. In addition, Bach2 constrained full effector differentiation within Th1, Th2 and Th17 cell lineages. These findings identify Bach2 as a key regulator of CD4+ T-cell differentiation that prevents inflammatory disease by controlling the balance between tolerance and immunity.
Bach2 is expressed in B cells where it acts as a transcriptional repressor of Blimp-1 and is critical for somatic hypermutation and class switch recombination9–11. Given the association of polymorphisms in the BACH2 locus with multiple inflammatory diseases in humans, we hypothesized an additional role for the transcription factor in the prevention of inflammation. To test this hypothesis, we characterized the phenotype of knockout (KO) mice in which the Bach2 gene had been disrupted9. While pups appeared normal at birth, they developed a progressive wasting disease (Fig. 1a and Supplementary Fig. 1a) that resulted in diminished survival compared to wildtype (WT) littermates (Fig. 1b). Sera from KO mice at 3 months of age contained elevated levels of anti-nuclear and anti-dsDNA autoantibodies (Fig. 1c). Gross examination revealed enlargement of the lungs (Fig. 1d and Supplementary Fig. 1b) with highly penetrant histopathological changes (Fig. 1e) including extensive perivascular and alveolar infiltration by lymphocytes and macrophages (Fig. 1f). Examination of the gut revealed less severe and incompletely penetrant inflammatory pathology of the small intestine and stomach also associated with lymphocytic and macrophage infiltration (Fig. 1g and Supplementary Fig. 2). Consistently, we measured elevated expression of the C-C chemokine receptors CCR4 and CCR9 on splenic CD4+ T cells, which guide migration to the lung and gut, respectively (Fig. 1h)12–13. Accordingly, we found a striking increase in the number of CD4+ T cells in the lungs of KO animals while peripheral lymphoid organs contained similar or decreased numbers (Fig. 1i and Supplementary Fig. 3). We also observed increased proportions of effector cells in both the spleen and lungs of KO animals (Supplementary Fig. 4a) and a substantial proportion of CD4+ T cells in lungs of KO animals expressed the acute activation marker CD69 (Fig. 1j and Supplementary Fig. 4b), a finding suggestive of their involvement in the inflammatory process affecting this organ. CD4+ T cells can be characterized into a number of functionally specialized subsets depending upon expression of lineage-specific transcription factors and cytokines14. Th2 cells play a central role in allergic inflammation and airway disease and are characterized by expression of the transcription factor Gata3 and cytokines such as interleukin (IL)-4 and IL-1315. Consistent with the presence of Th2 inflammation, there were increased proportions of Gata3+ CD4+ T cells in the spleen and lungs (Fig. 1k and Supplementary Fig. 5) and elevated expression of IL-13 and IL-4 in the spleen, lungs and lymph nodes (LN) of KO animals (Fig. 1l and Supplementary Fig. 6a). By contrast, we observed no differences in the frequency of IL-17A+ cells in these organs and only a minor increase in IFN-γ+ cells in the LN (Supplementary Fig. 6b).
Figure 1. Spontaneous lethal inflammation in Bach2 knockout animals.

a,b, Body weight at three months of age (a) and survival (b) of Bach2 knockout (KO) and wildtype (WT) littermate females. c, Titer of anti-dsDNA antibodies and anti-nuclear antibodies (ANA) in the sera of WT and KO animals. d, Gross morphology of lungs from WT and KO mice. e, Histopathology scoring of lung tissue from WT and KO mice (n = 7 per group). f, Haematoxylin and eosin (H+E) and immunohistochemical (IHC) stains of WT and KO lung tissue with hypertrophy of bronchial epithelium (B), eosinophilic crystals (C), perivascular lymphocytic infiltration (L) and macrophage infiltration (M). g, H+E and IHC stains of small intestinal tissue with hypertrophic crypts (C), lymphocytic infiltration (L) and macrophage infiltration (M). h, Expression of CCR4 and CCR9 on the surface of splenic CD4+ T cells. i, Quantification of CD4+ T cells in lungs of WT and KO animals. j, k, Percentage of CD4+ T cells expressing CD69 (j) and Gata3 (k) in the lungs and spleen. l, Flow cytometry of IFN-γ and IL-13 expression by CD4+ T cells from spleen, mLN and lungs. Mice were analyzed at 3 months of age unless otherwise specified. Data are representative of ≥2 independent experiments with ≥3 mice per genotype. Error bars s.e.m.; P values (Student’s t-test).
CD4+ T cells play a key role in both driving and constraining immune-mediated pathology. While effector (Teff) cells are often implicated in immune-mediated disease, FoxP3+ Treg cells suppress inflammatory reactions and play a non-redundant role in maintaining immune homeostasis16–17. Given dysregulated immune reactions in Bach2 KO animals, we measured expression of Bach2 mRNA in conventional and regulatory CD4+ T-cell subsets and their thymic precursors from FoxP3GFP reporter mice. Bach2 mRNA was expressed at high levels in both conventional FoxP3−and FoxP3+ (Treg) CD4SP thymocytes in addition to naïve (Tnai) and Treg cells in the spleen (Fig. 2a). Evaluation of conventional thymic maturation in KO animals revealed similar proportions of CD4SP, CD8SP and TCRβ+ cells (Supplementary Fig. 7). Given high levels of expression of Bach2 mRNA in CD4SP thymocytes, however, we wished to determine the cell-intrinsic function of Bach2 in regulating gene transcription within these cells. We therefore reconstituted Rag1−/− hosts with equal mixtures of lineage-depleted bone marrow cells (hereinafter, BM) from Ly5.1+ WT and Ly5.1− KO animals and measured global gene expression in WT and KO CD4SP cells that had developed within the same host (Supplementary Fig. 8 and Supplementary Table 1). Gene set enrichment analysis (GSEA) of this dataset (Supplementary Table 2) indicated the loss of genes known to be dependent upon FoxP32 or directly bound by FoxP3 (Supplementary Fig. 9a)18. Consistent with these observations, mRNA encoding FoxP3 itself exhibited the greatest fold-reduction in expression amongst all transcripts measured (Fig. 2b and Supplementary Fig. 9b). Consequently, we observed a near complete absence of FoxP3+ cells amongst KO CD4SP thymocytes in WT:KO mixed BM chimeric animals (Fig. 2c and Supplementary Fig. 10). In animals reconstituted with either KO BM alone (Fig. 2c) or equal mixtures of KO and FoxP3sf BM (Supplementary Fig. 11), however, FoxP3+ KO cells were present in both the thymus and spleen but at a lower frequency than amongst WT cells. Taken together, these findings indicated a cell-autonomous requirement for Bach2 in the formation of Treg cells in the thymus with an incomplete defect in non-competitive environments.
Figure 2. Bach2 is required for efficient formation of Treg cells.
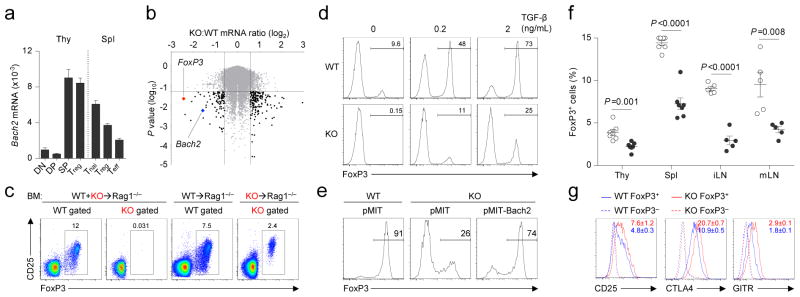
a, Expression of Bach2 mRNA in thymic FoxP3GFP− DP and CD4SP (SP), and FoxP3GFP+ CD4SP Treg cells and splenic CD4+ FoxP3GFP− Tnai and Teff, and FoxP3GFP+ Treg cells isolated from FoxP3GFP reporter mice relative to Actb mRNA. b, Volcano plot indicating differentially expressed genes in KO compared with WT CD4SP thymocytes from WT:KO mixed BM chimeric animals. c, Intracellular FoxP3 expression in CD4SP thymocytes from mice reconstituted with individual or mixed transfers of WT and KO BM. d, FoxP3 expression in WT and KO naïve splenic CD4+ T cells stimulated in the presence of indicated amounts of TGF-β in vitro. e, FoxP3 expression in Thy1.1+ (transduced) WT and KO naïve splenic CD4+ T cells stimulated in the presence of 2ng ml−1 TGF-β and transduced with indicated retroviruses. f, Ratio of FoxP3+ cells in thymic (gated on CD4SP) and extrathymic tissues (gated on CD3+CD4+cells) of 3 month old WT and KO littermates. g, Expression of CD25, CTLA4 and GITR on the surface of splenic FoxP3+ and FoxP3− CD4+ cells from WT and KO mice. Error bars s.e.m.; P values (Student’s t-test). All data are representative ≥2 independent experiments with ≥3 mice per genotype (a–c, f–g) or ≥ 4 experiments (d–e).
While a proportion of Treg cells found in peripheral tissues arise in the thymus (thymic Treg or tTreg)16, induced Treg (iTreg) cells develop from conventional FoxP3− CD4+ T cells in extrathymic tissues. To test whether Bach2 was required for efficient formation of iTreg cells, we tracked the fate of naïve CD4+ T cells upon transfer into Rag1−/− hosts. While a proportion of WT CD4+ T cells converted into FoxP3+ iTreg cells, significantly fewer KO cells underwent similar conversion (Supplementary Fig. 12). By contrast, KO cells exhibited similar stability of FoxP3 expression and survival upon transfer into Rag1−/− hosts over acute timepoints (Supplementary Fig. 13). Consistent with in vivo data, KO naïve CD4+ T cells were markedly impaired in their ability to induce FoxP3 mRNA and form FoxP3+ iTreg cells upon stimulation in the presence of TGF-β in vitro (Fig. 2d, Supplementary Fig. 14). Despite this, KO cells exhibited intact TGF-β and IL-2 signaling (Supplementary Fig. 15). Importantly, defective iTreg induction in KO cells was rescued by reconstitution with Bach2-expressing retroviruses (Fig. 2e), confirming that Bach2 is required during induction for the formation of iTreg cells. In addition, Bach2 overexpression in WT cells enhanced FoxP3 induction under suboptimal polarizing conditions (Supplementary Fig. 16). Taken together, our results demonstrated a requirement for Bach2 in the efficient generation of both tTreg and iTreg cells. Accordingly, analysis of primary and secondary lymphatic tissues from KO mice at three months of age revealed a deficiency in Treg cells resembling that in mice individually reconstituted with KO BM (Fig. 2f). Similar reduction in thymic Treg cell frequency was observed in neonatal mice prior to evidence of autoimmune disease (Supplementary Fig. 17). Furthermore, Treg cell formation was Bach2 gene-dose dependent since mice heterozygous for the KO allele had reduced frequencies of Treg cells (Supplementary Fig. 18). Thus, Treg cells are found at low frequencies in KO mice despite the presence of inflammation in these animals. Characterization of these cells revealed higher levels of expression of Treg cell suppressive molecules CD25, CTLA4 and GITR, the activation marker CD69 and the marker of terminal differentiation, KLRG1 (Fig. 2g and Supplementary Fig. 19; P<0.05)19. Consistent with this terminally differentiated phenotype, Treg cells from Bach2-deficient mice failed to prevent colitis in long-term assays despite possessing acute suppressive function (Supplementary Fig. 20a–e)19. Since Treg cells maintain immune homeostasis in an immunodominant fashion, disorders resulting from their deficiency are amenable to rescue by provision of wildtype Treg cells. To test whether failure to maintain immune homeostasis in the absence of Bach2 was a consequence of defective immunoregulatory capacity, we reconstituted lethally irradiated Rag1−/− mice with KO BM in the presence or absence of WT BM. Strikingly, while we observed massive induction of effector differentiation amongst KO CD4+ T cells and mucosal thickening of the large intestine accompanied by infiltration of KO cells, these changes were prevented by co-transfer of WT BM (Fig. 3a–b and Supplementary Fig. 21a). Consequently, animals reconstituted with KO BM exhibited profound weight-loss and diminished survival while co-transfer of WT BM prevented the induction of disease (Supplementary Fig 21b–c). The dominant immunoregulatory effect exerted by Bach2 sufficient (WT) BM was dependent upon FoxP3 since BM from mice which possess an intact Bach2 locus but lack functional FoxP3 protein (FoxP3Sf)20 could not rescue the phenotype induced by KO BM (Fig. 3c). Moreover, the lethal phenotype induced by KO BM was rescued by transfer of purified splenic CD4+ CD25+ Treg cells from WT mice. Thus, Bach2 is required for the prevention of lethal autoimmunity through its role in Treg cell formation.
Figure 3. Bach2 is required for suppression of lethal inflammation in a Treg-dependent manner.

a, CD44 and CD62L expression on splenic CD4+ T cells descended from KO BM 6 weeks following individual or mixed reconstitution of Rag1−/− mice with KO and WT BM. b, CD3 staining of large intestine and lung tissue from mice 6 weeks following reconstitution with indicated BM. Arrows indicate KO T cells (L). c, Mass of mice following individual or mixed reconstitution of Rag1−/− mice with BM from Scurfy (FoxP3sf), KO or WT mice with or without transfer of 4×105 purified splenic CD4+ CD25+ Treg cells. Data are representative of ≥3 independent experiments. Mass measurements were continued until <3 mice were remaining (c). Error bars s.e.m.; P values (Student’s t-test).
Taken together, these results demonstrated a non-redundant role for Bach2 in Treg-mediated immune homeostasis. We wished, however, to determine the molecular mechanism by which it plays this role. For transcriptional repression, Bach2 is dependent upon a DNA-binding basic leucine zipper region located near the C-terminus of the protein21. We found that overexpression of a truncation mutant deficient in this region (Bach2ΔZip) did not complement defective iTreg induction in KO CD4+ T cells (Fig. 4a) implicating its function as a transcriptional regulator in Treg cell formation. To identify genes whose expression is controlled by Bach2, we performed massively parallel RNA sequencing of KO naïve CD4+ T cells stimulated under iTreg polarizing conditions. Consistent with its role as a transcriptional repressor, a majority of differentially expressed genes were upregulated in Bach2-deficient cells (Supplementary Fig. 22). Strikingly, when we compared these genes with transcripts that were induced upon differentiation of naïve cells into effector-lineage Th1, Th2 or Th17 cells, we found that 31.8% (877) of all upregulated genes (2754) in Bach2-deficient cells were effector lineage-associated genes (Fig. 4b–c). To test whether Bach2 has a direct role in mediating these transcriptional differences, we measured genome-wide Bach2 binding in iTreg cells by chromatin immunoprecipitiation with massively parallel sequencing (ChIP-Seq), validating selected loci by qPCR (Supplementary Fig. 23, Supplementary Table 7). Bach2 bound 43.6% of all derepressed genes including 408 derepressed effector-lineage associated genes (Fig. 4b–c). Examples from this group of genes are provided (Fig. 4d and Supplementary Fig. 24a); notably, Bach2 bound and repressed Prdm1, which encodes Blimp-1, a transcription factor critical in driving full effector differentiation in CD4+ T cells.22 Bach2 also repressed genes with effector lineage-specific functions such as Gata3, Irf4 and Nfil3 and Il12rb1, Il12rb2, Map3k8 and Gadd45g which play important roles in Th2 and Th1 differentiation respectively23–27. Additionally, Bach2 repressed Ahr which is involved in Th17 differentiation28. Importantly, a number of effector-lineage associated genes repressed by Bach2 encode proteins that transduce signals antagonistic to Treg cell differentiation itself, including Il12rb1, Il12rb2 and Tnfsf429–30. Repression of Ccr4 and Ccr9 by Bach2 (Supplementary Fig. 24b) was also of interest since it provides insight into the predominance of lung and gut immunopathology in KO animals.
Figure 4. Bach2 represses effector programmes to stabilize iTreg cell development.

a, FoxP3 expression in GFP+ (transduced) WT and KO naïve splenic CD4+ T cells stimulated in the presence of 2ng ml−1 TGF-β and transduced with indicated retroviruses. b, Derepressed genes in KO compared with WT naïve CD4+ T cells stimulated under iTreg polarizing conditions. Proportion of effector-lineage associated transcripts (upregulated upon stimulation of naïve CD4+ T cells in Th1, Th2 or Th17 conditions respectively (pie chart) and genes that are directly bound by Bach2 in iTreg cells (outer arc) are shown. c, Heatmap indicating expression of effector-lineage associated transcripts derepressed in KO cells (iTreg conditions), their expression in wildtype Th1, Th2 and Th17 cells and binding at their respective gene loci by Bach2 (gene-body ±2kb). d, Alignments showing binding of Bach2 to selected genes and their mRNA expression in WT and KO cells cultured under iTreg conditions. e, Proliferation and effector cytokine expression in CFSE-labelled WT and KO naïve CD4+ T cells stimulated under iTreg conditions for 3 days. f, Transcription factor expression upon stimulation of WT and KO naïve CD4+ T cells under iTreg conditions for 3 days in the presence or absence of indicated anti-cytokine neutralizing antibodies. g, Proliferation and effector cytokine expression in CFSE-labelled WT and KO naïve CD4+ T cells stimulated under indicated polarizing conditions for 3 days. Data are representative of ≥2 independently repeated experiments (a,e–g).
These data indicated that an important aspect of the function of Bach2 is to repress the differentiation programmes of multiple effector lineages during iTreg cell development. Consistently, KO CD4+ T cells stimulated under iTreg conditions aberrantly expressed cytokines associated with effector lineages (Fig. 4e). To determine whether Bach2 stabilizes iTreg cell development through repression of effector differentiation, we tested whether blockade of effector cytokines, which play an important role in positive-reinforcement of effector cell differentiation, could restore iTreg induction in KO cells. While KO cells stimulated under iTreg conditions preferentially differentiated into FoxP3− cells expressing T-bet, Gata3 or RORγt, master regulators of the Th1, Th2 and Th17 differentiation programmes, respectively (Fig. 4f), addition of neutralizing antibodies against IFN-γ and IL-4 partially reverted this phenotype, preventing aberrant induction of T-bet and Gata3 and restoring FoxP3 expression. Interestingly, Ror t expression in KO cells increased in the presence of α-IFN-γ and α-IL-4 antibodies, consistent with the recognized ability of IFN-γ and IL-4 to block Th17 differentiation. Consequently, higher levels of IL-17A were expressed by KO cells under these conditions (Supplementary Fig. 25). These observations raised the possibility that Bach2 might also constrain full effector differentiation amongst conventional T-cell subsets. Strikingly, and consistent with this hypothesis, we observed increased IFN-γ, IL-13 and IL-17A expression when KO naïve CD4+ cells were stimulated under Th1, Th2 and Th17 conditions, respectively, indicating that additional to its role in Treg cell development, Bach2 limits full effector differentiation in conventional CD4+ T cells (Fig. 4g). During specification of a variety of tissues, negative regulators antagonistic to alternate fates often act in conjunction with positive regulators to stabilize lineage identity1. We have identified a function of Bach2 in repressing the differentiation programmes of multiple effector lineages in CD4+ T cells. By doing so, Bach2 stabilizes the development of Treg cells while limiting full effector differentiation in conventional T cell lineages. Thus, at both a cellular and molecular level, Bach2 functions to constrain immune activation enabling it to play a critical role in the maintenance of immune homeostasis. These findings help explain the emergence of Bach2 as a key node in human autoimmunity.
Methods
Plasmid DNA and cloning
For the generation of pMSCV-IRES-Bach2-Thy1.1, a fragment of Bach2 cDNA was amplified by PCR from pMSCV-IRES-Bach2-EGFP1 using the following primers: FW – 5′-GTATTAGCGG CCGCAGACCAT GGACTACAAGGACG ACGATGACAAG-3′ and RV – 5′-GATGAAATCG ATCTAGGCATA ATCTTTCC TGGGCTGT TCGTCCG-3′ and cloned into the NotI and ClaI sites within the multiple cloning site of pMSCV-IRES-Thy1.1-DEST (pMIT; Addgene 17442). pMIG-Bach2 and pMIG-Bach2ΔZip have been described previously10.
Cell culture
CD4+ T cells from spleens and lymph nodes of 6–8 week-old mice were purified by negative selection and magnetic separation (Miltenyi Biotec) followed by sorting of naïve CD4+CD62LhighCD44−CD25− cells using a FACSAria II sorter (BD). For isolation of Treg cells, CD4+GFP+ cells were sorted from FoxP3GFP reporter mice or CD4+CD25high cells. Naïve CD4+ T cells were activated by plate-bound anti-CD3 and soluble anti-CD28 (10 μg ml−1 each; eBioscience) in media for 3 days either under: Th0 conditions (media alone); Th1 conditions (IL-12 (20 ng ml−1, R&D Systems) and anti-IL-4 neutralizing antibodies (10 μg ml−1, BioXCell)); Th2 conditions (IL-4 (20 ng ml−1, R&D Systems) and anti-IFN-γ neutralizing antibodies (10 μg ml−1, BDPharmingen)); Th17 conditions (IL-6 (20 ng ml−1, R&D Systems) plus human TGF-β1 (2 ng ml−1, R&D Systems) and anti-IFN-γ neutralizing antibodies (10 μg ml−1) and anti-IL-4 neutralizing antibodies (10 μg ml−1)) or iTreg conditions (IL-2 (100 IU ml−1, R&D Systems) plus human TGF-β1 (5 ng ml−1)). Where indicated, purified naïve CD4+ T cells were labeled with carboxyfluorescein succinimidyl ester (CFSE, 1mM, Molecular Probes) for 8 minutes at room temperature. The labeling reaction was quenched by washing in FCS.
Retroviral transduction
20ug of retroviral plasmid DNA along with 6ug pCL-Eco plasmid DNA were transfected using 60 ul Lipofectamine 2000 in 3 ml OptiMEM (Invitrogen) for 8 hours in antibiotic-free media into Platinum-E ecotropic packaging cells (Cell Biolabs) plated a day prior on poly-D-lysine coated 10cm plates (Becton Dickinson) at a concentration of 6×106 cells per plate. Media were replaced 8 hours following transfection and cells were incubated for a further 48 hours. Retroviral supernatants were collected and spun at 2000 ×g for 2 hours at 32 °C onto 24 well non-tissue culture treated plates coated overnight in Retronectin (20 ug ml−1; Takara Bio) and 5 ug ml−1 anti-CD3 (2C11) and 5 ug ml−1 anti-CD28 (37.51) (eBioscience). Supernatant was discarded and cells one day following stimulation were applied to plates in triplicate wells for 24 hours in the presence of polarizing cytokines.
Antibodies and flow cytometry
The following fluorescent dye-conjugated antibodies against surface and intracellular antigens were used: anti-FoxP3 (FJK-16s), anti-IL13 (eBio13A), anti-IL17A clone eBio17B7 and anti-Gata3 clone TWAJ (eBioscience); anti-Thy1.1 (OX-7), anti-Ly5.1 (A20), anti-KLRG1 (2F1), anti-B220 (RA3-6B2), anti-NK1.1 (PK136), anti-CTLA4 (UC10-4F10-11), anti-CD4 (RM4-5), anti-CD25 (PC61), anti-CD62L (MEL-14), anti-IFNg (Cat 554413), anti-IL4 (Cat 554435), anti-CD44 (IM7) and anti-CD8a clone 53-6.7 (BD Biosciences); anti-GITR Cat. FAB5241A (R&D Systems) and anti-CD19 clone 6D5 (Biolegend). Cells were incubated with specific antibodies for 30min on ice in the presence of 2.4G2 monoclonal antibody to block Fc R binding. All samples were acquired with a Canto II flow cytometer (Becton Dickinson) and analyzed using FlowJo software (TreeStar). Intranuclear staining for FoxP3 was carried out using the FoxP3 staining kit (eBioscience). To determine cytokine expression, cellular suspensions containing T cells were stimulated with phorbol 12-myristate 13-acetate, ionomycin and brefeldin-A (Leukocyte activation cocktail with Golgiplug; BD biosciences) for 4 h. After stimulation, cells were stained an amine- reactive exclusion-based viability dye (Invitrogen) and with antibodies against cell-surface antigens, fixed and permeabilized followed by intracellular staining with specific anti-cytokine antibodies. Single cell suspensions from lung tissues were prepared by mechanical disruption (GentleMACS, Miltenyi). Countbright beads were spiked-in for the flow cytometric quantification of absolute cell number (Invitrogen).
Autoantibody enzyme-linked immunosorbent assay (ELISA)
For measurement of antinuclear antibodies (ANAs) ELISA assays were performed on mouse serum according to the manufacturer’s instructions (Alpha Diagnostic International). For the measurement of anti-dsDNA autoantiboties, dsDNA coated plates (Calbiotech, Inc.) were incubated with serum samples and anti-dsDNA titers were evaluated using a horseradish peroxidase-conjugated anti-mouse antibody (IgG, IgM, IgA) (Alpha Diagnostic International).
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
Cells were sorted or transferred into RNALater solution (Ambion) and stored at −80 °C. Total RNA from pelleted cells was isolated using the RNAeasy mini kit (Qiagen). First-strand cDNA synthesis was performed using random priming using the high-capacity cDNA synthesis kit (Applied Biosystems) in the presence of SuperaseIn RNase inhibitor (Ambion). cDNA was used as a template for quantitative PCR reactions using the following Taqman primer-probes (Applied Biosystems): Actb (mm00607939_s1), Bach2 (mm00464379_m1) and Foxp3 (mm00475162_m1). Reactions were performed using Fast Universal PCR Mastermix (Applied Biosystems) according to manufacturer’s instructions and thermocycled in quadruplicate 10uL reactions in 384-well plates using the Applied Biosystems. Signals in the FAM channel were normalized to ROX intensity, and ct values were calculated using automatically determined threshold values using SDS software (Applied Biosystems).
Bone marrow chimeras and Treg cell rescue experiments
For bone marrow reconstitution experiments, Rag1−/− mice were administered 1000Gy total-body γradiation from a 137Cs source prior to intravenous injection of BM cells depleted of mature lineages from single-cell bone-marrow preparatons using antibody-coupled magnetic beads (Miltenyi). BM from 6–10 week old mice donor mice were used except from Scurfy mice where 12 day old pups were used as donors. Where indicated, 4 × 105FACS purified CD4+ CD25+ T cells were transferred intravenously into mice 1 day following transfer of BM cells.
In vivo iTreg induction
Rag1−/− mice were injected intravenously with 4 × 105 CD4+CD25−CD45RBhigh cells from wild type or Bach2 deficient mice. On day 21 to 23, mononuclear cells were isolated from whole blood and analyzed for FoxP3 expression.
In vitro suppression assay
Varying numbers of WT and KO CD4+CD25+ Treg cells were cultured in 96-well round-bottom plates with 5 × 104 CFSE-labeled naïve CD4+CD62L+CD44low responder (Tresp) cells along with 1 ×104 MACS isolated CD11c+ dendritic cells used as antigen-presenting cells (Miltenyi). Cells were stimulated with 1 μg ml−1 anti-CD3 antibody (BD Biosciences) for 72 h at 37°C and 5% CO2. Tresp cell proliferation was detected by flow cytometry.
In vivo suppression assay
In vivo suppression assays were done as previously described31. Briefly, Rag2−/− mice were injected intravenously with 1 × 105 CFSE-labeled naive CD4+CD25−CD45Rbhi cells from CD45.1 mice with or without 1 × 105 wild type or Bach2-deficient CD4+ FoxP3GFP+ Treg cells. Mice were analysed on day seven for effector T-cell proliferation by flow cytometry.
Transfer colitis model
The transfer colitis model has been described previously32. Briefly, Rag1−/− mice were injected intravenously with 4 × 105 FACS-sorted naive CD4+CD25−CD45RBhigh cells from CD45.1+ mice with or without 1 × 105 WT or KO CD4+CD25high Treg cells. Mice were monitored weekly for weight loss and signs of disease, and killed at week 6. Sections of the proximal, mid-, and distal colon were fixed in buffered 10% formalin and stained with hematoxylin and eosin (H&E).
RNA Sequencing
RNA Sequencing was performed and analyzed as described previously33. Total RNA was prepared from approximately 1 million cells by using mirVana miRNA Isolation Kit (AM1560, ABI). 200 ng of total RNA was subsequently used to prepare RNA-seq library by using TruSeq SRRNA sample prep kit (FC-122-1001, Illumina) by following manufacturer’s protocol. The libraries were sequenced for 50 cycles (single read) with HiSeq 2000 (Illumina). Sequence reads from each cDNA library were mapped onto the mouse genome build mm9 by using tophat, and the mappable data were then processed by Cufflinks (Trapnell BA Nat. Biotechnol 2010). The obtained data were normalized based on RPKM (reads per kilobase exon model per million mapped reads). To find differentially regulated genes, we used a 1.5-fold change difference between genotypes and 4-fold change difference between different lineages.
Chromatin Immunoprecipitation
T cells were chemically crosslinked and sonicated cells to generate fragmented genomic DNA. Chromatin immunoprecipitation was performed using an anti-Bach2 antibody (N-2; Tohoku University). For sequencing of immunoprecipitated DNA, DNA fragments were blunt-end ligated to the Illumina adaptors, amplified, and sequenced by using the Hi-Seq 2000 (Illumina, San Diego, CA). Sequence reads of 50 bps were obtained by using the Illumina Analysis Pipeline. All reads were mapped to the mouse genome (mm9), and only uniquely matching reads were retained. After removal of redundant reads, enriched peaks were called using ChIP-Seq analysis tool MACS34. Around 20000 peaks were detected at the p-value level of less than 10e-05 and FDR of less than 5%. Peaks in +/− 2kb vicinity of genes bodies were assigned to genes to identify the bound target genes. For PCR-based confirmation of Bach2 binding, chromatin immunoprecipitation was performed as described above, and qPCR reactions were carried out on input and immunoprecipitated DNA using the Power SYBR Green kit (Applied Biosystems) and primers as specified in Supplementary Table 7.
Microarray analysis
100 ng of total RNA extracted as previously described was amplified using Ovation® Pico WTA System V2 (NuGEN) according to the manufacturer’s instructions. Briefly, first strand cDNA was synthesized using the SPIA tagged random and oligo dT primer mix in 10ul reaction after denaturation and incubated at 65 °C for 2 min and priming at 4 °C followed by extension at 25 °C for 30 min, 42 °C for 15 min and 77 °C for 15min. Second strand cDNA synthesis of fragmented RNA was performed using DNA polymerase at 4 °C for 1min, 25 °C for 10 min, 50 °C for 30 min and 80 °C for 20 min. 5′ SPIA tagged double stranded cDNA was used as the template for isothermal single strand cDNA amplification using the SPIA primer following a cycle of SPIA DNA/RNA primer binding, DNA replication, strand displacement and RNA cleavage at 4 °C for 1 min, 47 °C for 75 min and 95 °C for 5min in total 100 ul reaction. 5.5 ug of amplified sscDNA from 12–13 ug total amplification were used for fragmentation and biotinilation using Encore™ Biotin Module (NuGEN) according to the manufacturer’s instructions. Biotinylated cDNA was then hybridized to Mouse Gene 1.0 ST arrays (Affymetrix) overnight at 45 °C and stained on a Genechip Fluidics Station 450 (Affymetrix), all according to the respective manufacturers’ instructions. Arrays were scanned on a GeneChip Scanner 3000 7G (Affymetrix). Global gene expression profiles rank ordered by relative fold-change values were analyzed by using Gene set enrichment analysis software (Broad Institute, MIT). P values were calculated using Student’s t-test using Partek Genomic Suite after RMA normalization.
Statistical analysis
Student’s t-test was used unless otherwise specified to calculate statistical significance of the difference in mean values and P values are provided. For calculation of statistical significance of differences in clinical histopathology scores, the Wilcoxon rank-sum test was used.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Programs of the US National Institutes of Health, National Cancer Institute, National Institute of Arthritis, Musculoskeletal and Skin Diseases and the JSPS Research Fellowship for Japanese Biomedical and Behavioural Researchers at NIH (K.H.). We thank D.N. Roychoudhuri, D.C. Macallan, G.E. Griffin, Y. Ji, D. Palmer, M. Sukumar, G. Fabozzi, K Hanada, E. Lugli, J.H. Pan and N.Van Panhuys for discussions, A. Mixon and S. Farid for cell sorting, G. McMullen for mouse handling and Y. Luo, Y. Wakabayashi, J. Zhu, G. Gutierrez-Cruz and H.W. Sun for help with sequencing and analysis.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author contributions
R.R., K.H., J.O’S. and N.P.R. wrote the manuscript and designed experiments; R.R., K.H., K.M., D.C., M.B., G.S., Y.K., B.D., Z.Y., H.T. and H.L carried out experiments; R.R., H.Z., G.V., E.W., V.S., J.O’S and N.P.R. analyzed experiments; V.H. performed histopathological evaluations; G.P., A.N., A.M. and K.I contributed reagents; C.A.K., M.R., P.M., J.G.C., J.R., D.B., A.N., A.M., F.M., L.G., V.S. and K.I edited the manuscript.
Reprints and permissions information is available at www.nature.com/reprints. Massively parallel RNA and ChIP sequencing data have been deposited to the Gene Expression Omnibus under the accession number GSE45975.
The authors declare no competing financial interests.
References
- 1.Rothenberg EV, Scripture-Adams DD. Competition and collaboration: GATA-3, PU.1, and Notch signaling in early T-cell fate determination. Semin Immunol. 2008;20:236–246. doi: 10.1016/j.smim.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferreira MA, et al. Identification of IL6R and chromosome 11q13.5 as risk loci for asthma. Lancet. 2011;378:1006–1014. doi: 10.1016/S0140-6736(11)60874-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franke A, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christodoulou K, et al. Next generation exome sequencing of paediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes. Gut. 2012 doi: 10.1136/gutjnl-2011-301833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dubois PC, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin Y, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet. 2012;44:676–680. doi: 10.1038/ng.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.International Multiple Sclerosis Genetics C et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper JD, et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat Genet. 2008;40:1399–1401. doi: 10.1038/ng.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muto A, et al. The transcriptional programme of antibody class switching involves the repressor Bach2. Nature. 2004;429:566–571. doi: 10.1038/nature02596. [DOI] [PubMed] [Google Scholar]
- 10.Ochiai K, et al. Plasmacytic transcription factor Blimp-1 is repressed by Bach2 in B cells. J Biol Chem. 2006;281:38226–38234. doi: 10.1074/jbc.M607592200. [DOI] [PubMed] [Google Scholar]
- 11.Muto A, et al. Bach2 represses plasma cell gene regulatory network in B cells to promote antibody class switch. EMBO J. 2010;29:4048–4061. doi: 10.1038/emboj.2010.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sigmundsdottir H, Butcher EC. Environmental cues, dendritic cells and the programming of tissue-selective lymphocyte trafficking. Nat Immunol. 2008;9:981–987. doi: 10.1038/ni.f.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lloyd CM, et al. CC chemokine receptor (CCR)3/eotaxin is followed by CCR4/monocyte-derived chemokine in mediating pulmonary T helper lymphocyte type 2 recruitment after serial antigen challenge in vivo. J Exp Med. 2000;191:265–274. doi: 10.1084/jem.191.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327:1098–1102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med. 1985;161:72–87. doi: 10.1084/jem.161.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gavin MA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- 18.Zheng Y, et al. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445:936–940. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- 19.Cheng G, et al. IL-2 receptor signaling is essential for the development of Klrg1+ terminally differentiated T regulatory cells. J Immunol. 2012;189:1780–1791. doi: 10.4049/jimmunol.1103768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brunkow ME, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 21.Oyake T, et al. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol Cell Biol. 1996;16:6083–6095. doi: 10.1128/mcb.16.11.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. 2010;11:114–120. doi: 10.1038/ni.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rengarajan J, et al. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med. 2002;195:1003–1012. doi: 10.1084/jem.20011128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 25.Murphy KM, et al. Signaling and transcription in T helper development. Annu Rev Immunol. 2000;18:451–494. doi: 10.1146/annurev.immunol.18.1.451. [DOI] [PubMed] [Google Scholar]
- 26.Watford WT, et al. Tpl2 kinase regulates T cell interferon-gamma production and host resistance to Toxoplasma gondii. J Exp Med. 2008;205:2803–2812. doi: 10.1084/jem.20081461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kashiwada M, Cassel SL, Colgan JD, Rothman PB. NFIL3/E4BP4 controls type 2 T helper cell cytokine expression. EMBO J. 2011;30:2071–2082. doi: 10.1038/emboj.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veldhoen M, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 29.Xiao X, et al. OX40 signaling favors the induction of T(H)9 cells and airway inflammation. Nat Immunol. 2012;13:981–990. doi: 10.1038/ni.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oldenhove G, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pesu M, et al. T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance. Nature. 2008;455:246–250. doi: 10.1038/nature07210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Powrie F, Carlino J, Leach MW, Mauze S, Coffman RL. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J Exp Med. 1996;183:2669–2674. doi: 10.1084/jem.183.6.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vahedi G, et al. STATs shape the active enhancer landscape of T cell populations. Cell. 2012;151:981–993. doi: 10.1016/j.cell.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.