

Abstract
Nemaline myopathy (NEM) is a common congenital myopathy. At the very severe end of the NEM clinical spectrum are genetically unresolved cases of autosomal-recessive fetal akinesia sequence. We studied a multinational cohort of 143 severe-NEM-affected families lacking genetic diagnosis. We performed whole-exome sequencing of six families and targeted gene sequencing of additional families. We identified 19 mutations in KLHL40 (kelch-like family member 40) in 28 apparently unrelated NEM kindreds of various ethnicities. Accounting for up to 28% of the tested individuals in the Japanese cohort, KLHL40 mutations were found to be the most common cause of this severe form of NEM. Clinical features of affected individuals were severe and distinctive and included fetal akinesia or hypokinesia and contractures, fractures, respiratory failure, and swallowing difficulties at birth. Molecular modeling suggested that the missense substitutions would destabilize the protein. Protein studies showed that KLHL40 is a striated-muscle-specific protein that is absent in KLHL40-associated NEM skeletal muscle. In zebrafish, klhl40a and klhl40b expression is largely confined to the myotome and skeletal muscle, and knockdown of these isoforms results in disruption of muscle structure and loss of movement. We identified KLHL40 mutations as a frequent cause of severe autosomal-recessive NEM and showed that it plays a key role in muscle development and function. Screening of KLHL40 should be a priority in individuals who are affected by autosomal-recessive NEM and who present with prenatal symptoms and/or contractures and in all Japanese individuals with severe NEM.
Introduction
Nemaline myopathy (NEM) is a common form of nondystrophic congenital myopathy and is defined clinically by skeletal-muscle dysfunction and pathologically by the presence of nemaline bodies within myofibers.1,2 Typical clinical symptoms include hypotonia, muscle weakness of proximal dominance, respiratory insufficiency, and feeding problems. Congenital onset is usual, but a wide variation in age of onset and disease severity is recognized. Mutations in seven genes are known to cause NEM (NEM1–NEM7).1,2 Six of these encode sarcomere-thin-filament proteins or associated proteins: ACTA1 (MIM 102610),3 CFL2 (MIM 601443),4 NEB (MIM 161650),5 TNNT1 (MIM 191041),6 TPM2 (MIM 190990),7 and TPM3 (MIM 191030);8 the seventh, KBTBD13 (kelch-repeat- and BTB-[POZ]-domain-containing 13 [MIM 613727])9 is involved in the ubiquitin proteasome pathway.10 Nevertheless, some forms of NEM remain genetically unsolved.
One such subtype, which has long been recognized,11,12 has apparent autosomal-recessive inheritance and is characterized by severe weakness, in utero presentation of fetal akinesia or hypokinesia and associated abnormalities, and muscle biopsy often showing numerous small nemaline bodies, sometimes only visible by electron microscopy and frequently with virtually no normal myofibrils remaining (“miliary NEM” Figure 1A and Figure S1, available online). We aimed to identify genetic causes of these severe NEM cases by using a combination of linkage analysis, or homozygosity mapping, SNP array, and whole-exome sequencing (WES) in selected families. We have identified loss-of-function mutations in KLHL40 as a frequent cause of severe NEM and have shown through functional studies that KLHL40 is crucial for myogenesis and skeletal-muscle maintenance.
Figure 1.
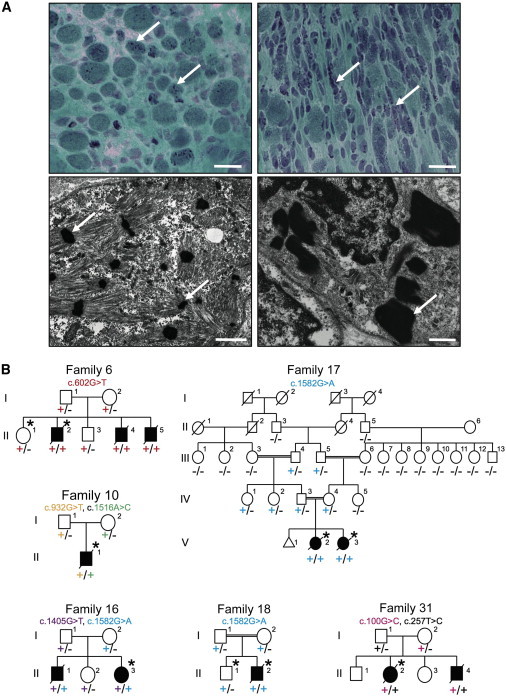
Family Pedigrees and Light and Electron Microscopy of Muscle Biopsies
(A) Modified Gomori trichrome (upper) and electron microscopy (lower) of muscle biopsies from affected individuals of families 15 (right) and 20 (left). Abnormal variation in fiber size, together with many small myofibers and sometimes increased connective tissue, and the presence of numerous red- or purple-stained nemaline bodies (arrows) can be seen (upper panels). Numerous nemaline bodies with varying sizes and shapes and a lack of normal myofibrils are visible by electron microscopy (arrows). Scale bars represent 20 μm for modified Gomori trichrome and 1 μm for electron microscopy.
(B) Pedigrees for the families in which exome sequencing and analysis were performed on the probands. Asterisks indicate the individuals whose DNA was analyzed by exome sequencing. Segregation of the mutations identified in each pedigree is shown.
Subjects and Methods
Subject Details and Ethics
We recruited 143 genetically unresolved severe-NEM-affected families from large congenital-myopathy cohorts in major centers around the world (Boston, Helsinki, Perth, and Tokyo). All individuals within the cohorts were diagnosed with NEM on the basis of muscle-biopsy findings.
Written informed consent was obtained for participation in this study, which was approved by the Human Research Ethics Committee of the University of Western Australia (UWA), the ethics committee of the Children’s Hospital of the University of Helsinki, Yokohama City University School of Medicine, and the Boston Children’s Hospital institutional review board. The UWA Animal Ethics Committee approved animal studies.
Microscopy
Light microscopy and electron microscopy of biopsies was performed as previously described.13
Whole-Genome SNP Genotyping, Linkage Analysis, and WES
Genotyping was performed for families 6 and 18 with the use of the HumanOmniExpress BeadChip Kit (Illumina) and Infinium II Assay Workflow (Illumina) at the Institute for Molecular Medicine Finland (FIMM). Data were analyzed with PLINK v.1.07. Multiple large homozygous regions were identified, but none included known myopathy-associated genes. WES was performed on one healthy and one affected sibling from family 6 and the proband from family 18 with the SeqCap EZ Human Exome Library v.2.0 exome system (Nimblegen, Roche Diagnostics). Coverage depths were 31- to 62-fold. Variant quantification was performed with the FIMM Variant Calling Pipeline v.1.0 and the Integrative Genomics Viewer (IGV, Broad Institute of MIT and Harvard). All known and heterozygous SNPs were excluded. Healthy siblings’ genotypes were used for the exclusion of shared homozygous variants.
Five individuals from family 16 were genotyped with the Human Mapping 10K XbaI 142 2.0 array (Affymetrix) and GeneChip Genotyping Analysis Software (Gtypev4.1). Parametric linkage analysis was performed with Allegro v.2 with a fully penetrant autosomal-recessive model. WES was performed on the proband with the use of the SureSelect Human All Exon 50 Mb Kit (Agilent Technologies) and sequenced in one lane on a GAIIx platform (Illumina) with 108 bp paired-end reads. Reads were aligned to the UCSC Genome Browser (GRCh37/hg19) with Novoalign (Novocraft Technologies). Mean coverage depth was 59-fold. Single-nucleotide variants and small indels were identified with GATK UnifiedGenotyper and filtered according to the Broad Institute’s Best Practices guidelines v.3. Variants registered in dbSNP132 were filtered. The filter-passed variants were annotated with ANNOVAR. Only genes with homozygous variants or more than two variants located in the candidate linkage regions were included.
Family 17 was genotyped with the HumanCytoSNP-12 BeadChip (Illumina). MERLIN was used for performing linkage analysis on a subset of 14,514 SNPs.14 WES was performed for the proband from family 10 and for both siblings from family 17 as described.15 Coverage depth was 61- to 97-fold. Variants were called with LifeScope 2.5 (Life Technologies) and filtered with ANNOVAR16 against ENCODE GENCODE v.11 (October 2011 freeze, GRCh37).17 Two custom variant-filtering steps were used: (1) one against the 1000 Genomes database (February 2012 release) (variants with a minor allele frequency > 0.5% were excluded) and (2) one against the dbSNP135 common database.
Family 31 (BOS74) was one in a cohort of 59 NEM-affected families who underwent WES by the Intellectual and Developmental Disabilities Research Center Core Next-Gen Sequencing Facility of Boston Children’s Hospital and Harvard Medical School in collaboration with Axeq Technologies, Complete Genomics, Integrated Genetics (LabCorp), and the Boston Children’s Hospital Gene Partnership. Exome sequencing was performed with the Illumina HiSeq 2000 platform. Reads were mapped with the Burrows-Wheeler Aligner (v.0.5.8). SNPs and indels were called with SAMtools (v.0.1.7). Data analysis and variant calling were performed with the Broad GATK Best Practices for identification of SNPs and small indels. Annotated variants were filtered against dbSNP135, the 1000 Genomes Project database (October 2011 edition), and the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server (EVS).
Sequencing
Bidirectional Sanger sequencing of KLHL40 (RefSeq accession number NM_152393.2) was performed on biobanked DNA from additional probands with severe NEM and their family members in Boston, Helsinki, Perth, Yokohama, and Tokyo. Identified variants were then screened in all available family members. Primer sequences and conditions are available upon request. For detection of the c.1582G>A (p.Glu528Lys) mutation in normal Japanese controls, high-resolution melting (HRM) analysis with and without the spike-in method18 was performed on LightCycler 480 System II (Roche Diagnostics). If samples showed any aberrant melting patterns, Sanger sequencing was performed for confirmation of the mutation.
LOD Scores
Where possible, MERLIN was used for calculating LOD scores for individual families.14
Expression Analysis on Human cDNAs
TaqMan quantitative real-time PCR analyses were performed with cDNAs of human adult (Human MTCPanel I, #636742, Clontech Laboratories) and fetal (Human Fetal MTC Panel, #636747, Clontech Laboratories) tissues.19 Predesigned TaqMan probe sets for human KLHL40 (KBTBD5, Hs00328078_m1, Applied Biosystems) and human β-actin (ACTB, 4326315E, Applied Biosystems) were used. PCR was performed on a Rotor-Gene Q (QIAGEN) (conditions are available upon request) and analyzed with the Rotor-Gene Q Series Software by the 2−ΔΔCt method. Relative concentrations of cDNA were normalized to concentrations obtained from the hearts.
Calculations of the Free-Energy Change upon Amino Acid Substitutions
Molecular structures were drawn with PyMOL. FoldX v.3.0 beta20 was used through a graphics interface as a plugin for the YASARA molecular viewer.21 Crystal structures of the kelch domain of human KLHL40 (Protein Data Bank [PDB] code 4ASC) and the BTB (bric-a-brac, tram-track, broad-complex)-BACK (BTB and C-terminal kelch) domain of human KHLH11 (PDB code 3I3N) were energy-minimized with the RepairPDB command implemented in FoldX and subsequently with the BuildModel command for mutagenesis. Protein stabilities were calculated by the Stability command, and the free-energy changes were estimated by subtraction of the free-energy value of the wild-type protein from those of the altered proteins. The procedure was repeated three times for each substitution, and the resultant data were presented as an average value with SDs.
Immunoblotting and Immunohistochemistry
SDS-PAGE and immunoblotting were performed as described.22,23 For protein studies, C2C12 myoblasts and myotubes were grown and prepared for immunoblotting and immunofluorescence as described.23 For KLHL40 immunoblots, the Human Protein Atlas (HPA) rabbit polyclonal KLHL40 (KBTBD5) antibody from Sigma was used (HPA024463 [1:2,500 dilution]). Immunostaining of human and mouse muscle samples was performed as described13,23 with a KLHL40 antibody (KBTBD5; HPA024463 [1:100 dilution]).
Zebrafish Studies
In Situ Hybridization
Digoxigenin probes for klhl40a and klhl40b were generated by cDNA amplification of 1,340 and 694 bp sequences, respectively (Table S1). In situ hybridizations were performed as described previously.24
Morpholino Microinjection
Antisense translation-blocking morpholinos (Table S1) for klhl40a (klhl40a-MO) and klhl40b (klhl40b-MO and klhl40b-MO2) were coinjected into 1- to 2-cell-stage embryos at a final concentration of 0.25 or 0.5 mM. Morpholino efficacies were tested by immunoblotting for Klhl40.
Zebrafish Immunohistochemistry
Immunohistochemistry of zebrafish embryos was performed as described24,25 with myosin heavy chain (MHC) antibody (F59 [1:20 dilution] or A4.1025 [1:10 dilution]; Developmental Studies Hybridoma Bank) and α-actinin (1:100 dilution; Sigma) and filamin C (1:100 dilution; Sigma) antibodies, and Alexa-Fluor-488-conjugated phalloidin (1:100 dilution; Molecular Probes) was used for labeling F-actin. Immunoreactivity was detected with an Alexa-Fluor-594-conjugated anti-mouse secondary antibody diluted in blocking buffer (1:200).
Statistical Analyses
Statistical analyses of clinical features were carried out with SPSS Statistics 19 (IBM) software. Individuals for whom information for a clinical feature was not available were excluded from the analysis of that feature. Either Chi-square tests or Fisher’s exact tests were applied for comparing each phenotypic variable between different genotypes. p < 0.05 was considered statistically significant.
Results
WES identified homozygous or compound-heterozygous mutations in KLHL40 (kelch-like family member 40; also known as KBTBD5 [kelch-repeat- and BTB-(POZ)-domain-containing 5] and SYRP [sarcosynapsin]) in six NEM-affected families (families 6, 10, 16–18, and 31; Figure 1B and Table 1). Subsequent screening of KLHL40 by Sanger sequencing in additional probands with severe NEM resulted in the identification of a total of 19 variants (4 frameshifts, 12 missense mutations, 2 nonsense mutations, and 1 splice site) in 28 (19.6%) apparently unrelated families (Table 1) from the cohort of 143 families affected by severe NEM. In addition, 129 probands with milder NEM were screened, but no KLHL40 mutations were identified in this cohort, confirming that KLHL40 mutations are most likely exclusive to cases of severe NEM.
Table 1.
KLHL40 Mutations by Family, Individual LOD Scores, Ethnicity, and Population-wide Incidence
| Family | Exon(s) |
Mutation |
LOD Score | Ethnicity | Incidence from EVS (1st; 2nd) | Incidence from 1000 Genomes (1st; 2nd) | |
|---|---|---|---|---|---|---|---|
| Nucleotide Change | Amino Acid Change | ||||||
| Family 31a | 1 | c.[100G>C];[257T>C] | p.[Asp34His];[Leu86Pro] | 0.6 | Vietnamese | ND; ND | ND; ND |
| Family 2 | 1 | c.[134delC];[134delC] | p.[Pro45Argfs∗19];[Pro45Argfs∗19] | NA | Italian | NA | ND |
| Family 3 | 1 | c.[270C>G];[270C>G] | p.[Tyr90∗];[Tyr90∗] | NA | Turkish | ND | ND |
| Family 5 | 1 | c.[581T>A];[581T>A] | p.[Val194Glu];[Val194Glu] | 0.6 | Israeli | ND | ND |
| Family 6a | 1 | c.[602G>T];[602G>T] | p.[Trp201Leu];[Trp201Leu] | 1.454 | Turkish | ND | ND |
| Family 7 | 1 | c.[602G>A];[602G>A] | p.[Trp201∗];[Trp201∗] | NA | Norwegian | ND | ND |
| Family 9 | 1 | c.[790delC];[790delC] | p.[Arg264Alafs∗59];[Arg264Alafs∗59] | 0.25 | Turkish | NA | ND |
| Family 10a | 1 and 4 | c.[932G>T];[1516A>C] | p.[Arg311Leu];[Thr506Pro] | NA | Chinese | ND; ND | ND; ND |
| Family 34 | 2 and 6 | c.[1190C>T];[1762G>A] | p.[Pro397Leu];[Glu588Lys] | NA | Turkish | ND; ND | ND; A = 2 and G = 2,184 |
| Family 12 | 2 and 4 | c.[1270_1272delinsAGATCAAGGT];[1582G>A] | p.[Asp424Argfs∗23];[Glu528Lys] | NA | Japanese | NA; ND | ND; ND |
| Family 13 | 2 and 4 | c.[1281_1294delCTGCCTGGACTCGG];[1582G>A] | p.[Cys428Hisfs∗12];[Glu528Lys] | NA | Korean | NA; ND | ND; ND |
| Family 14 | 3 | c.[1364A>G];[1364A>G] | p.[His455Arg];[His455Arg] | NA | Turkish | ND | ND |
| Family 15 | 3 | c.[1405G>T];[1405G>T] | p.[Gly469Cys];[Gly469Cys] | NA | Japanese | ND | ND |
| Family 16a | 3 and 4 | c.[1405G>T];[1582G>A] | p.[Gly469Cys];[Glu528Lys] | 0.727 | Japanese | ND; ND | ND; ND |
| Family 17a | 4 | c.[1582G>A];[1582G>A] | p.[Glu528Lys];[Glu528Lys] | 1.654 | Turkish | ND | ND |
| Family 18a | 4 | c.[1582G>A];[1582G>A] | p.[Glu528Lys];[Glu528Lys] | 0.125 | Kurdish | ND | ND |
| Family 19 | 4 | c.[1582G>A];[1582G>A] | p.[Glu528Lys];[Glu528Lys] | 0.25 | Kurdish | ND | ND |
| Family 20 | 4 | c.[1582G>A];[1582G>A] | p.[Glu528Lys];[Glu528Lys] | NA | Japanese | ND | ND |
| Family 21 | 4 | c.[1582G>A];[1582G>A] | p.[Glu528Lys];[Glu528Lys] | NA | Japanese | ND | ND |
| Family 22 | 4 | c.[1582G>A];[1582G>A] | p.[Glu528Lys];[Glu528Lys] | NA | Japanese | ND | ND |
| Family 23 | 4 | c.[1582G>A];[1582G>A] | p.[Glu528Lys];[Glu528Lys] | NA | Japanese | ND | ND |
| Family 24 | 4 | c.[1582G>A];[1582G>A] | p.[Glu528Lys];[Glu528Lys] | NA | Japanese | ND | ND |
| Family 25 | 4 | c.[1582G>A];[1582G>A] | p.[Glu528Lys];[Glu528Lys] | NA | Japanese | ND | ND |
| Family 26 | 4 | c.[1582G>A];[1582G>A] | p.[Glu528Lys];[Glu528Lys] | NA | Japanese | ND | ND |
| Family 27 | 4 | c.[1582G>A];[1582G>A] | p.[Glu528Lys];[Glu528Lys] | NA | Japanese | ND | ND |
| Family 28 | 4 | c.[1582G>A];[1582G>A] | p.[Glu528Lys];[Glu528Lys] | NA | Japanese | ND | ND |
| Family 29 | 4/5 | c.[1608–1G>A];[1608–1G>A] | NA | NA | Turkish | ND | ND |
| Family 30 | 5 | c.[1612G>C];[1612G>C] | p.[Ala538Pro];[Ala538Pro] | NA | Turkish | ND | ND |
The individual pedigree LOD scores are given where possible. This table also shows the incidence of the mutations reported within the NHLBI EVS and the 1000 Genomes browser. Abbreviations are as follows: NA, not available; and ND, not detected.
Families for whom WES was performed.
In all cases where it was possible to test unaffected parents, siblings, and extended family, the mutations cosegregated with disease in an autosomal-recessive fashion (Figure 1B), giving a combined LOD score of 5.66 (Table 1). All mutations were either absent from the NHLBI EVS and the 1000 Genomes database26 or present at low frequencies in the heterozygous state (Table 1). In five additional NEM-affected families, only single KLHL40 variants were identified (Table S2); the significance of these variants in these individuals remains unclear.
In Japanese persons, KLHL40 mutations are the most common cause of this severe form of NEM (13/47 [∼28%]) as a result of a founder effect with the c.1582G>A mutation. Given that this mutation was present in Turkish, Kurdish, and Japanese families, we completed a haplotype analysis of Japanese and Turkish families (families 16 and 17) but did not identify a common haplotype between them (Figure S2). HRM with confirmatory Sanger sequencing of 510 normal Japanese individuals revealed a heterozygous c.1582G>A mutation in one individual. Therefore, the mutant-allele frequency in the Japanese population was estimated to be 0.0098. According to the equation described by Kimura and Ota27 and under the assumption of 25 years per generation, the age of this mutation is calculated to be 4,900 years old.
The identified KLHL40 mutations were scattered throughout all exons (Table 1 and Figure 2A) encoding mostly conserved residues (Figure S3). To investigate disease mechanisms, all substitutions except p.Arg311Leu were mapped to the crystal structures of the kelch domain of human KLHL40 and the BTB-BACK domain of human kelch-like protein 11 (KLHL11; Figures 2B and 2C and Figure S4). p.Arg311Leu (c.932G>T) was predicted to be in the structurally flexible region, a linker of nonconserved amino acids connecting the BACK and kelch domains (Figure S7D), and was therefore excluded from structural consideration. All the modeled substituted residues are involved in intramolecular interactions, and thus the substitutions would most likely destabilize the hydrophobic cores of the BTB-BACK domain (p.Leu86Pro [c.257T>C], p.Val194Glu [c.581T>A], and p.Trp201Leu [c.602G>A]), the kelch domain (p.Pro397Leu [c.1190C>T], p.His455Arg [c.1364A>G], and p.Gly469Cys [c.1405G>T]), the β sheet (p.Thr506Pro [c.1516A>C] and p.Ala538Pro [c.1612G>C]), or the hydrogen bonds between the main chain and side chain (p.Asp34His [c.100G>C] and p.Glu528Lys [c.1582G>A]) or between side chains (p.Glu588Lys [c.1762G>A]) (Figures S5–S7). The p.Pro397Leu and p.Glu588Lys substitutions appear to be conservative for the hydrophobic core and hydrogen bonding, respectively. The former substitution is predicted to affect the polyproline II helix conformation (residues 396–399; Figure S6A). The calculated free-energy change for most substitutions was estimated to be over 2.0 kcal/mol (Figure 2D), which is typically associated with destabilization of domain folds.28 These analyses suggested that most KLHL40 missense mutations impair protein stability.
Figure 2.
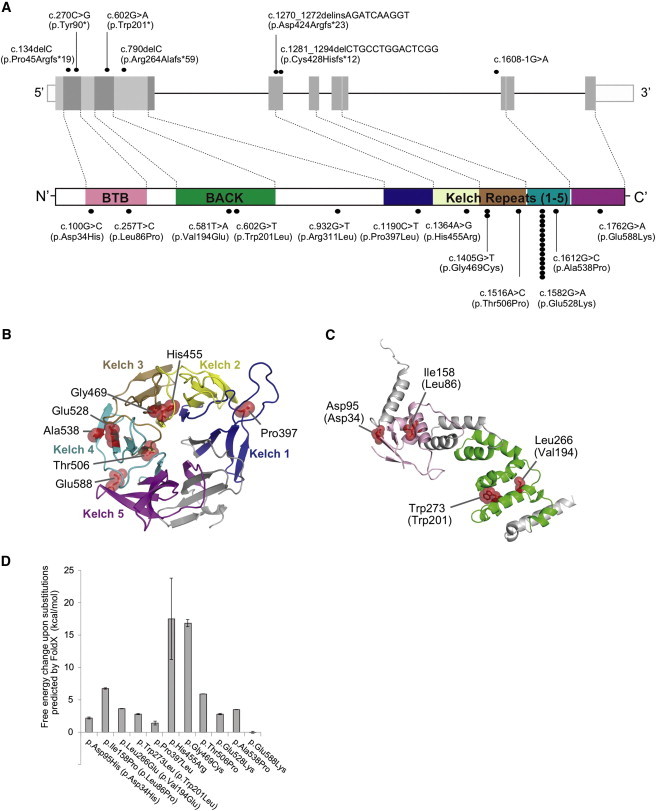
Mutations Identified in Our Cohort and the Structural Modeling of the Missense KLHL40 Substitutions
(A) Schematic presentation of the genomic structure of KLHL40 (upper) and its encoded protein, KLHL40, with the BTB-BACK domain and kelch repeats (lower). The localization of mutations and substitutions identified is depicted with dots, and the number of dots for each mutation or substitution indicates the number of times it was found. Most substitutions occurred at conserved amino acids. The dots above KLHL40 indicate truncating mutations, and those below KLHL40 indicate missense mutations.
(B and C) Structural modeling of the missense KLHL40 substitutions. The crystal structures of the (B) kelch domain of KLHL40 and the (C) BTB-BACK domain of KLHL11 and the location of the substitutions are shown. p.Pro397Leu, p.His455Arg, p.Glu469Cys, p.Thr506Pro, p.Glu528Lys, p.Ala538Pro, and p.Glu588Lys map to the kelch repeats (B), p.Asp34His and p.Leu86Pro map to the BTB domain, and p.Val194Lys and p.Trp201Leu map to the BACK domain (C). The side chains of the mutated residues are shown as sticks with space-filling spheres in red. α helices, β sheets, and loops are drawn as ribbons, arrows, and threads, respectively. Each kelch repeat (B) is color coded in the kelch domain, and the BTB and BACK domains (C) are colored pink and green, respectively. Molecular structures were drawn with PyMOL.
(D) The calculated free-energy changes resulting from the missense substitutions in the kelch domain of human KLHL40 and the BTB-BACK domain of human KLHL11 were predicted by FoldX. Data are presented as the mean ± SD. Residue numbers used in (C) and (D) refer to human KLHL11, and those corresponding to human KLHL40 are in parentheses.
To investigate KLHL40 expression and KLHL40 abundance, we performed quantitative RT-PCR and immunoblotting of human and mouse tissues. KLHL40 transcripts and their encoded proteins were exclusive to developing and adult skeletal muscle (Figures 3A–3C) and more abundant in fetal muscle than in postnatal muscle (Figure 3C). Confocal microscopy suggested that KLHL40 might localize to the sarcomeric A-band (Figure 3D and Figure S8), a region not previously linked to NEM. Immunoblotting showed that KLHL40 is absent or of low abundance in KLHL40-associated NEM muscle (Figure 3E), even for persons harboring two missense mutations (F10 and F17). Immunohistochemistry confirmed that KLHL40 was absent or very scarce in KLHL40-associated NEM myofibers (Figure 3F).
Figure 3.
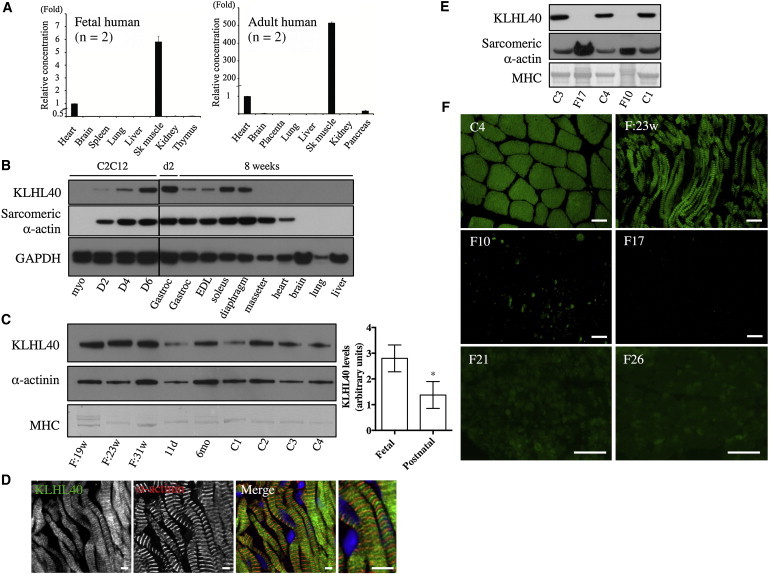
KLHL40 Expression in Human and Mouse Tissues
(A) Taqman quantitative real-time PCR analysis of cDNA from adult or fetal human tissues. Error bars represent the SD. The following abbreviation is used: Sk, skeletal.
(B) KLHL40 levels in C2C12 cells and mouse tissues (HPA, top panel) and immunoblotting for sarcomeric α-actin (clone 5C5, middle panel) and GAPDH (lower panel). Lanes are as follows: myo, C212 myoblasts; D2, myotubes on day 2 of differentiation; D4, myotubes on day 4 of differentiation; D6, myotubes on day 6 of differentiation; Gastroc (left), C57BL/6 postnatal day 2 (d2) gastrocnemius; Gastroc (right), C57BL/6 8-week-old gastrocnemius; and EDL (extensor digitorum longus) to liver, C57BL/6 8-week-old tissues. For all mouse tissue lysates, samples were pooled from three different mice.
(C) On the left is KLHL40 expression in human skeletal muscle (HPA, top panel), immunoblotting for α-actinin (clone EA-53, middle panel), and Coomassie staining of MHC band (bottom panel). Lanes are as follows: F:19w, 19-week-old fetus; F:23w, 23-week-old fetus; F:31w, 31-week-old fetus; 11d, 11-day-old neonate; 6mo, 6-month-old baby; and C1–C4, healthy adult controls of 19–42 years of age. On the right, KLHL40 intensity normalized to MHC for fetal muscle is 3.34 ± 0.92 (n = 3) versus 1.37 ± 0.21 (n = 6) for postnatal skeletal muscle. ∗p = 0.023, unpaired two-tailed t test. Error bars represent the SEM.
(D) Single Z-plane confocal microscopy showing localization of KLHL40 (green) and α-actinin (red) in a longitudinal section of skeletal muscle from a 31-week-old fetus; costaining with Hoechst (blue) is also shown (Merge). Scale bars represent 5 μm.
(E) Immunoblotting shows that KLHL40 is absent in KLHL40-associated NEM muscle (II-1 from family 10 [F10] and V-2 from family 17 [F17]) compared with healthy control muscle (C1, C3, and C4). Coomassie staining of the MHC band (bottom panel) and immunoblotting for sarcomeric α-actin (clone 5C5, middle panel) indicate similar or greater loading for the KLHL40-associated NEM samples compared with control samples.
(F) Immunofluorescence for KLHL40 in a human 23-week-old fetal skeletal muscle sample (F:23w), an adult healthy control (C4), and KLHL40-associated NEM muscle biopsies (II-1 from family 10 [F10], V-2 from family 17 [F17], family 21 [F21], and family 26 [F26]). Scale bars represent 50 μm.
We further investigated Klhl40 function in zebrafish. The zebrafish genome contains two orthologs of KLHL40: klhl40a and klhl40b, which have 57% (klhl40a) and 55.7% (klhl40b) amino acid similarity to human KLHL40. RT-PCR demonstrated expression of both klhl40 genes at 24 and 48 hr postfertilization (hpf) (Figure S9A). In adult zebrafish, klhl40a was most abundant in the skeletal muscle and heart and klhl40b was most abundant in the skeletal muscle (Figure S9A). At the 16 and 24 hpf stages, expression of both genes was restricted to the muscle precursor cells in the somites (Figure 4A). We knocked down zebrafish klhl40a and klhl40b with antisense morpholino oligonucleotides (klhl40a-MO, klhl40b-MO, and klhl40b-MO2) (Figures S9B and S10A). Embryos injected with klhl40a-MO, klhl40b-MO, and klhl40a-MO/40b-MO (double morpholinos) showed a curved trunk and small head at 48 hpf (Figures 4B and 4C). A normal phenotype resulted from 5 bp mismatched morpholinos (5mis-MOs). We analyzed slow myofibers in more detail by immunostaining slow myosin heavy chains (Figure 4D, upper panels). klhl40 morphants showed disruption of muscle patterning with an irregular, wavy appearance of the striated myofibers and extensive gaps between the myofibers (Figures 4D and 4E and Figure S10B) and a greatly diminished birefringence (Figure S10C). Isolated myofibers from klhl40a-MO/40b-MO fish, coimmunostained with phalloidin and an α-actinin antibody (Z-disk), showed disorganized and irregular patterns with small aggregates of α-actinin, suggesting nemaline bodies (Figure 4F). Aggregation of Z-disk material was also confirmed by immunostaining for filamin C in klhl40a-MO/40b-MO fish (Figure S11). Electron-microscopic analysis revealed disarranged myofibrils with widened Z-disks (Figure 4G). Fish injected with klhl40a-MO, klhl40b-MO, klhl40b-MO2, or klhl40a-MO/40b-MO2 (double morpholinos) exhibited sporadic muscle tremors, and coordinated swimming behavior was not observed (Movies S1 and S2). These results suggest that Klhl40a and Klhl40b are required for muscle development and function and that loss of either isoform in the early embryo is sufficient to impair normal mobility.
Figure 4.
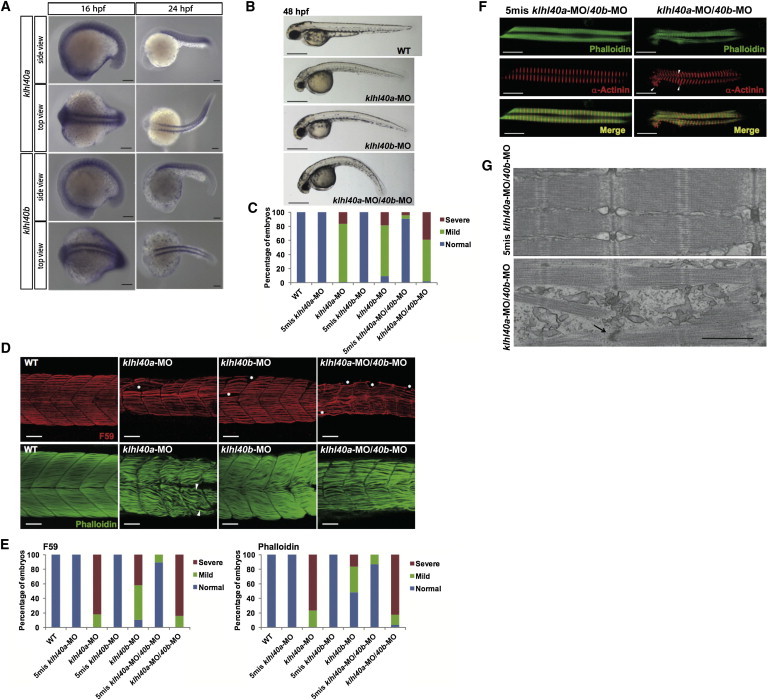
Expression and Function of klhl40 in Zebrafish
(A) In situ hybridization demonstrates that expression of both klhl40a and klhl40b is restricted to the skeletal muscle at 16 and 24 hpf.
(B) Gross morphology of uninjected embryos (WT) and embryos injected with klhl40a-MO, klhl40b-MO, and klhl40a-MO/40b-MO. Lateral views of MO-injected embryos (4 ng) at 48 hpf are shown. Scale bars represent 500 μm.
(C) Percentage of embryos categorized in phenotypic classes after injection with the 5mis-MO control, klhl40a-MO, klhl40b-MO, or klhl40a-MO/40b-MO. We categorized the phenotypes at 48 hpf into normal (normal appearance), mild (curved trunk), and severe (tail defect and severe development delay) (n = 111–130).
(D) Knockdown of klhl40a, klhl40b, or both resulted in severe disruption of the skeletal muscle: fibers appeared wavy, and there were extensive gaps between fibers in contrast to the densely packed and aligned fibers of the controls. Maximum-intensity projection images from a confocal image series followed immunolabeling with a myosin antibody (F59, upper panels) at 36 hpf and F-actin (lower panels) at 72 hpf.
(E) Embryos injected with 5mis-MO, klhl40a-MO, klhl40b-MO, or klhl40a-MO/40b-MO were categorized phenotypically on the basis of the presence of myofiber detachment affecting one to two somites (mild) or multiple (three or more) somites (severe) (n = 25–44).
(F) Double-labeled immunofluorescence was performed on isolated myofibers from 72 hpf embryos with the use of phalloidin (green) and α-actinin (red). Frequent areas of aberrant α-actinin accumulation were detected in klhl40a-MO/40b-MO myofibers (arrowheads).
(G) Electron microscopy of 72 hpf myofibers. A 5mis-MO-injected embryo shows correctly aligned sarcomeres and T-tubules (upper panel). A klhl40a-MO/40b-MO-injected embryo (lower panel) shows disarranged myofibrils with widened Z-disks (arrow), but thin filament lengths are unchanged. The scale bar represents 0.7 μm.
Detailed clinical records were collected and analyzed for 32 affected individuals from the 28 unrelated kindreds afflicted with KLHL40 mutations. These individuals were from various ethnicities, such as European, Middle and Near Eastern, or Asian. Clinical features of individuals with KLHL40 mutations were severe and distinctive (Table 2 and Table S3). Eighty-three percent of affected individuals showed prenatal symptoms, and 76% displayed fetal akinesia or hypokinesia. Most persons had severe respiratory compromise (97%), and approximately a third required ventilatory support (38%). Almost all affected individuals (96%) also had swallowing problems, and half required tube feeding or gastrostomy. Muscle weakness was severe. Forty-five percent of individuals had no spontaneous antigravity movement. Seventeen percent of affected individuals were also noted to have ophthalmoparesis, a relatively rare symptom in NEM. Multiple joint contractures and pathological bone fracture were other common features. Dysmorphic facial features and deformities of the chest, spine, fingers, and feet were also frequent. The average age of death was 5 months. Many families, including a previously described family (family 30 herein, cases 2–6 in Lammens et al.),11 were consanguineous.
Table 2.
Summary of Clinical Features of NEM Individuals with KLHL40 Mutations
|
Individuals with KLHL40 Mutations (n = 32 Cases from 28 Families) |
||
|---|---|---|
| Total | Percentage | |
| Family history | 17/28 | 60.7% |
| Consanguinity | 10/28 | 35.7% |
| Prenatal Period | ||
| Prenatal symptoms | 24/29 | 82.8% |
| Fetal akinesia or hypokinesia | 16/21 | 76.2% |
| Polyhydramnios | 14/29 | 48.3% |
| Neonatal Period | ||
| Respiratory function | ||
| respiratory failure | 28/29 | 96.6% |
| requiring ventilation | 11/29 | 37.9% |
| Facial involvement | 26/26 | 100% |
| weakness | 23/23 | 100% |
| ophthalmoparesis | 4/23 | 17.4% |
| mild dysmorphology | 15/15 | 100% |
| Dysphagia | 23/24 | 95.8% |
| with tube feeding or gastrostomy | 13/24 | 54.2% |
| Muscle weakness | 29/29 | 100.0% |
| with no spontaneous antigravity movements | 13/29 | 44.8% |
| Contracture(s) | 24/27 | 88.9% |
| Pathological fracture(s) | 10/19 | 52.6% |
| Average age at death | 5 months (n = 14) | |
| Average gestation age at birth | 37 weeks (n = 27) | |
| Average birth weight | 2,558 g (n = 26) | |
Total numbers were calculated as the number of individuals with the clinical features over the total number of individuals whose medical records were available for each category.
We further evaluated whether there are any genotype-phenotype correlations in KLHL40-associated NEM. We compared the clinical features of individuals according to the type of mutation they had (either two truncating mutations, one truncating mutation and one missense mutation, or two missense mutations) and the pattern of mutations (homozygous or compound heterozygous). No significant differences in frequencies of these clinical features were observed (data not shown). We also compared the clinical features of persons with the recurrent c.1582G>A genotype (either with this mutation [genotype G/A or A/A as group A] or without [genotype G/G as group G]). Prenatal symptoms, including fetal akinesia or hypokinesia, were frequently observed (73.3% in group A versus 92.9% in group G). Respiratory failure was common in both groups (100% in group A versus 92.9% in group G), but there were significantly fewer individuals requiring ventilation in group A than in group G (20.0% in group A versus 57.1% in group G; p = 0.047). Dysphagia was also common in both groups (100% in group A versus 90.0% in group G), but there were fewer persons requiring tube feeding or gastrostomy in group A than in group G, although the difference was not significant (42.9% in group A versus 70.0% in group G; p = 0.127). Facial weakness was observed in all affected individuals in both groups, but fewer individuals in group A had ophthalmoparesis (7.7% in group A versus 30.0% in group G; p = 0.281). All persons also had muscle weakness, but significantly fewer individuals in group A had the most severe form of muscle weakness with no antigravity movements (20.0% in group A versus 71.4% in group G; p = 0.018). Significantly fewer affected individuals in group A were deceased at the time of study than in group G (23.5% in group A versus 71.4% in group G; p = 0.012; odds ratio = 8.125; 95% confidence interval = 1.62–40.75) (Figure 5). We further compared the clinical features of individuals of different ethnicities (either European or Asian descent) according to the c.1582G>A genotype, and similar tendencies were demonstrated (data not shown). There was, however, great variation in severity for individuals with or without the c.1582G>A genotype.
Figure 5.
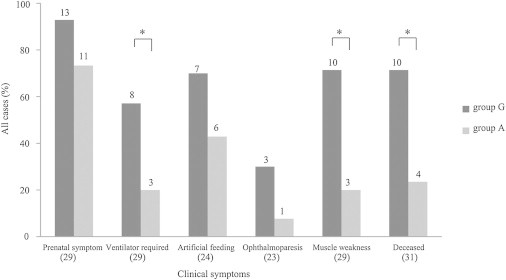
Correlation between the c.1582G>A (p.Glu528Lys) Mutation and Clinical Features
The clinical characteristics of NEM are shown for the two groups of affected individuals (32 total), either with the c.1582G>A (p.Glu528Lys) mutation (as group A) or without it (as group G). The numbers of total affected individuals with clinical records regarding either the presence or the absence of each characteristic are indicated below the bars, and the numbers of affected individuals in each group are indicated above the respective bars. Labels on the x axis are as follows: prenatal symptoms, individuals demonstrating either fetal akinesia or hypokinesia, polyhydramnios, or fetal edema or effusion; ventilator required, individuals with respiratory failure requiring ventilation; artificial feeding, dysphagia-affected persons requiring tube feeding or gastrostomy; ophthalmoparesis, individuals with ophthalmoparesis along with facial weakness; muscle weakness, individuals with the most severe form of muscle weakness and demonstrating no antigravitory movement; and deceased, individuals who were deceased at the time of study. Asterisks indicate that statistical significance was observed.
Discussion
We have described the identification of recessive KLHL40 mutations in individuals with severe NEM from 28 unrelated families of various ethnicities. The c.1582G>A mutation was the most frequently detected mutation and was found in Japanese, Kurdish, and Turkish persons. However, comparison of haplotypes between a Japanese family and a Turkish family suggested that the mutation arose independently in these ethnic groups. We have shown several lines of evidence of the pathogenicity of the KLHL40 mutations. The missense mutations occurred mostly in conserved functional domains within KLHL40, and they were predicted to destabilize the intramolecular interactions and thus impair protein stability. This was corroborated by the absence of KLHL40 even in the skeletal muscle of individuals harboring two missense mutations. We have established a locus-specific database for KLHL40 mutations at the Leiden Muscular Dystrophy Pages.
Expression of KLHL40 in fetal and adult skeletal muscle indicates that KLHL40 plays a role in both myogenesis and mature muscle. KLHL40 appears to be more abundant in fetal skeletal muscle than in postnatal skeletal muscle and most likely accounts for the prevalence of in utero presentations in this NEM cohort. Perhaps KLHL40 is more important for myogenesis than for muscle maintenance; this could account for the fact that the disease ranges so much in severity, from some individuals’ dying within hours of being born to others’ surviving into adolescence. Our zebrafish studies have demonstrated that Klhl40a and Klhl40b are not required for the specification of muscle cells but rather for muscle patterning and function and that loss of either isoform in the early embryo is sufficient to impair normal mobility, supporting the involvement of KLHL40 in NEM-associated fetal akinesia. It has previously been suggested that KLHL40 is also important for muscle maintenance through the process of degeneration and regeneration.29,30 Klhl40 is upregulated in myogenic precursors after cardiotoxin injury of mouse skeletal muscle, supporting a role for Klhl40 in the response to muscle damage.29 Studies of cattle muscle have shown increased Klhl40 expression in another catabolic process, undernutrition, further suggesting a role for KLHL40 in the stress response.30
KLHL40 belongs to the superfamily of kelch-repeat-containing proteins that form characteristic β-propeller structures,31 which bind substrate proteins and are involved in a wide variety of functions. In humans, 71 kelch-repeat-containing proteins have been identified.31 The majority contain an N-terminal BTB domain (also known as the POZ [poxvirus and zinc finger] domain) and a BACK motif. Proteins containing both a BTB domain and a kelch repeat have previously been implicated in neuromuscular disease. A dominant KLHL9 mutation causes an early-onset distal myopathy (distal myopathy 1 [MIM 160500]),32 and dominant KBTBD13 mutations cause nemaline myopathy with cores (MIM 609273).9 We now show that KLHL40, encoding KLHL40, which contains both a BTB domain and a kelch repeat, is associated with autosomal-recessive neuromuscular disease. BTB domains function as substrate-specific adaptors for cullin 3 (Cul3),33,34 a component of the E3-ubiquitin-ligase complex. Both KLHL9 and KBTBD13 bind Cul3.10,32 MuRF1, an E3-ubiquitin ligase, is known to be recruited to M-line titin and is thought to modulate myofibrillar turnover and the trophic state of muscle.35 KLHL40 appears to be present at the A-band and might be similarly involved through the ubiquitin-proteasome pathway.
We have characterized the severe and distinctive features of this disease as fetal akinesia or hypokinesia during the prenatal period, respiratory failure and swallowing difficulty at birth, contractures and fractures along with dysmorphic features, and in most cases, early death. We have also shown that persons with the recurrent c.1582G>A mutation tend to have relatively milder symptoms compared to those of individuals without c.1582G>A. However, the severity of the disease in persons with or without the c.1582G>A genotype varied greatly (for example, from death at 20 days to still being alive at 11 years for persons homozygous for the c.1582G>A genotype), suggesting modifying factors.
Fetal akinesias are clinically and genetically heterogeneous, and the majority of cases still remain genetically unsolved.36 Primary muscle diseases account for up to 50% of such syndromes.37 On the basis of our study, KLHL40 mutations cause a significant proportion of severe NEM cases of fetal akinesia sequence and the disease shows worldwide prevalence. KLHL40 should be considered when a clinician encounters an individual presenting with prenatal symptoms, such as fetal akinesia or hypokinesia, or clinical features and/or pathology of severe NEM at birth (especially miliary NEM, which was present in at least 20% of our KLHL40-mutation cases), along with an autosomal-recessive pattern of family history. Fractures are a relatively frequent presentation within this cohort, unlike other NEM cohorts, and should also be used for directing genetic screening of KLHL40. We show that KLHL40 immunohistochemistry, immunoblotting, or genetic screening will identify the disease and thus allow genetic counseling for the affected individual’s family.
In conclusion, this study associates loss-of-function KLHL40 mutations with severe, often in utero, NEM. Many probands who do not harbor KLHL40 mutations present with NEM in utero, suggesting further genetic heterogeneity. Clarification of KLHL40 function and interactions might lead to a greater understanding of the pathogenesis of disease, the identification of other candidates for this severe form of NEM, and the investigation of possible therapies.
Acknowledgments
This research was supported by the National Health and Medical Research Council of Australia (fellowships APP1035955 to G.R. and APP1002147 to N.G.L. and grant APP1022707) and Association Francaise contre les Myopathies (AFM; AFM15734). E.T. and K.S.Y. are supported by university postgraduate awards. This work received grants from the Ministry of Health, Labour, and Welfare (N. Miyake, H.S., and N. Matsumoto), Japan Science and Technology Agency (N. Matsumoto), Strategic Research Program for Brain Sciences (E.K. and N. Matsumoto), and Takeda Science Foundation (N. Miyake and N. Matsumoto) and Grants-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (N. Miyake and N. Matsumoto) and for Scientific Research from the Japan Society for the Promotion of Science (N. Miyake, H.S., and N. Matsumoto). The A.H.B. laboratory was supported by the National Institutes of Health (R01-AR044345) and the Muscular Dystrophy Association (MDA201302). O.C. is a Dubai Harvard Foundation for Medical Research Fellow and a grantee of the Schlumberger Foundation Faculty for the Future Program. E.B. is supported by grants from Telethon (GUP08005) and the Ministry of Health on Congenital Myopathies. F.M. is supported by the Great Ormond Street Children’s Charity and National Specialist Commissioning Group. P.V. and V.-L.L. were supported by grants to C.W.-P. by the AFM, Sigrid Jusélius Foundation, Academy of Finland, Finska Läkaresällskapet, and Medicinska Understödsföreningen Liv och Hälsa r.f. R.V. is supported by a Monash Graduate Research Scholarship and a Faculty of Science Dean’s International Postgraduate Research Scholarship.
Contributor Information
Naomichi Matsumoto, Email: naomat@yokohama-cu.ac.jp.
Nigel G. Laing, Email: nigel.laing@uwa.edu.au.
Supplemental Data
Time-lapse imaging of a control dye-injected zebrafish at 72 hpf shows a normal startle response with rapid swimming away from contact.
Time-lapse imaging of a klhl40a-MO/40b2-MO2-injected zebrafish at 72 hpf shows limited movement and loss of swimming ability compared to control fish (Movie S1).
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
Leiden Open Variation Database, www.LOVD.nl/KLHL40
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PyMOL, http://www.pymol.org
References
- 1.Nance J.R., Dowling J.J., Gibbs E.M., Bönnemann C.G. Congenital myopathies: an update. Curr. Neurol. Neurosci. Rep. 2012;12:165–174. doi: 10.1007/s11910-012-0255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nowak K.J., Davis M.R., Wallgren-Pettersson C., Lamont P.J., Laing N.G. Clinical utility gene card for: nemaline myopathy. Eur. J. Hum. Genet. 2012;20 doi: 10.1038/ejhg.2012.70. Published online April 18, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nowak K.J., Wattanasirichaigoon D., Goebel H.H., Wilce M., Pelin K., Donner K., Jacob R.L., Hübner C., Oexle K., Anderson J.R. Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat. Genet. 1999;23:208–212. doi: 10.1038/13837. [DOI] [PubMed] [Google Scholar]
- 4.Agrawal P.B., Greenleaf R.S., Tomczak K.K., Lehtokari V.L., Wallgren-Pettersson C., Wallefeld W., Laing N.G., Darras B.T., Maciver S.K., Dormitzer P.R., Beggs A.H. Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am. J. Hum. Genet. 2007;80:162–167. doi: 10.1086/510402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lehtokari V.L., Pelin K., Sandbacka M., Ranta S., Donner K., Muntoni F., Sewry C., Angelini C., Bushby K., Van den Bergh P. Identification of 45 novel mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Hum. Mutat. 2006;27:946–956. doi: 10.1002/humu.20370. [DOI] [PubMed] [Google Scholar]
- 6.Johnston J.J., Kelley R.I., Crawford T.O., Morton D.H., Agarwala R., Koch T., Schäffer A.A., Francomano C.A., Biesecker L.G. A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am. J. Hum. Genet. 2000;67:814–821. doi: 10.1086/303089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Donner K., Ollikainen M., Ridanpää M., Christen H.J., Goebel H.H., de Visser M., Pelin K., Wallgren-Pettersson C. Mutations in the beta-tropomyosin (TPM2) gene—a rare cause of nemaline myopathy. Neuromuscul. Disord. 2002;12:151–158. doi: 10.1016/s0960-8966(01)00252-8. [DOI] [PubMed] [Google Scholar]
- 8.Laing N.G., Wilton S.D., Akkari P.A., Dorosz S., Boundy K., Kneebone C., Blumbergs P., White S., Watkins H., Love D.R. A mutation in the alpha tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathy. Nat. Genet. 1995;9:75–79. doi: 10.1038/ng0195-75. [DOI] [PubMed] [Google Scholar]
- 9.Sambuughin N., Yau K.S., Olivé M., Duff R.M., Bayarsaikhan M., Lu S., Gonzalez-Mera L., Sivadorai P., Nowak K.J., Ravenscroft G. Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with cores. Am. J. Hum. Genet. 2010;87:842–847. doi: 10.1016/j.ajhg.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sambuughin N., Swietnicki W., Techtmann S., Matrosova V., Wallace T., Goldfarb L., Maynard E. KBTBD13 interacts with Cullin 3 to form a functional ubiquitin ligase. Biochem. Biophys. Res. Commun. 2012;421:743–749. doi: 10.1016/j.bbrc.2012.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lammens M., Moerman P., Fryns J.P., Lemmens F., van de Kamp G.M., Goemans N., Dom R. Fetal akinesia sequence caused by nemaline myopathy. Neuropediatrics. 1997;28:116–119. doi: 10.1055/s-2007-973683. [DOI] [PubMed] [Google Scholar]
- 12.Lacson A.G., Donaldson G., Barness E.G., Ranells J.D., Pomerance H.H. Infant with high arched palate, bell-shaped chest, joint contractures, and intrauterine fractures. Pediatr. Pathol. Mol. Med. 2002;21:569–584. doi: 10.1080/pdp.21.6.569.584. [DOI] [PubMed] [Google Scholar]
- 13.Ravenscroft G., Jackaman C., Bringans S., Papadimitriou J.M., Griffiths L.M., McNamara E., Bakker A.J., Davies K.E., Laing N.G., Nowak K.J. Mouse models of dominant ACTA1 disease recapitulate human disease and provide insight into therapies. Brain. 2011;134:1101–1115. doi: 10.1093/brain/awr004. [DOI] [PubMed] [Google Scholar]
- 14.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 15.Ravenscroft G., Thompson E.M., Todd E.J., Yau K.S., Kresoje N., Sivadorai P., Friend K., Riley K., Manton N.D., Blumbergs P. Whole exome sequencing in foetal akinesia expands the genotype-phenotype spectrum of GBE1 glycogen storage disease mutations. Neuromuscul. Disord. 2013;23:165–169. doi: 10.1016/j.nmd.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 16.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrow J., Denoeud F., Frankish A., Reymond A., Chen C.K., Chrast J., Lagarde J., Gilbert J.G., Storey R., Swarbreck D. GENCODE: producing a reference annotation for ENCODE. Genome Biol. 2006;7(Suppl 1):S4.1–S4.9. doi: 10.1186/gb-2006-7-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garritano S., Gemignani F., Voegele C., Nguyen-Dumont T., Le Calvez-Kelm F., De Silva D., Lesueur F., Landi S., Tavtigian S.V. Determining the effectiveness of High Resolution Melting analysis for SNP genotyping and mutation scanning at the TP53 locus. BMC Genet. 2009;10:5. doi: 10.1186/1471-2156-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doi H., Yoshida K., Yasuda T., Fukuda M., Fukuda Y., Morita H., Ikeda S., Kato R., Tsurusaki Y., Miyake N. Exome sequencing reveals a homozygous SYT14 mutation in adult-onset, autosomal-recessive spinocerebellar ataxia with psychomotor retardation. Am. J. Hum. Genet. 2011;89:320–327. doi: 10.1016/j.ajhg.2011.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33(Web Server issue):W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Durme J., Delgado J., Stricher F., Serrano L., Schymkowitz J., Rousseau F. A graphical interface for the FoldX forcefield. Bioinformatics. 2011;27:1711–1712. doi: 10.1093/bioinformatics/btr254. [DOI] [PubMed] [Google Scholar]
- 22.Nowak K.J., Ravenscroft G., Jackaman C., Filipovska A., Davies S.M., Lim E.M., Squire S.E., Potter A.C., Baker E., Clément S. Rescue of skeletal muscle alpha-actin-null mice by cardiac (fetal) alpha-actin. J. Cell Biol. 2009;185:903–915. doi: 10.1083/jcb.200812132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ravenscroft G., Nowak K.J., Jackaman C., Clément S., Lyons M.A., Gallagher S., Bakker A.J., Laing N.G. Dissociated flexor digitorum brevis myofiber culture system—a more mature muscle culture system. Cell Motil. Cytoskeleton. 2007;64:727–738. doi: 10.1002/cm.20223. [DOI] [PubMed] [Google Scholar]
- 24.Ruparelia A.A., Zhao M., Currie P.D., Bryson-Richardson R.J. Characterization and investigation of zebrafish models of filamin-related myofibrillar myopathy. Hum. Mol. Genet. 2012;21:4073–4083. doi: 10.1093/hmg/dds231. [DOI] [PubMed] [Google Scholar]
- 25.Zeller J., Schneider V., Malayaman S., Higashijima S., Okamoto H., Gui J., Lin S., Granato M. Migration of zebrafish spinal motor nerves into the periphery requires multiple myotome-derived cues. Dev. Biol. 2002;252:241–256. doi: 10.1006/dbio.2002.0852. [DOI] [PubMed] [Google Scholar]
- 26.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura M., Ota T. The age of a neutral mutant persisting in a finite population. Genetics. 1973;75:199–212. doi: 10.1093/genetics/75.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guerois R., Nielsen J.E., Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 29.Embree, E.J. (2007). The identification and characterization of MKRP, a novel kelch related protein. PhD Thesis, Graduate School of Biomedical Sciences, The University of Texas Southwestern Medical Center at Dallas, Dallas, TX. http://repositories.tdl.org/utswmed-ir/bitstream/handle/2152.5/226/embreelaurence.pdf?sequence=3.
- 30.Lehnert S.A., Byrne K.A., Reverter A., Nattrass G.S., Greenwood P.L., Wang Y.H., Hudson N.J., Harper G.S. Gene expression profiling of bovine skeletal muscle in response to and during recovery from chronic and severe undernutrition. J. Anim. Sci. 2006;84:3239–3250. doi: 10.2527/jas.2006-192. [DOI] [PubMed] [Google Scholar]
- 31.Prag S., Adams J.C. Molecular phylogeny of the kelch-repeat superfamily reveals an expansion of BTB/kelch proteins in animals. BMC Bioinformatics. 2003;4:42. doi: 10.1186/1471-2105-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cirak S., von Deimling F., Sachdev S., Errington W.J., Herrmann R., Bönnemann C., Brockmann K., Hinderlich S., Lindner T.H., Steinbrecher A. Kelch-like homologue 9 mutation is associated with an early onset autosomal dominant distal myopathy. Brain. 2010;133:2123–2135. doi: 10.1093/brain/awq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Furukawa M., He Y.J., Borchers C., Xiong Y. Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat. Cell Biol. 2003;5:1001–1007. doi: 10.1038/ncb1056. [DOI] [PubMed] [Google Scholar]
- 34.Canning P., Cooper C.D., Krojer T., Murray J.W., Pike A.C., Chaikuad A., Keates T., Thangaratnarajah C., Hojzan V., Marsden B.D. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J. Biol. Chem. 2013;288:7803–7814. doi: 10.1074/jbc.M112.437996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mrosek M., Labeit D., Witt S., Heerklotz H., von Castelmur E., Labeit S., Mayans O. Molecular determinants for the recruitment of the ubiquitin-ligase MuRF-1 onto M-line titin. FASEB J. 2007;21:1383–1392. doi: 10.1096/fj.06-7644com. [DOI] [PubMed] [Google Scholar]
- 36.Ravenscroft G., Sollis E., Charles A.K., North K.N., Baynam G., Laing N.G. Fetal akinesia: review of the genetics of the neuromuscular causes. J. Med. Genet. 2011;48:793–801. doi: 10.1136/jmedgenet-2011-100211. [DOI] [PubMed] [Google Scholar]
- 37.Quinn C.M., Wigglesworth J.S., Heckmatt J. Lethal arthrogryposis multiplex congenita: a pathological study of 21 cases. Histopathology. 1991;19:155–162. doi: 10.1111/j.1365-2559.1991.tb00006.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-lapse imaging of a control dye-injected zebrafish at 72 hpf shows a normal startle response with rapid swimming away from contact.
Time-lapse imaging of a klhl40a-MO/40b2-MO2-injected zebrafish at 72 hpf shows limited movement and loss of swimming ability compared to control fish (Movie S1).