

Abstract
To better understand different molecular mechanisms by which mutations lead to various human diseases, we classified 82,833 disease-associated mutations according to their inheritance modes (recessive versus dominant) and molecular types (in-frame [missense point mutations and in-frame indels] versus truncating [nonsense mutations and frameshift indels]) and systematically examined the effects of different classes of disease mutations in a three-dimensional protein interactome network with the atomic-resolution interface resolved for each interaction. We found that although recessive mutations affecting the interaction interface of two interacting proteins tend to cause the same disease, this widely accepted “guilt-by-association” principle does not apply to dominant mutations. Furthermore, recessive truncating mutations in regions encoding the same interface are much more likely to cause the same disease, even for interfaces close to the N terminus of the protein. Conversely, dominant truncating mutations tend to be enriched in regions encoding areas between interfaces. These results suggest that a significant fraction of truncating mutations can generate functional protein products. For example, TRIM27, a known cancer-associated protein, interacts with three proteins (MID2, TRIM42, and SIRPA) through two different interfaces. A dominant truncating mutation (c.1024delT [p.Tyr342Thrfs∗30]) associated with ovarian carcinoma is located between the regions encoding the two interfaces; the altered protein retains its interaction with MID2 and TRIM42 through the first interface but loses its interaction with SIRPA through the second interface. Our findings will help clarify the molecular mechanisms of thousands of disease-associated genes and their tens of thousands of mutations, especially for those carrying truncating mutations, often erroneously considered “knockout” alleles.
Introduction
Understanding genotype-to-phenotype relationships has been a central theme of human genetics.1 In the past few decades, great progress has been made in identifying and characterizing disease-associated genes underlying many Mendelian disorders.2,3 Advances in next-generation-sequencing technologies and genome-wide association studies have further facilitated the identification of allelic variants associated with complex genetic diseases.4,5 However, it is often unclear how these mutations translate into complex disease phenotypes. Furthermore, disease-associated mutations can be classified into different categories on the basis of their inheritance modes (dominant or recessive) and molecular types (missense or nonsense). Mutations in different categories might cause disease through completely different mechanisms at the molecular level (e.g., loss of function or gain of function).3,6
Given that the cell functions as an intricate molecular network, disease mutations not only cause aberrations of single genes but could also perturb the broader network and lead to the observed phenotype.7–10 Various network-based approaches have been employed for exploring genotype-to-phenotype relationships.7–11 Goh et al. and Feldman et al. found that protein products of genes associated with similar diseases are more likely to physically interact and form disease-specific functional modules.8,9 On the basis of this commonly accepted “guilt-by-association” principle,12 many methods have been developed for the prediction of novel disease-associated genes with the use of the protein interactome network.13–15 However, none of these methods have considered the potential differences in the molecular mechanisms leading to the corresponding disorders for mutations of different inheritance modes and molecular types. Zhong et al. found that disease mutations could lead to two types of perturbations at the network level: node removal (loss of all known interactions of a protein) or edgetic perturbation (loss of specific interactions of a protein).11 They also found that a higher fraction of mutations associated with autosomal-dominant diseases are in-frame, tend to affect structural proteins, and are likely to affect exposed residues.11
The functional consequences of different classes of disease mutations can be better characterized by the consideration of the three-dimensional (3D) structures of proteins. Recent studies have shown that incorporating structural information with the protein-protein interaction network provides mechanistic understanding of disease-associated genes and mutations at the molecular level.6,16 In a previous study, we established a high-quality 3D protein interactome network with structurally resolved interfaces for each interaction.16 We analyzed in-frame disease mutations affecting this 3D interactome network and found that disease specificity of in-frame mutations can be explained by the locations they affect within the corresponding interaction interfaces.16
However, to date, no systematic analysis has been done to examine the widely-used guilt-by-association principle on disease mutations with different inheritance modes and molecular types, which is the focus of this study. We compiled a comprehensive set of disease-associated mutations from the Human Gene Mutation Database (HGMD)17,18 and the Catalogue of Somatic Mutations in Cancer (COSMIC).19,20 We then annotated the inheritance modes of disease mutations on the basis of manually curated inheritance information. We further expanded the 3D protein interactome network with 668 additional high-quality binary interactions.16 Structural details of protein interactions provide a tool for examining the effects of different types of disease mutations at atomic resolution. Here, we applied this approach to systematically analyze disease mutations of different inheritance modes and molecular types, which can be divided into four categories (i.e., dominant in-frame, dominant truncating, recessive in-frame, and recessive truncating). First, we examined how the effects of disease mutations in different categories were distributed in proteins with respect to interaction interfaces. We then investigated to what extent the guilt-by-association principle can be applied to pairs of mutations that are in different categories and that affect different locations in the corresponding proteins. We found that the guilt-by-association principle does not apply to dominant disease mutations (both in-frame and truncating). Furthermore, we found that 61% of recessive truncating mutation pairs in regions encoding the same interaction interface cause the same disease; this percentage is significantly higher than that for mutations in regions encoding different interfaces (12%). This analysis was not performed in our previous study,16 and our results indicate that a significant fraction of truncating mutations can generate protein products that retain at least some of the wild-type functions, contrary to the common belief that truncating mutations are often complete loss-of-function mutations.21–24
Material and Methods
Compiling a High-Quality List of Disease-Associated Genes and Mutations
Somatic mutations and their associated cancers were obtained from COSMIC19,20 (version 56). To remove putative passenger mutations, we only included mutations in genes in the Cancer Gene Census.25–28 Germline mutations and their associated diseases were obtained from HGMD17,18 (professional version 2010.12). Only “disease-causing mutations” and “disease-associated polymorphisms of functional significance” were selected for further analyses. Each mutation and its flanking sequence was translated into an amino acid sequence and mapped onto the corresponding protein sequence. Protein sequences used were obtained from SwissProt29 (release 57.6).
The nomenclatures of diseases are not standardized between the two databases. We compiled a comprehensive disease-gene association map based on the Online Mendelian Inheritance in Man (OMIM)2 and HGMD databases and gave unique disease IDs to each phenotypically distinct disorder. To standardize the nomenclature, we mapped all disease names to our disease IDs through bioinformatic processing and manual curation.
Constructing the 3D Protein Interactome Network
The human 3D protein interactome network was constructed as previously described in Wang et al.16 Since the publication, the 3D protein interactome network has been continuously updated.30 New binary protein interactions have been incorporated.31 Furthermore, in Wang et al.,16 binary protein interactions that are supported by only one cocrystal structure and have no other supporting evidence in the literature were excluded from the 3D protein interactome network for quality assurance. Here, we modified our filtering criteria to include all binary protein interactions supported by cocrystal structures in 3did32 or iPfam,33 given that cocrystal structures are usually considered gold-standard evidence that these interactions exist. Our homology-modeling approach assigns each interface domain with specific interactions of that protein, and one interaction could have multiple interface domains. If two proteins interact through multiple domains, all domains involved in the interaction are considered to be the interaction interface. If each of the two interacting proteins has other domains that interact with other proteins, these different domains are classified as different interfaces for different interactions. To evaluate the performance of our homology-modeling approach, we carried out 3-fold cross-validation by using the 1,456 human interaction pairs with known cocrystal structures. We found that over 94% of these interactions were correctly predicted with corresponding interaction interfaces, indicating the high accuracy of our approach.16 Currently, our homology-modeling approach cannot account for protein-peptide interactions, given that it is extremely difficult to predict protein-peptide interactions with high accuracy.34
Annotating the Inheritance Modes
Inheritance information of disease-associated genes was obtained from two sources: Zhong et al.11 and the Cancer Gene Census.35 Each unique gene-disease pair was assigned either autosomal-dominant or autosomal-recessive inheritance. Gene-disease pairs with other inheritance patterns, e.g., sex-linked inheritance, were discarded. Gene-disease pairs with conflicting annotations in the two data sets were removed. In total, we collected inheritance patterns for 1,794 unique gene-disease pairs. Next, we separated mutations into either autosomal-dominant or autosomal-recessive inheritance on the basis of the genes in which they reside and the disease with which they are associated. A total of 38,497 disease-associated mutations with either autosomal-dominant or autosomal-recessive inheritance were obtained.
Statistical Analysis: Distribution of Mutations with Respect to Regions Encoding Interaction Interfaces
Proteins affected by at least one mutation and with at least one interaction domain were chosen for the mutation enrichment calculation. Each protein sequence was divided into three regions: “in interaction interface,” “in other domain,” and “outside domains.” The total number of amino acids and the total number of mutations affecting each region were counted. If mutations are randomly distributed, the fraction of mutations affecting each region should be proportional to the relative length of each region. We calculated the expected fraction of mutations affecting each region (p2) by dividing the sum of the sequence length of each region in all proteins by the sum of the total sequence length of all proteins. We calculated the observed fraction of mutations affecting each region (p1) by adding mutations affecting each region of all proteins and dividing the sum by the total number of mutations. The odds ratios (ORs) were calculated on the basis of these expected and observed fractions:
Z scores and the 95% confidence intervals for the ORs36 were calculated as follows:
where N0.975 is the 97.5th percentile value of the standard normal distribution, nmut is the number of mutations, and nres is the total number of residues.
Statistical Analysis: Locus Heterogeneity Calculations
Mutation Pairs Affecting Interaction Partners
Genes with at least one mutation affecting an interaction interface and encoding proteins with at least one interaction interface were selected for this calculation. Of all mutations in these genes, only mutations affecting interaction interfaces were used and mutations not affecting interaction interfaces were discarded. From this list of genes, all possible pairs of genes in which at least one of the two genes encodes a protein with more than one interaction interface were selected.
For each gene pair, all possible mutations pairs with one mutation affecting an interaction interface encoded by gene A and another mutation affecting an interaction interface encoded by gene B were considered. We divided all mutation pairs into three categories: (1) if genes A and B encode interacting proteins in the 3D protein network and both mutations affect the interaction interface responsible for the interaction between the proteins encoded by genes A and B, we considered the mutation pair to “affect the same interface;” (2) if at least one of the two mutations does not affect the interaction interface between the proteins encoded by genes A and B, we considered the mutation pair to “affect other interaction interfaces;” and (3) if genes A and B do not encode interacting proteins, we considered the mutation pair to be “noninteracting.” We then calculated the percentage of mutation pairs causing the same disease for each category. The statistical significance of the comparisons between categories was evaluated by the cumulative binomial distribution
where N is the total number of mutation pairs, co is the number of observed pairs causing the same disease, and π is the fraction of pairs causing the same disease in the control sample. In all calculations, the binomial test was performed twice, and the test and control groups were swapped in the second test. The least significant p value was used.
Effect of Mutation Location on Locus Heterogeneity
All genes with at least one mutation were selected. All possible pairs of genes encoding proteins that interact in the 3D protein network were used. For each gene pair, gene A was divided into three equal parts and mutations on each third were paired with all gene B mutations affecting the corresponding interaction interface. All mutation pairs were classified into two categories: (1) if the mutation in gene A also affects the corresponding interaction interface with the protein encoded by gene B, the mutation pair was considered to “affect the interaction interface,” or (2) the mutation pair was classified as “other.” For each gene pair, both genes were used once as gene A and once as gene B. The statistical significance of the difference between the two categories for each third was evaluated by the cumulative binomial distribution as described above.
Statistical Analysis: Enrichment of Truncating Mutations in Sequences Encoding the Interdomain Regions
In this study, an interdomain region was defined as a region between two different interaction interfaces on a protein. Two interaction interfaces on a protein were considered different if at least one protein interacts with one interface, but not the other, in the 3D protein interaction network. Genes with at least one truncating mutation and encoding proteins with at least one interdomain region were selected for further analyses. The enrichment of dominant and recessive truncating mutations in sequences encoding the interdomain regions was measured by an OR as detailed above. Sample sizes in all the calculations are listed in Table S1, available online.
Identification of Loss-of-Function Dominant Mutations
Huang et al.37 have made a genome-wide prediction of the probability of genes to exhibit haploinsufficiency. On the basis of their predicted probability scores of being haploinsufficient (HI), p(HI), we considered the top 10% of genes with the highest p(HI) as HI genes. To validate the predicted HI gene set, we checked whether the predicted HI genes tend to be dominantly inherited. Among 583 dominantly inherited genes, 161 were predicted to be HI genes, whereas only 32 out of 515 recessively inherited genes were predicted to be HI. The enrichment of dominantly inherited genes in the HI gene set verified the prediction accuracy. We classified all dominant mutations on HI genes as “HI mutations” and all dominant mutations not on HI genes as “non-HI mutations.”
Selection of Proof-of-Principle Example for Experimental Validation
To experimentally validate our hypothesis that protein products of alleles with truncating mutations in sequences encoding the interdomain regions can retain some of their original functions or interactions, we searched for dominant truncating mutations satisfying the following criteria: (1) The mutation has to be located between regions encoding two different interaction interfaces. (2) Dominant truncating mutations have to be enriched in the sequence encoding the interdomain region. (3) To test the conservation and loss of specific interactions of the truncated protein with yeast two-hybrid (Y2H) assays, the interactions between the wild-type protein and its interactors at different interaction interfaces must be detectable by our Y2H pipeline. TRIM27 (MIM 602165) c.1024delT (p.Tyr342Thrfs∗30) was chosen for experimental validation because it satisfies all the above requirements and we have existing clones of TRIM27, SIRPA (MIM 602461), MID2 (MIM 300204), and TRIM42.
Determination of Interaction Interfaces with the Use of the 3D Protein Interaction Network and Structural Interface Matching
Using a combination of the 3D protein interaction network and structural interface matching, we determined domains mediating interactions between TRIM27 and SIRPA, TRIM27 and MID2, and TRIM27 and TRIM42. Structural interface matching comprised two steps—rigid body docking and flexible docking.38 For putative interacting domains, crystal structures were obtained from the Protein Data Bank (PDB) (see Web Resources).39 Only high-resolution (<2.5Å) X-ray-diffraction structures were used. Rigid body docking was performed with Patchdock40,41 with default parameters. This was followed by backbone refinement of the two proteins with the use of normal-mode analysis.42 Finally, both the side chain and the backbone conformations were refined with the computationally efficient FiberDock algorithm with default parameters.43,44 We found that a SPRY domain on TRIM27 and a C1-set domain on SIRPA mediate the interaction between TRIM27 and SIRPA. We found an energetically feasible solution by docking the SPRY domain (PDB ID 2YYO, crystallized by the RIKEN Structural Genomics/Proteomics Initiative) and the C1-set domain (PDB ID 2WNG).45 Both the TRIM27-MID2 and TRIM27-TRIM42 interactions are mediated by zf-B box domains on the corresponding proteins. The energetic feasibility of dimerization of the zf-B box domain is demonstrated by a cocrystal structure (PDB ID 2YVR, crystallized by the RIKEN Structural Genomics/Proteomics Initiative).
Construction of Plasmids and Disease Mutant Clones
Wild-type TRIM27, MID2, TRIM42, and SIRPA entry clones are from the hORFeome 3.1 collection.46 To generate disease mutant clones, we performed PCR mutagenesis as previously described.11,16,47 In brief, wild-type TRIM27 in an activation domain (AD) vector was used as the template in PCR reactions for the generation of N- and C-terminal fragments, each containing the region affected by the desired mutation in their overlapping region. BP recombination reactions were performed according to the manufacturer’s manual (Gateway BP Clonase II Enzyme Mix, catalog number 11789-020) for moving mutant clones into the entry vector.
Y2H
Y2H was performed as previously described.48 In brief, wild-type and mutant TRIM27 were transferred into AD vectors. Wild-type MID2, TRIM42, and SIRPA were transferred into DNA-binding (DB) vectors. AD and DB constructs were transformed into Y2H strains MATa Y8800 and MATα Y8930, respectively. Transformed yeast were spotted onto YPD plates and incubated at 30°C for ∼20 hr before replica plating onto synthetic complete (SC) plates lacking Leu and Trp. Yeast cells were allowed to grow at 30°C for 24 hr before replica plating onto each of the four selection plates (SC-Leu-Trp-His, SC-Leu-His+CYH, SC-Leu-Trp-Ade, and SC-Leu-Ade+CYH). At 72 hr after replicating, plates were evaluated for protein interactions.
Results
Mapping the Effects of Disease Mutations onto the 3D Protein Interactome Network
Here, we have compiled a comprehensive list of human disease mutations, including 68,789 germline mutations in 2,781 genes associated with 2,244 phenotypically distinct Mendelian diseases from HGMD17,18 and 14,044 somatic cancer mutations in 366 genes associated with 112 cancers from COSMIC.19,20 Because COSMIC includes results from whole-genome-sequencing experiments, it is likely that some mutations identified are passenger mutations that do not cause the cancer phenotype. To remove putative passenger mutations from the COSMIC data set, we included only mutations in genes in the Cancer Gene Census,35 a literature-curated list of known cancer-associated genes.
Disease mutations can be dominant or recessive at the cellular level. For dominant mutations, a single mutated allele can lead to pathogenesis, whereas for recessive mutations, both alleles need to be mutated for disease to occur. To examine the potential differences between dominant and recessive mutations, we compiled a list of genes with manually curated inheritance and disease information from published data sets.11,35 Disease mutations were then classified as autosomal dominant or autosomal recessive according to the inheritance mode of the respective gene and the disease they are associated with. In total, we annotated the inheritance modes of 38,497 disease-associated mutations.
Using our recently developed homology-modeling approach16 and incorporating newly published binary protein-protein interactions, we generated a high-quality 3D atomic-resolution protein interactome network comprising 4,890 structurally resolved interactions involving 3,174 proteins (Figure 1A). A total of 11,290 dominant mutations and 8,702 recessive mutations were mapped onto their corresponding proteins in the 3D protein interactome network.
Figure 1.
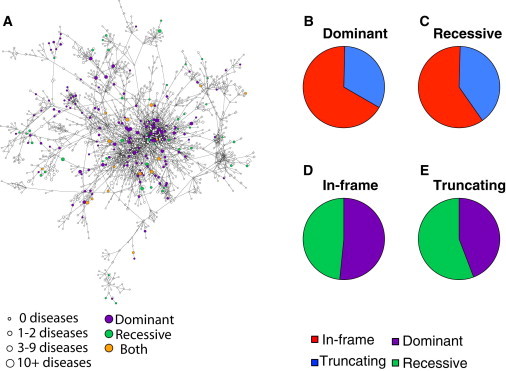
Disease-Associated Genes and Mutations Affecting the 3D Protein Interactome Network
(A) Network representation of the structurally resolved protein interactome.
(B) Proportions of in-frame and truncating mutations among all dominant mutations.
(C) Proportions of in-frame and truncating mutations among all recessive mutations.
(D) Proportions of dominant and recessive mutations among all in-frame mutations.
(E) Proportions of dominant and recessive mutations among all truncating mutations.
Different Molecular Mechanisms between Dominant and Recessive Mutations
All disease mutations can be further divided into two broad classes according to their molecular types and effects on the translated protein products: missense point mutations and in-frame insertions or deletions are classified as in-frame mutations; nonsense mutations and frameshift insertions or deletions are classified as truncating mutations.11,16 In-frame alleles are likely to produce full-length protein products with local defects, whereas truncating alleles, which are also called “complete loss-of-function (LoF)” alleles,21 are often assumed not to produce any functional protein products, especially in many current whole-exome- and whole-genome-sequencing studies.22–24,49
Among the dominant mutations, 67% are in-frame, whereas 60% of the recessive mutations are in-frame (Figures 1B and 1C). Conversely, 52% of in-frame mutations are dominant, whereas only 44% of truncating mutations are dominant (Figures 1D and 1E). The results agree with our current knowledge of the mechanisms of the action of dominant mutations. Other than the case of haploinsufficiency, dominance is most often a result of gain-of-function mutations or dominant-negative mutations, where the altered protein product is activated for a specific function or interferes with the normal function(s) of the wild-type protein.11,50 Therefore, most dominant mutations should translate into specific localized changes in the protein, which can be more easily achieved with in-frame mutations than with truncating mutations.
Most proteins carry out their functions through interactions with other proteins. Our recent study has demonstrated that the disruption of specific interactions of a protein is an important mechanism for pathogenesis of many human disease-associated genes and their mutations.16 To further investigate the differences between dominant and recessive disease mutations, we mapped the mutations onto their corresponding proteins in the 3D protein interactome and examined the locations they affect with respect to interaction interfaces and other functional protein domains. We found that for recessive mutations, both in-frame and truncating mutations are significantly enriched in regions encoding interaction interfaces (OR = 3.3, p < 10−20 by Z-test and OR = 1.9, p < 10−20 by Z-test; respectively, Figure 2B). Because recessive mutations are more likely to be loss-of-function mutations,6,51 our results demonstrate that the disruption of protein interaction interfaces is a common mechanism leading to the loss of specific functions. Dominant in-frame mutations are also enriched in regions encoding interaction interfaces (OR = 1.5, p < 10−20 by Z-test, Figure 2A). Because a significant fraction of dominant mutations are gain-of-function mutations,6,51 the results suggest that changes in protein interaction interfaces not only lead to the loss of specific interactions but also have the potential to generate new ones. Remarkably, the dominant truncating mutations are enriched in regions encoding sequences outside of functional domains (OR = 3.6, p < 10−20 by Z-test, Figure 2A) and are depleted in regions encoding protein interaction interfaces. This shows that the molecular mechanisms of dominant truncating mutations tend to be distinct from their recessive counterparts. To further assess the differences between dominant and recessive mutations, we next investigated the disease specificity of mutations in different categories.
Figure 2.
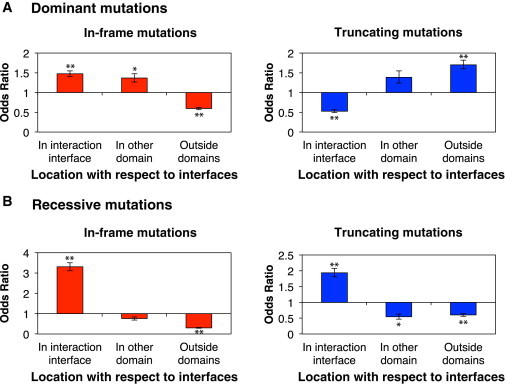
Distribution of Recessive and Dominant Disease Mutations with Respect to Regions Encoding Interaction Interfaces
(A) ORs of the distributions of dominant in-frame (left) and truncating (right) mutations in sequences encoding different protein regions.
(B) ORs of the distribution of recessive in-frame (left) and truncating (right) mutations in sequences encoding different protein regions. ∗∗p < 10−20, ∗p < 10−10. The p values were calculated with Z-tests for the log OR. Error bars represent 95% confidence intervals of ORs.
The Guilt-by-Association Principle Does Not Apply to Dominant Mutations
Many genetic diseases show locus heterogeneity, whereby a disease is associated with mutations on more than one gene. Understanding how different genes converge functionally to associate with the same disorder has important implications in the search for novel disease-associated genes and drug targets. Previous studies have shown that interacting protein pairs are more functionally similar and tend to be associated with the same diseases.12,52 More specifically, it has recently been shown that two in-frame mutations affecting the corresponding interaction interfaces of two interacting proteins tend to cause the same disease.16 This provides a higher-resolution explanation for the guilt-by-association principle: mutations affecting the interaction interface of two interacting proteins disrupt the same interaction in the cellular network and therefore abolish the same function and cause the same disorder.
To investigate whether the guilt-by-association principle holds for both dominant and recessive mutations, we examined the likelihood that in-frame mutation pairs affecting two different proteins cause the same disease. Among recessive in-frame mutations, 88% of mutation pairs affecting the corresponding interfaces of two interacting proteins cause the same disease; this percentage is significantly higher than that of mutation pairs affecting interaction interfaces that are not responsible for the interaction between the two proteins (21%, p < 10−20 by cumulative binomial test; Figure 3A, left). In contrast, among dominant in-frame mutations, only 10.1% of mutation pairs affecting the corresponding interfaces of interacting proteins cause the same disorder. Furthermore, the probability that two dominant in-frame mutations affecting interacting proteins cause the same disease does not depend on whether the two mutations affect the corresponding interaction interface responsible for the interaction between the two proteins or on other interaction interfaces (10.1% or 10.6%, respectively; Figure 3B, right). To further investigate the possible mechanisms of truncating mutations, we repeated the above calculation with truncating mutations. Interestingly, the observed difference between dominant and recessive in-frame mutations can also be seen among truncating mutations (Figure 3B). The likelihood that recessive truncating mutation pairs affecting interacting proteins cause the same disease depends on their location relative to the region encoding the interaction interface (Figure 3B, left). In contrast, dominant truncating mutation pairs affecting interacting proteins are less likely to cause the same disease, regardless of their location (Figure 3B, right). Just like to in-frame mutations, the guilt-by-association principle applies well to recessive truncating mutations, but not to dominant ones.
Figure 3.
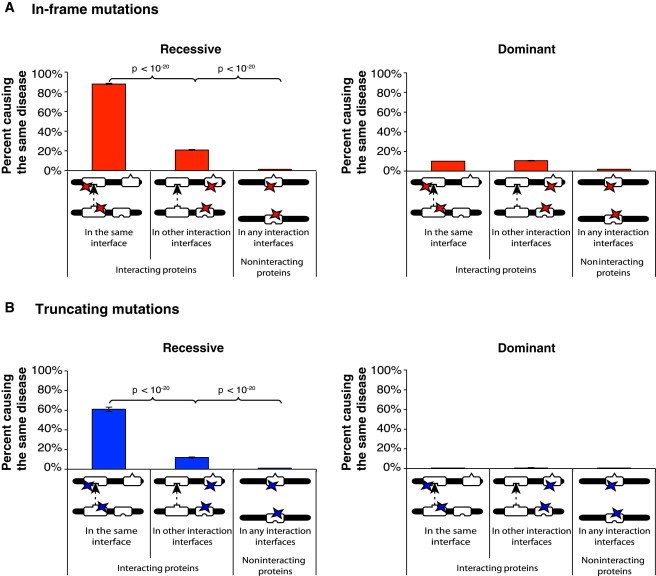
Analysis of Locus Heterogeneity among Dominant and Recessive Disease Mutations
(A) Percentage of recessive (left) or dominant (right) in-frame mutation pairs that affect two different proteins and cause the same disease.
(B) Percentage of recessive (left) or dominant (right) truncating mutation pairs that affect two different proteins and cause the same disease. Error bars represent ± SE. The p values were calculated with cumulative binomial tests.
An interesting example of the guilt-by-association principle can be observed in Glanzmann thrombasthenia (MIM 273800), which is associated with recessive mutations affecting the corresponding interaction interfaces of both ITGA2B and ITGB3. On the other hand, dominant mutations affecting the interaction interface of two proteins are often associated with different diseases. For example, dominant mutations affecting the calcium-binding epidermal growth factor domains of FBN1 are associated with Marfan syndrome (MIM 154700), whereas dominant mutations affecting the corresponding interaction interface of FBN2 are associated with contractural arachnodactyly (MIM 121050). Although Marfan syndrome and contractural arachnodactyly are related diseases, they have distinct clinical phenotypes.53
Our guilt-by-association analysis demonstrates that although recessive mutations that affect two different proteins and disrupt the same interaction tend to cause the same disorder, the same principle cannot be extended to dominant mutations. A likely explanation for these results is that loss-of-function mutations affecting two interacting proteins often cause the same disease by disrupting the same edge in the interaction network, but gain-of-function mutations affecting interacting proteins are less likely to cause the same disease because mutations in two different genes rarely gain the same function. Whereas recessive mutations are more likely to be loss-of-function mutations, dominant mutations can be gain-of-function, dominant-negative, or loss-of-function mutations (in the case of haploinsufficiency). To differentiate the molecular mechanisms of different classes of dominant mutations, we divided all dominant mutations into two categories—those likely to cause disease through haploinsufficiency (haploinsufficient [HI] mutations) and those not likely to cause disease through haploinsufficiency (non-HI mutations)—on the basis of a genome-wide prediction of HI genes.37 We found that compared to two non-HI in-frame mutations affecting the corresponding interfaces of interacting proteins, two HI in-frame mutations affecting the corresponding interaction interfaces between interacting proteins are significantly more likely to cause the same disease (p < 10−20 by cumulative binomial test; Figure 4A). Our results support the idea that because a large fraction of dominant mutations are gain-of-function variants, the guilt-by-association principle does not apply to these mutations. A similar calculation could not be performed on truncating mutations because of the small sample size. However, we found a clear distinction in the distribution patterns of HI and non-HI truncating mutations on their corresponding proteins. Similar to recessive truncating mutations, HI truncating mutations are enriched in regions encoding protein interaction interfaces. In contrast, non-HI truncating mutations are highly enriched in regions encoding sequences outside of functional domains (Figure 4B). This suggests that truncating mutations can also cause loss or gain of specific functions through distinct molecular mechanisms. Furthermore, the mode of action of truncating mutations can be inferred from their locations with respect to the regions encoding the interaction interfaces.
Figure 4.
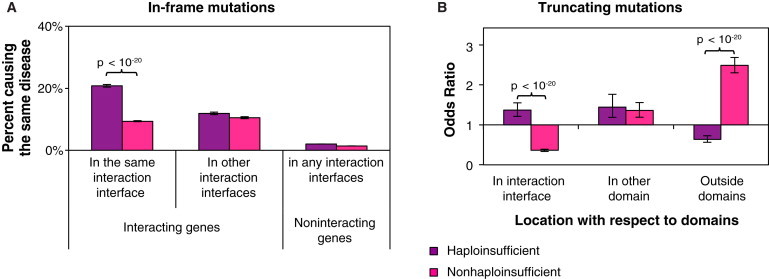
Analysis of Different Molecular Mechanisms of Dominant Mutations
(A) Percentage of HI and non-HI in-frame mutation pairs that affect two different proteins and cause the same disease.
(B) ORs of the distribution of HI and non-HI truncating mutations in sequences encoding different regions of proteins. Error bars represent ± SE. The p values were calculated with cumulative binomial tests.
Truncating Alleles Can Give Rise to Functional Products
Currently, truncating mutations are most often regarded as “knockout” mutations leading to absent or nonfunctional protein fragments.11,16 This is because mRNAs harboring premature stop codons are known to be selectively degraded by nonsense-mediated mRNA decay (NMD),54,55 and furthermore, even if the mRNA is translated, the resultant protein fragment is unlikely to fold into a stable product. If most of the truncating mutations lead to the loss of protein product, truncating mutations should be randomly distributed across protein-coding regions. However, in Figure 2 we observe that recessive truncating mutations are specifically enriched and dominant truncating mutations are specifically depleted in regions encoding protein interaction interfaces. Figure 3 further shows that truncating mutations with different inheritance modes have different patterns of disease association and that pairs of recessive truncating mutations in regions encoding the same interaction interface are much more likely to cause the same disease than those in regions encoding different interfaces. These results suggest that, contrary to common belief, a significant portion of truncating mutations are translated into functional protein products.
Truncating mutations in regions encoding sequences near the N terminus delete larger fractions of the wild-type protein. Therefore, it is generally believed that alleles carrying truncating mutations in regions encoding sequences near the N terminus are even less likely to produce functional products. In this study, we investigated how location with respect to the N terminus affects the functional consequences of truncating mutations. We first classified all truncating mutations into three categories: (1) mutations that are in regions encoding sequences near the N terminus and that truncate more than two-thirds of the wild-type protein, (2) mutations that are in regions encoding sequences near the C terminus and that truncate less than one-third of the wild-type protein, and (3) mutations that are in regions encoding the middle of the protein and that truncate between one-third and two-thirds of the wild-type protein. Then, for each pair of interacting proteins, we calculated in each category the percentage of truncating mutations that cause the same disease as mutations in regions encoding the corresponding interaction interfaces of the interaction partner (Figure 5 and Figure S5). If most truncating mutations in regions encoding sequences near the N terminus cause complete loss of function, all pairs of these mutations should have the same likelihood of causing the same disease, irrespective of whether they are in regions encoding the same interacting interface or not. However, we found that regardless of their location relative to the region encoding the N terminus, recessive truncating mutations in regions encoding the corresponding interaction interfaces of two proteins are always more likely to cause the same disease than are those that are not in regions encoding the corresponding interaction interfaces (Figure 5). Furthermore, we also found that irrespective of their location relative to the region encoding the N terminus, dominant truncating mutations are always enriched in regions encoding sequences outside of interaction interfaces and that recessive truncating mutations are always enriched in regions encoding interaction interfaces (Figure S6). These results show that truncating mutations’ location relative to the region encoding the N terminus does not significantly alter the proportion of truncated proteins that retain specific functions. These results, together with our observations in Figures 2–4, confirm that a significant fraction of alleles carrying truncating mutations, even those in regions encoding sequences near the N terminus, can be translated into proteins with specific functions.
Figure 5.
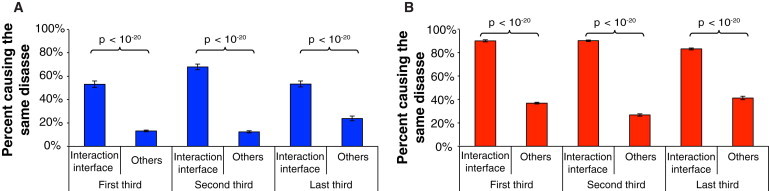
Disease Specificity of Truncating Mutations in Sequences Encoding Different Regions of the Protein
Percentage of truncating (A) or in-frame (B) mutations that are in regions encoding different parts of the protein and that cause the same disease as mutations affecting its interaction partner. Error bars represent ± SE. The p values were calculated with cumulative binomial tests.
To further characterize the molecular mechanisms underlying dominant and recessive truncating mutations, we calculated the enrichment of disease-associated truncating mutations that occur in regions encoding sequences between two different interaction interfaces. We found that dominant truncating mutations are enriched in regions encoding sequences located between interaction interfaces (OR = 1.7, p < 10−20 by Z-test) and that recessive truncating mutations are depleted in regions encoding sequences located between interaction interfaces (OR = 0.74, p < 10−5 by Z-test; Figure 6A and Figure S7). This result confirms that dominant truncating mutations tend to preserve specific interactions while losing others.
Figure 6.
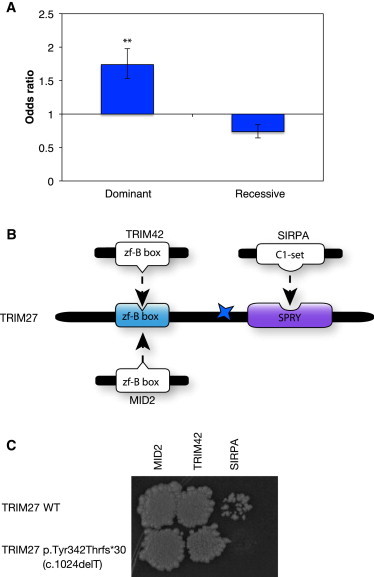
Enrichment of Truncating Mutations in Regions Encoding Sequences between Two Interaction Interfaces
(A) ORs of dominant and recessive truncating mutations affecting regions between interaction interfaces. ∗∗p < 10−20, ∗p < 0.05. The p values were calculated with Z-tests for the log OR. Error bars represent 95% confidence intervals of ORs.
(B) Illustration of TRIM27 and its interaction interfaces with MID2, TRIM42, and SIRPA. The colored star indicates the location of the experimentally tested mutation (c.1024delT [p.Tyr342Thrfs∗30]).
(C) Y2H-tested effects of truncating mutation c.1024delT (p.Tyr342Thrfs∗30) on the interactions of TRIM27.
To experimentally validate our conclusions, we tested the interactions of the protein product of tripartite-motif-containing 27 (TRIM27), a known cancer-associated gene that acts dominantly in oncogenesis.35,56 A frameshift deletion (p.Tyr342Thrfs∗30 [c.1024delT]) occurring just before the SPRY domain of TRIM27 was found to be associated with ovarian carcinoma (MIM 167000).20,57 Using a combination of the 3D protein interaction network and structural interface matching, we found that TRIM27 interacts with three other proteins, MID2, TRIM42, and SIRPA, of which only SIRPA interacts exclusively with the SPRY domain in TRIM27 (Figure 6B). None of the three interaction partners of TRIM27 were previously known to be involved in ovarian cancer. Here, we tested the interactions of wild-type TRIM27 and truncated TRIM27 by using Y2H. Because the truncating mutation occurs after the interaction interfaces with MID2 and TRIM42 but before the interaction interface with SIRPA, we hypothesized that the truncated TRIM27 would lose its interaction with SIRPA while retaining the other two interactions. The Y2H results confirm that the truncating mutation only disrupts the TRIM27-SIRPA interaction and leaves the other two interactions unaffected (Figure 6C). This supports our hypothesis that truncating mutations can retain specific interactions or functions. This result also suggests that abolition of the interaction between TRIM27 and SIRPA might contribute to the cancer phenotype and that SIRPA might be associated with ovarian carcinoma.
Discussion
One challenge in deciphering the molecular basis of genetic diseases is that disease phenotypes are often associated with multiple mutations that are in different genes and that have variable associated risks. Studies have found that genes associated with the same disease tend to cluster in functional modules within biological networks.8,9 This guilt-by-association principle has been widely applied for the identification of novel disease-associated genes.58 However, the accuracy of these predictions is still relatively low.59 Here, we systematically dissected the guilt-by-association principle on the basis of the molecular types and inheritance modes of over 20,000 mutations. Although recessive disease mutations affecting the corresponding interaction interfaces of interacting proteins tend to cause the same disease, the same does not apply to dominant disease mutations. Although current tools that predict disease-associated genes have integrated the protein-protein interactome network with disease phenotypic information to improve the accuracies of predictions,13–15 none of the current prediction models incorporate the difference in inheritance modes of disease-associated genes. By pointing out that the guilt-by-association principle only applies to recessive mutations, our findings could significantly improve the accuracy of current prediction methods for disease-associated genes.
Furthermore, truncating mutations, also called LoF mutations, are often regarded as knockout mutations in large-scale mutational screens and genome-sequencing projects.21–24,49 However, there are instances reported where mRNAs harboring truncating mutations escape NMD and are translated into proteins with dominant-negative activities.60,61 One particularly interesting case study involving SOX10 (MIM 602229) demonstrated that truncating mutations in different regions of SOX10 confer distinct neurological phenotypes. Among all SOX10 alleles harboring nonsense or frameshift mutations, transcripts with mutations in exons 3 and 4 are targeted by NMD, causing a neurological phenotype called Waardenburg-Shah syndrome (MIM 277580). On the other hand, transcripts with mutations in exon 5 escape NMD and lead to a more severe phenotype as a result of the dominant-negative effects of the translated protein.61 Furthermore, a recent publication revealed that, contrary to common belief, only a small percentage (16.3%) of LoF alleles show significant evidence of NMD.21 Our results further suggest that it is overly simplistic to consider all truncating mutations as null mutations, given that a significant fraction of them do generate functional protein products. Interestingly, our results show that truncating mutations that lead to functional products are not limited to the extreme C-terminal region of proteins; many proteins can lose more than two-thirds of their length and still retain specific functions.
All results that we discussed above are robust to the removal of protein hubs and domain hubs (Figures S1–S4), confirming that these results are not biased by overrepresented proteins or domain families. Moreover, although filtering COSMIC mutations within Cancer Gene Census genes enriches for cancer-causing mutations (Figure S10) and this filtering scheme is often used for selecting a high-confidence set of cancer mutations,25–28 some of the filtered mutations might still be passenger mutations. Therefore, we repeated our calculations by using only the HGMD mutations, and all results remained the same (Figures S8 and S9). These results indicate that although cancer is a complex disease, cancer-causing mutations are likely to disrupt normal protein functions through similar biophysical and/or biochemical mechanisms at the molecular level as are Mendelian mutations.
In recent years, large numbers of mutations have been discovered from whole-genome- and whole-exome-sequencing studies. Popular tools such as PolyPhen-2,62 SIFT,63 and MutationTaster64 estimate the impact of amino acid substitutions on the respective protein and are frequently used for prioritizing variants discovered from exome-sequencing projects. Our method could potentially be used in conjunction with these tools for the generation of hypotheses regarding the molecular mechanisms of the deleterious variants discovered. Moreover, it might be interesting to consider the penetrance and expressivity of the disease mutations in future analyses,65,66 when sufficient information is available.
In conclusion, by integrating inheritance information with atomic-resolution structural details of protein interactions, our analysis provides an approach to predicting functional consequences at the molecular level for both in-frame and truncating mutations, especially those discovered by various ongoing genome-sequencing efforts.
Acknowledgments
The authors wish to thank Nicolas A. Cordero for critical reading of the manuscript. This work was supported by National Institutes of Health grants R01 GM104424 (to H.Y. and S.M.L.), R01 CA167824 (to H.Y. and S.M.L.), and R01 HG003229 (to A.G.C.); by the Weill Cornell Medical College Clinical and Translational Science Center Pilot Award (to H.Y. and S.M.L.); by the Cornell University Seed Grant for Collaborations between Cornell University-Ithaca and Weill Cornell Medical College Faculty (to H.Y. and S.M.L.); by a donation from Matthew Bell (to S.M.L.); by the Cornell Presidential Life Sciences Fellowship (to Y.G.); and by the Tata Graduate Fellowship (to J.D.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Catalogue of Somatic Mutations in Cancer (COSMIC), http://www.sanger.ac.uk/genetics/CGP/cosmic/
Centre for Cancer Systems Biology, http://ccsb.dfci.harvard.edu/web/www/ccsb/
High-Quality Interactomes (HINT), http://hint.yulab.org/
Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/ac/index.php
INstruct, http://instruct.yulab.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Protein Data Bank, http://www.rcsb.org/pdb/home/home.do
References
- 1.Botstein D., Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat. Genet. 2003;33(Suppl):228–237. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- 2.Amberger J., Bocchini C.A., Scott A.F., Hamosh A. McKusick’s Online Mendelian Inheritance in Man (OMIM) Nucleic Acids Res. 2009;37(Database issue):D793–D796. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jimenez-Sanchez G., Childs B., Valle D. Human disease genes. Nature. 2001;409:853–855. doi: 10.1038/35057050. [DOI] [PubMed] [Google Scholar]
- 4.Altshuler D., Daly M.J., Lander E.S. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCarthy M.I., Abecasis G.R., Cardon L.R., Goldstein D.B., Little J., Ioannidis J.P., Hirschhorn J.N. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 6.Schuster-Böckler B., Bateman A. Protein interactions in human genetic diseases. Genome Biol. 2008;9:R9. doi: 10.1186/gb-2008-9-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barabási A.L., Gulbahce N., Loscalzo J. Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feldman I., Rzhetsky A., Vitkup D. Network properties of genes harboring inherited disease mutations. Proc. Natl. Acad. Sci. USA. 2008;105:4323–4328. doi: 10.1073/pnas.0701722105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goh K.I., Cusick M.E., Valle D., Childs B., Vidal M., Barabási A.L. The human disease network. Proc. Natl. Acad. Sci. USA. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vidal M., Cusick M.E., Barabási A.L. Interactome networks and human disease. Cell. 2011;144:986–998. doi: 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong Q., Simonis N., Li Q.R., Charloteaux B., Heuze F., Klitgord N., Tam S., Yu H., Venkatesan K., Mou D. Edgetic perturbation models of human inherited disorders. Mol. Syst. Biol. 2009;5:321. doi: 10.1038/msb.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliver S. Guilt-by-association goes global. Nature. 2000;403:601–603. doi: 10.1038/35001165. [DOI] [PubMed] [Google Scholar]
- 13.Lage K., Karlberg E.O., Størling Z.M., Olason P.I., Pedersen A.G., Rigina O., Hinsby A.M., Tümer Z., Pociot F., Tommerup N. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007;25:309–316. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- 14.Wu X., Jiang R., Zhang M.Q., Li S. Network-based global inference of human disease genes. Mol. Syst. Biol. 2008;4:189. doi: 10.1038/msb.2008.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X., Liu Q., Jiang R. Align human interactome with phenome to identify causative genes and networks underlying disease families. Bioinformatics. 2009;25:98–104. doi: 10.1093/bioinformatics/btn593. [DOI] [PubMed] [Google Scholar]
- 16.Wang X., Wei X., Thijssen B., Das J., Lipkin S.M., Yu H. Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat. Biotechnol. 2012;30:159–164. doi: 10.1038/nbt.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stenson P.D., Ball E.V., Mort M., Phillips A.D., Shiel J.A., Thomas N.S., Abeysinghe S., Krawczak M., Cooper D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 18.Stenson P.D., Mort M., Ball E.V., Howells K., Phillips A.D., Thomas N.S., Cooper D.N. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forbes S.A., Bhamra G., Bamford S., Dawson E., Kok C., Clements J., Menzies A., Teague J.W., Futreal P.A., Stratton M.R. The Catalogue of Somatic Mutations in Cancer (COSMIC) Curr. Protoc. Hum. Genet. 2008;Chapter 10 doi: 10.1002/0471142905.hg1011s57. Unit 10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forbes S.A., Bindal N., Bamford S., Cole C., Kok C.Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39(Database issue):D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., 1000 Genomes Project Consortium A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agrawal N., Frederick M.J., Pickering C.R., Bettegowda C., Chang K., Li R.J., Fakhry C., Xie T.X., Zhang J., Wang J. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ernst T., Chase A.J., Score J., Hidalgo-Curtis C.E., Bryant C., Jones A.V., Waghorn K., Zoi K., Ross F.M., Reiter A. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010;42:722–726. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 24.Seal S., Thompson D., Renwick A., Elliott A., Kelly P., Barfoot R., Chagtai T., Jayatilake H., Ahmed M., Spanova K., Breast Cancer Susceptibility Collaboration (UK) Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat. Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 25.Kaminker J.S., Zhang Y., Watanabe C., Zhang Z. CanPredict: a computational tool for predicting cancer-associated missense mutations. Nucleic Acids Res. 2007;35(Web Server issue):W595–W598. doi: 10.1093/nar/gkm405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaminker J.S., Zhang Y., Waugh A., Haverty P.M., Peters B., Sebisanovic D., Stinson J., Forrest W.F., Bazan J.F., Seshagiri S., Zhang Z. Distinguishing cancer-associated missense mutations from common polymorphisms. Cancer Res. 2007;67:465–473. doi: 10.1158/0008-5472.CAN-06-1736. [DOI] [PubMed] [Google Scholar]
- 27.Stephens P.J., Tarpey P.S., Davies H., Van Loo P., Greenman C., Wedge D.C., Nik-Zainal S., Martin S., Varela I., Bignell G.R., Oslo Breast Cancer Consortium (OSBREAC) The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pajkos M., Mészáros B., Simon I., Dosztányi Z. Is there a biological cost of protein disorder? Analysis of cancer-associated mutations. Mol. Biosyst. 2012;8:296–307. doi: 10.1039/c1mb05246b. [DOI] [PubMed] [Google Scholar]
- 29.UniProt Consortium Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40(Database issue):D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyer M.J., Das J., Wang X., Yu H. INstruct: a database of high-quality 3D structurally resolved protein interactome networks. Bioinformatics. 2013 doi: 10.1093/bioinformatics/btt181. Published online May 17, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das J., Yu H. HINT: High-quality protein interactomes and their applications in understanding human disease. BMC Syst. Biol. 2012;6:92. doi: 10.1186/1752-0509-6-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stein A., Panjkovich A., Aloy P. 3did Update: domain-domain and peptide-mediated interactions of known 3D structure. Nucleic Acids Res. 2009;37(Database issue):D300–D304. doi: 10.1093/nar/gkn690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finn R.D., Marshall M., Bateman A. iPfam: visualization of protein-protein interactions in PDB at domain and amino acid resolutions. Bioinformatics. 2005;21:410–412. doi: 10.1093/bioinformatics/bti011. [DOI] [PubMed] [Google Scholar]
- 34.Petsalaki E., Stark A., García-Urdiales E., Russell R.B. Accurate prediction of peptide binding sites on protein surfaces. PLoS Comput. Biol. 2009;5:e1000335. doi: 10.1371/journal.pcbi.1000335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris J.A., Gardner M.J. Calculating confidence intervals for relative risks (odds ratios) and standardised ratios and rates. Br. Med. J. (Clin. Res. Ed.) 1988;296:1313–1316. doi: 10.1136/bmj.296.6632.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang N., Lee I., Marcotte E.M., Hurles M.E. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tuncbag N., Gursoy A., Nussinov R., Keskin O. Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM. Nat. Protoc. 2011;6:1341–1354. doi: 10.1038/nprot.2011.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duhovny D., Nussinov R., Wolfson H.J. Efficient unbound docking of rigid molecules. Lecture Notes in Computer Science. 2002;2452:185–200. [Google Scholar]
- 41.Schneidman-Duhovny D., Inbar Y., Nussinov R., Wolfson H.J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33(Web Server issue):W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tirion M.M. Large Amplitude Elastic Motions in Proteins from a Single-Parameter, Atomic Analysis. Phys. Rev. Lett. 1996;77:1905–1908. doi: 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- 43.Mashiach E., Nussinov R., Wolfson H.J. FiberDock: Flexible induced-fit backbone refinement in molecular docking. Proteins. 2010;78:1503–1519. doi: 10.1002/prot.22668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mashiach E., Nussinov R., Wolfson H.J. FiberDock: a web server for flexible induced-fit backbone refinement in molecular docking. Nucleic Acids Res. 2010;38(Web Server issue):W457–W461. doi: 10.1093/nar/gkq373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hatherley D., Graham S.C., Harlos K., Stuart D.I., Barclay A.N. Structure of signal-regulatory protein alpha: a link to antigen receptor evolution. J. Biol. Chem. 2009;284:26613–26619. doi: 10.1074/jbc.M109.017566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lamesch P., Li N., Milstein S., Fan C., Hao T., Szabo G., Hu Z., Venkatesan K., Bethel G., Martin P. hORFeome v3.1: a resource of human open reading frames representing over 10,000 human genes. Genomics. 2007;89:307–315. doi: 10.1016/j.ygeno.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suzuki Y., Kagawa N., Fujino T., Sumiya T., Andoh T., Ishikawa K., Kimura R., Kemmochi K., Ohta T., Tanaka S. A novel high-throughput (HTP) cloning strategy for site-directed designed chimeragenesis and mutation using the Gateway cloning system. Nucleic Acids Res. 2005;33:e109. doi: 10.1093/nar/gni103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu H., Braun P., Yildirim M.A., Lemmens I., Venkatesan K., Sahalie J., Hirozane-Kishikawa T., Gebreab F., Li N., Simonis N. High-quality binary protein interaction map of the yeast interactome network. Science. 2008;322:104–110. doi: 10.1126/science.1158684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Haaften G., Dalgliesh G.L., Davies H., Chen L., Bignell G., Greenman C., Edkins S., Hardy C., O’Meara S., Teague J. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009;41:521–523. doi: 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Veitia R.A. Exploring the molecular etiology of dominant-negative mutations. Plant Cell. 2007;19:3843–3851. doi: 10.1105/tpc.107.055053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lodish H.F. W.H. Freeman and Co.; New York: 2013. Molecular cell biology. [Google Scholar]
- 52.Yu H., Jansen R., Stolovitzky G., Gerstein M. Total ancestry measure: quantifying the similarity in tree-like classification, with genomic applications. Bioinformatics. 2007;23:2163–2173. doi: 10.1093/bioinformatics/btm291. [DOI] [PubMed] [Google Scholar]
- 53.Wang M., Clericuzio C.L., Godfrey M. Familial occurrence of typical and severe lethal congenital contractural arachnodactyly caused by missplicing of exon 34 of fibrillin-2. Am. J. Hum. Genet. 1996;59:1027–1034. [PMC free article] [PubMed] [Google Scholar]
- 54.Chang Y.F., Imam J.S., Wilkinson M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 55.Maquat L.E. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 2004;5:89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- 56.Hatakeyama S. TRIM proteins and cancer. Nat. Rev. Cancer. 2011;11:792–804. doi: 10.1038/nrc3139. [DOI] [PubMed] [Google Scholar]
- 57.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X., Gulbahce N., Yu H. Network-based methods for human disease gene prediction. Brief Funct Genomics. 2011;10:280–293. doi: 10.1093/bfgp/elr024. [DOI] [PubMed] [Google Scholar]
- 59.Oti M., Snel B., Huynen M.A., Brunner H.G. Predicting disease genes using protein-protein interactions. J. Med. Genet. 2006;43:691–698. doi: 10.1136/jmg.2006.041376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fan S., Yuan R., Ma Y.X., Meng Q., Goldberg I.D., Rosen E.M. Mutant BRCA1 genes antagonize phenotype of wild-type BRCA1. Oncogene. 2001;20:8215–8235. doi: 10.1038/sj.onc.1205033. [DOI] [PubMed] [Google Scholar]
- 61.Inoue K., Khajavi M., Ohyama T., Hirabayashi S., Wilson J., Reggin J.D., Mancias P., Butler I.J., Wilkinson M.F., Wegner M., Lupski J.R. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 2004;36:361–369. doi: 10.1038/ng1322. [DOI] [PubMed] [Google Scholar]
- 62.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 64.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 65.Niemann S., Müller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000;26:268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 66.Zlotogora J. Penetrance and expressivity in the molecular age. Genet. Med. 2003;5:347–352. doi: 10.1097/01.gim.0000086478.87623.69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.