

Abstract
Hereditary spastic paraplegias (HSPs) form a heterogeneous group of neurological disorders. A whole-genome linkage mapping effort was made with three HSP-affected families from Spain, Portugal, and Tunisia and it allowed us to reduce the SPG26 locus interval from 34 to 9 Mb. Subsequently, a targeted capture was made to sequence the entire exome of affected individuals from these three families, as well as from two additional autosomal-recessive HSP-affected families of German and Brazilian origins. Five homozygous truncating (n = 3) and missense (n = 2) mutations were identified in B4GALNT1. After this finding, we analyzed the entire coding region of this gene in 65 additional cases, and three mutations were identified in two subjects. All mutated cases presented an early-onset spastic paraplegia, with frequent intellectual disability, cerebellar ataxia, and peripheral neuropathy as well as cortical atrophy and white matter hyperintensities on brain imaging. B4GALNT1 encodes β-1,4-N-acetyl-galactosaminyl transferase 1 (B4GALNT1), involved in ganglioside biosynthesis. These findings confirm the increasing interest of lipid metabolism in HSPs. Interestingly, although the catabolism of gangliosides is implicated in a variety of neurological diseases, SPG26 is only the second human disease involving defects of their biosynthesis.
Main Text
Hereditary spastic paraplegias (HSPs) constitute a clinically and genetically heterogeneous group of neurodegenerative conditions. They are characterized by a slowly progressive spasticity of the lower extremities resulting from the axonal degeneration and/or dysfunction observed in long axons of the corticospinal tracts.1,2 HSPs are classified according to the following criteria: (1) absence (uncomplicated or pure HSP) or presence (complicated or complex HSP) of additional neurological signs and symptoms, including intellectual disability, cerebellar ataxia, peripheral neuropathy, retinopathy, cataract, epilepsy, and ichthyosis; and (2) mode of inheritance in the case of familial forms, which can be autosomal-dominant, autosomal-recessive (AR), mitochondrial, or X-linked.3 To date, more than 55 HSP loci (denoted SPG) have been mapped. Among them, mutations have been found in ∼33 genes; the proteins encoded by these genes are often involved in intracellular trafficking, lipid metabolism, or mitochondrial functions.2,4–8
In 2005, Wilkinson et al. mapped the SPG26 locus (MIM 609195) to a 22.8 cM region flanked by markers D12S59 and D12S1676 (34.2 Mb) on chromosome 12p11.1–12q14 in a Kuwaiti family with AR complicated HSP.9 In the present study, we linked additional HSP families to the SPG26 locus, refined the locus region, identified the segregating mutations in seven of these families, and described the associated phenotype.
We selected three families in which diagnosis of AR HSP was established according to Harding’s criteria and careful exclusion of alternative disorders.10 Blood samples and clinical assessments were performed after informed consent and after local ethics approvals. The disease was not caused by mutations in any of the frequently involved genes previously linked to HSPs. All available affected (n = 4) and unaffected (n = 5) members of a Tunisian family (TUN34) were subjected to a genome-wide linkage mapping that used 6,090 SNP markers (LINKAGE_24 Illumina) and an additional 30 microsatellite markers, as previously described.11 All four affected individuals shared a single region of homozygosity of 22.5 cM (24 Mb) on chromosome 12 flanked by markers D12S1632 (56,415,415 bp) and D12S2074 (80,431,457 bp) (Figure S1 available online). Pairwise LOD scores reached the significance value of +3 at 19 consecutive markers with a multipoint LOD score of +4.45 (data not shown). In three affected and three unaffected subjects of a Spanish family (FSP112), the genome-wide scan was performed with 428 microsatellite markers, including the ABI Mapping set (Applied Biosystems), as described.12 In a single homozygous region of 33 cM (41 Mb), a maximal multipoint LOD score of +2.53 reached the maximal expected value in this pedigree, flanked by markers D12S1617 and D12S1686 (Figure S1). Both homozygous regions in families TUN34 and FSP112 overlapped with the SPG26 candidate interval and allowed its reduction from 34.2 Mb (27 cM) to 9.3 Mb (6.9 cM) between D12S1632 and D12S1686 (Figure S1). In a third family, of Portuguese origin without known consanguinity (FSP995), the same strategy was employed using the Illumina SNP panel and identified 10 regions with positive LOD score values ranging from +0.2 to +2.25, including a large portion of chromosome 12 containing SPG26.
Exome sequencing was performed on affected subjects of families FSP112, FSP995, and TUN34 as well as in two additional families, IHG25297 from Brazil and THI26004 from Germany (Figure 1). Coding exons and flanking intronic sequences were enriched with the SureSelect Human All Exon 50 Mb kit (Agilent) according to the manufacturer’s standard protocol. Enriched samples were prepared for the Hiseq2000 instrument (Illumina) and paired-end reads of 100 bp length were produced. The Burrows-Wheeler algorithm was applied to align sequence reads to the UCSC Genome Browser hg19 version of the human genome and variants were called via the GATK software package.13 Data were then imported into dedicated analysis toolsets, including the online GEnomes Management application (GEM.app)14 and Eris (Integragen), for further analysis. In families TUN34, FSP995, and FSP112, the variants were filtered according to their quality, functional class (nonsynonymous and/or affecting splicing), presence in chromosomal regions with putative or nonexcluded linkage, and frequency ≤1% in publically available genomic databases. Together, these criteria helped to reduce the list of variants to two to five variants per family (Table S1). Based on conservation and variant class, we restricted this list to single mutations for each family that were all in the same gene, B4GALNT1 (MIM 601873; RefSeq accession number NM_001478.3): c.898C>T (p.Arg300Cys), c.358C>T (p.Gln120∗), and c.395delC (p.Pro132Glnfs∗7), respectively. In family IHG25397, filtering of exome variants under a recessive model identified three candidate variants, one being a homozygous stop mutation in B4GALNT1 (c.682C>T [p.Arg228∗]). Similarly, a single homozygous variant in B4GALNT1 remained under the same filters in family THI26004 (c.1298A>C [p.Asp433Ala]) (Table S1).
Figure 1.
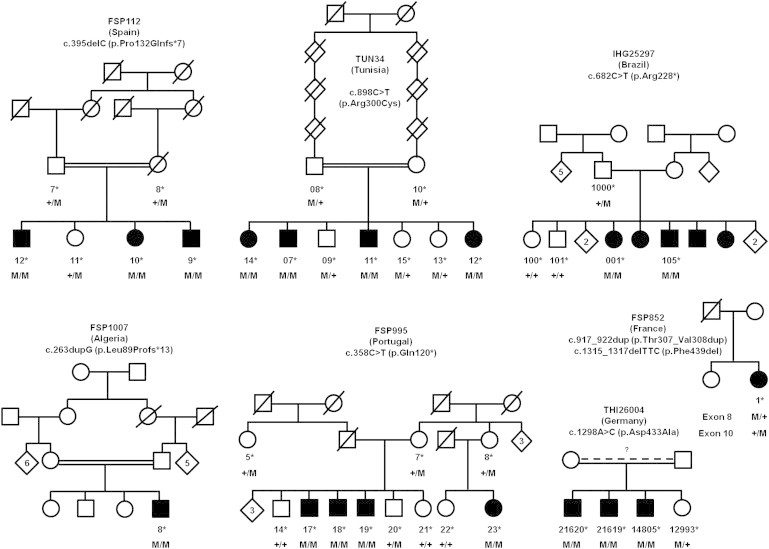
SPG26 Pedigrees and Segregation of the Mutations Detected in B4GALNT1
Square symbols indicate males and circles indicate females. Filled symbols indicate affected individuals. The numbers are an internal reference for each sampled individual. Stars indicate sampled subjects. Abbreviations are as follows: M, mutation; +, wild-type.
We then screened a series of 65 index cases of HSP families compatible with an AR mode of inheritance and found three other mutations in two simplex cases. One homozygous truncating mutation was identified in a consanguineous Algerian subject (FSP1007-8: c.263dupG [p.Leu89Profs∗13]). A French HSP case harbored compound heterozygote changes: one codon deletion affecting a conserved amino acid and one in-frame duplication of two codons (FSP852-1: c.1315_1317delTTC [p.Phe439del] and c.917_922dup [p.Thr307_Val308dup]). For the latter case, it was not possible to verify whether they segregated in cis or in trans (Figures 1 and 2).
Figure 2.
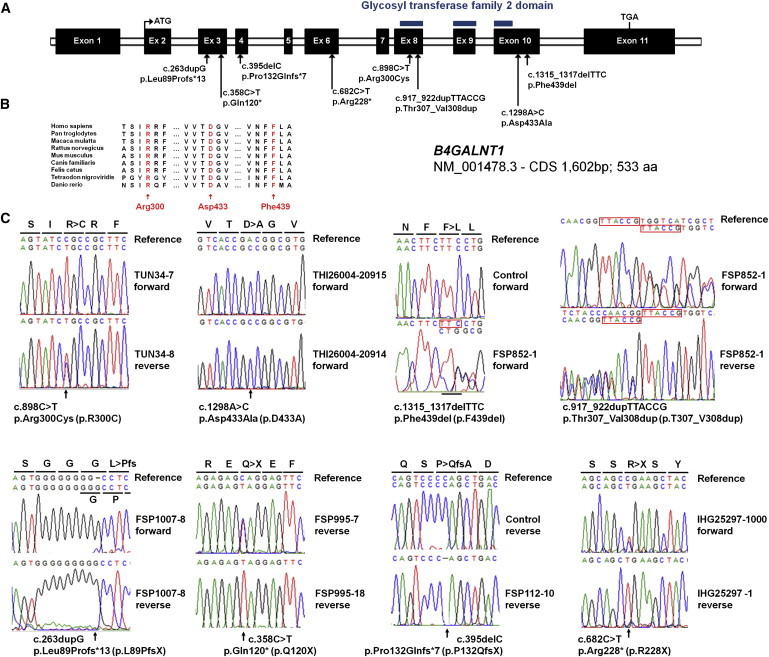
Schematic Representation of B4GALNT1/SPG26 and Location of the Mutations
(A) Exon-intron structure of B4GALNT1, with positions of mutations identified in seven SPG26-affected families. Exons are indicated as black boxes. The region encoding a functional domain is indicated by blue bars.
(B) Phylogenetic conservation of three amino acids mutated in SPG26-affected individuals.
(C) Electropherograms of the mutations identified. Mutation nomenclature is in agreement with ALAMUT 2.2 and Mutalyzer software with transcript NM_001478.3.
These mutations all segregated with the disease in their respective pedigree, when it could be tested (Figure 1), and they were also absent from the Exome Variant Server (EVS; 13,006 chromosomes) and our local databases (>3,340 chromosomes). The two missense variants and the codon deletion affected strongly conserved amino acids (Figure 2) in B4GALNT1 and they were all predicted to be pathogenic when examined with algorithms designed to assess the impact of genetic variations (Polyphen, MutationTaster, Sift).
B4GALNT1 encodes β-1,4-N-acetyl-galactosaminyl transferase 1 (GM2/GD2 synthase; EC 2.4.1.92), an enzyme involved in the biosynthesis of complex gangliosides (G), which are mono- (M), di- (D), and tri- (T) sialic acid-containing glycosphingolipids generated by sequential glycosylations (Figure 3).15 Gangliosides are part of the larger family of glycosphingolipids and are components of the synaptic plasma membrane involved in synaptic plasticity, signal transduction, and endocytosis and then are critical for CNS development. B4GALNT1 catalyzes the transfer of N-acetyl-galactosamine into GM3, GD3, and globotriaosylceramide by a β-1,4 linkage (Figure 3). The reverse reaction is performed by β-hexosaminidase and its cofactor (GM2 activator). Molecular defects in the degradation of glycosphingolipids led to well-known lysosomal storage diseases, including Gaucher disease caused by GBA (MIM 606463) mutations,16,17 Fabry disease (MIM 301500), metachromatic leukodystrophy (MIM 250100 and 249900), Tay-Sachs AB variant/GM2 gangliosidosis (MIM 272750), Sandhoff disease (MIM 268800), and GM1 gangliosidosis (MIM 230500) (Figure 3). Conversely, to our knowledge, a single proven case of a human disorder resulting from disruption of ganglioside biosynthesis has been described.18 Mutations in ST3GAL5 (also known as SIAT9 [MIM 604402]) coding for GM3 synthase, also called lactosylceramide α-2,3 sialyltransferase, resulted in infantile epileptic encephalopathy (MIM 609056) with severe developmental delay and blindness. ST3GAL5-mutated individuals displayed a complete lack of plasma GM3 and its downstream biosynthetic derivatives but had increased levels of plasma lactosylceramide and increased flux through the globoside and paragloboside pathways.18 Therefore, SPG26 resulting from B4GALNT1 mutations is the second ganglioside biosynthesis defect and is expected to lead to the accumulation of globotriaosylceramide and gangliosides G[M/D/T]3. Of note, another complex AR HSP, SPG46 (MIM 614409), is caused by loss-of-function mutations in GBA2 (MIM 609471), coding for an enzyme acting on glucosylceramides, substrates of lactosylceramides (Figure 3).8
Figure 3.
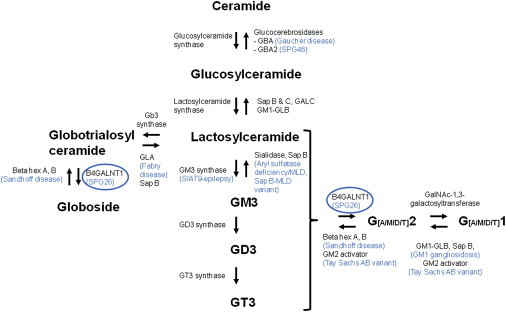
Simplified Representation of Ganglioside Metabolism and Their Related Disorders
Arrows indicate the orientation of the enzymatic reactions and the corresponding enzymes are indicated in black. Metabolic diseases are indicated in blue at the corresponding altered reaction. Ganglioside formation is performed in the endoplasmic reticulum and Golgi by successive glycosylations. Their degradation takes place in lysosomes. Abbreviations are as follows: hex, hexosaminidase; Gb3, globotrioaosylceramide; GBA, glucocerebrosidase; GD, disialic ganglioside; GALC, galactosylceramide-beta-galactosidase; GLA, alpha galactosidase; GLB, beta galactosidase; GM, monosialic ganglioside; GT, trisialic ganglioside; GT3 synthase, alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase; MLD, metachromatic leukodystrophy; Sap, saposin.
The clinical features of 18 affected individuals from the 7 SPG26 families identified in this study are summarized in Table S2. Gait difficulties occurred early, from 2 to 19 years of age. All subjects had predominant spasticity of the lower limbs (with variable spastic involvement of the upper limbs in 33%), increased reflexes, and bilateral Babinski sign. Pseudobulbar dysarthria was present in 47% (7/15). After a mean disease duration of 32.2 ± 19.5 years (range 1–60), four subjects were confined to a wheelchair or bedridden. Severity was variable: one was bedridden after 13 years (TUN34-11) whereas another was confined to a wheelchair after 47 years of disease evolution (FSP995-17). Lower motor neuron involvement was variably present, indicated by lower limb muscle wasting (11/18, 61%) at later disease stages, decrease or absence of lower limb tendon reflexes (11/18, 61%) during the course of the disease (as in family THI26004), and peripheral neuropathy predominantly of the axonal type in four families (7/11, 64%). Mild to moderate cognitive impairment and developmental delay was noted in all individuals, often preceding motor deficits. Cognitive dysfunction was noted to worsen with time in some subjects. Cerebellar signs (10/18, 55%) and extrapyramidal signs (8/18, 44%), such as facial dyskinesia and dystonia, were also noted. Further commonly observed signs and symptoms included bladder disturbances (6/18, 33%), peripherally decreased vibration sense (9/15, 60%), scoliosis (10/15, 67%), pes cavus (9/16, 56%), and strabismus (4/18, 22%). Four subjects had psychiatric features (the three German cases and one Brazilian individual). In addition, several signs were confined to one family: mild dysmorphic features in the Brazilian subjects and congenital posterior capsular cataract in the three Spanish individuals. Brain magnetic resonance imaging (MRI) was either normal or, after long disease durations, showed cortical atrophy (n = 5/10), subcortical atrophy (n = 1/10), and/or white matter hyperintensities (WMHs) on FLAIR sequence (n = 4/10) (Figure S2). In subject FSP995-19, WMHs worsen with time, as observed in two brain MRIs performed 12 years apart. We noted a slightly enlarged corpus callosum in 4/10 subjects; some of them with otherwise unremarkable MRIs. Finally, serum testosterone was reduced or at the lower limit in four of the six young men tested (67%), and one of them (IHG25297-105) had hypogonadism and delayed puberty.
Interestingly, the B4galnt1−/− mouse19 shows progressive gait disorder reminiscent of clinical features of SPG26. This phenotype could result from decreased central myelination in dorsal spinal cord and increased numbers of unmyelinated fibers,20,21 increased astrocyte/microglia proliferation, or alteration of lipid content in glycolipid-enriched membrane (raft) domains.22,23 Cerebellar granule cells of B4galnt1−/− mice are also more prone to degeneration in depolarizing conditions, very probably due to an alteration of calcium homeostasis.24 In addition, knockout males have progressive testicular atrophy with the presence of diffuse vacuoles in Sertoli cells and, despite almost normal testosterone levels per mg of testis, a severe reduction of serum testosterone.25 All these phenotypes could be due to the accumulation of globotriaosylceramide and simple gangliosides and/or the lack of complex gangliosides.19,24 Interestingly, GM3 accumulation has been reported in a simplex case with liver and brain disease.26,27 Whether the disease in this subject is the result of a mutation in B4GALNT1 remains unknown.
In conclusion, we used a combined mapping and exome sequencing approach to identify B4GALNT1 variants as the cause of an AR complicated form of HSP. This was achieved with 18 SPG26-affected individuals that belonged to 7 unrelated families from Europe, South America, and North Africa (indicating the world-wide distribution of this particular form of HSP). In accordance with previous reports,9 the phenotype of SPG26 consisted in early-onset and slowly progressive spastic paraparesis and cognitive impairment, complicated by cerebellar ataxia. We extended this clinical spectrum to include extrapyramidal involvement and peripheral neuropathy as well as cortical atrophy with white matter hyperintensities at brain MRI and low levels of testosterone in males. Disease progression seemed slow: most affected subjects were still able to walk after long disease durations but heterogeneity in disease progression was frequent among families. Altogether, SPG26 is the second human disorder of ganglioside biosynthesis. Abnormal glycosphingolipid profile may be detectable in peripheral samples from SPG26 subjects: this would hasten clinical diagnosis and facilitate monitoring in therapeutic trials as for other HSPs, such as SPG5A (MIM 270800) associated with 25- and 27-hydroxycholesterol accumulation28 and X-linked adrenoleukodystrophy (MIM 300100).
Acknowledgments
The authors are grateful to the family members who participated in this study, to M.A.M. Salih and M. Koenig for family referral, and to S. Trefouret and P. Ribai as well as the DNA and cell Bank of CRICM and the Centre National de Genotypage (Evry, France) for their help. This work was financially supported by the French-Tunisian Cooperation Project (to A. Brice and C.M.) led by INSERM (France) and DGRSRT (Tunisia), the VERUM Foundation (to A. Brice), the French Agency for Research (ANR) (to G.S. and A.D.), the Association Française contre les Myopathies (“LIGENAX” to G.S.), the Strumpell-Lorrain association (to the SPATAX network), the Deutsches Zentrum für Neurodegenerative Erkrankungen (to L.S.), the Interdisziplinären Zentrums für Klinische Forschung University of Tübingen (grant 1970-0-0 to R.S.), and the European Community with the ANR (“Eurospa” project to A. Brice and L.S.; 7th Framework Programme Neuromics to A. Brice). This study also benefited from funding from the program “Investissements d’avenir” ANR-10-IAIHU-06 (to the Brain and Spine Institute, Paris), a Canadian Institutes of Health Research grant (#119191) entitled “Emerging Team to identify and characterize novel and existing Hereditary Spastic Paraplegia (HSP) disease genes,” and a National Institutes of Health grant (R01NS072248 to S.Z.).
Contributor Information
Alexis Brice, Email: alexis.brice@upmc.fr.
Giovanni Stevanin, Email: giovanni.stevanin@upmc.fr.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Ensembl Genome Browser, http://www.ensembl.org/index.html
GEM.app, https://genomics.med.miami.edu
Mutalyzer, https://mutalyzer.nl/index
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Behan W.M., Maia M. Strümpell’s familial spastic paraplegia: genetics and neuropathology. J. Neurol. Neurosurg. Psychiatry. 1974;37:8–20. doi: 10.1136/jnnp.37.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stevanin G., Ruberg M., Brice A. Recent advances in the genetics of spastic paraplegias. Curr. Neurol. Neurosci. Rep. 2008;8:198–210. doi: 10.1007/s11910-008-0032-z. [DOI] [PubMed] [Google Scholar]
- 3.Fink J.K. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- 4.Schüle R., Schöls L. Genetics of hereditary spastic paraplegias. Semin. Neurol. 2011;31:484–493. doi: 10.1055/s-0031-1299787. [DOI] [PubMed] [Google Scholar]
- 5.Finsterer J., Löscher W., Quasthoff S., Wanschitz J., Auer-Grumbach M., Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 2012;318:1–18. doi: 10.1016/j.jns.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 6.Tesson C., Nawara M., Salih M.A.M., Rossignol R., Zaki M.S., Al Balwi M., Schule R., Mignot C., Obre E., Bouhouche A. Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am. J. Hum. Genet. 2012;91:1051–1064. doi: 10.1016/j.ajhg.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuurs-Hoeijmakers J.H.M., Geraghty M.T., Kamsteeg E.-J., Ben-Salem S., de Bot S.T., Nijhof B., van de Vondervoort I.I.G.M., van der Graaf M., Nobau A.C., Otte-Höller I., FORGE Canada Consortium Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2012;91:1073–1081. doi: 10.1016/j.ajhg.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin E., Schüle R., Smets K., Rastetter A., Boukhris A., Loureiro J.L., Gonzalez M.A., Mundwiller E., Deconinck T., Wessner M. Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am. J. Hum. Genet. 2013;92:238–244. doi: 10.1016/j.ajhg.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilkinson P.A., Simpson M.A., Bastaki L., Patel H., Reed J.A., Kalidas K., Samilchuk E., Khan R., Warner T.T., Crosby A.H. A new locus for autosomal recessive complicated hereditary spastic paraplegia (SPG26) maps to chromosome 12p11.1-12q14. J. Med. Genet. 2005;42:80–82. doi: 10.1136/jmg.2004.020172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. doi: 10.1016/s0140-6736(83)92879-9. [DOI] [PubMed] [Google Scholar]
- 11.Boukhris A., Feki I., Elleuch N., Miladi M.I., Boland-Augé A., Truchetto J., Mundwiller E., Jezequel N., Zelenika D., Mhiri C. A new locus (SPG46) maps to 9p21.2-q21.12 in a Tunisian family with a complicated autosomal recessive hereditary spastic paraplegia with mental impairment and thin corpus callosum. Neurogenetics. 2010;11:441–448. doi: 10.1007/s10048-010-0249-2. [DOI] [PubMed] [Google Scholar]
- 12.Stevanin G., Bouslam N., Thobois S., Azzedine H., Ravaux L., Boland A., Schalling M., Broussolle E., Dürr A., Brice A. Spinocerebellar ataxia with sensory neuropathy (SCA25) maps to chromosome 2p. Ann. Neurol. 2004;55:97–104. doi: 10.1002/ana.10798. [DOI] [PubMed] [Google Scholar]
- 13.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez M.A., Lebrigio R.F., Van Booven D., Ulloa R.H., Powell E., Speziani F., Tekin M., Schule R., Zuchner S. GEnomes Management Application (GEM.app): A New Software Tool for Large-Scale Collaborative Genome Analysis. Hum. Mutat. 2013;34:860–863. doi: 10.1002/humu.22305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu Y.-H., Barnes S., Sun Y., Grabowski G.A. Multi-system disorders of glycosphingolipid and ganglioside metabolism. J. Lipid Res. 2010;51:1643–1675. doi: 10.1194/jlr.R003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuji S., Choudary P.V., Martin B.M., Stubblefield B.K., Mayor J.A., Barranger J.A., Ginns E.I. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher’s disease. N. Engl. J. Med. 1987;316:570–575. doi: 10.1056/NEJM198703053161002. [DOI] [PubMed] [Google Scholar]
- 17.Cox T.M. Gaucher disease: understanding the molecular pathogenesis of sphingolipidoses. J. Inherit. Metab. Dis. 2001;24(Suppl 2):106–121. doi: 10.1023/a:1012496514170. discussion 87–88. [DOI] [PubMed] [Google Scholar]
- 18.Simpson M.A., Cross H., Proukakis C., Priestman D.A., Neville D.C.A., Reinkensmeier G., Wang H., Wiznitzer M., Gurtz K., Verganelaki A. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet. 2004;36:1225–1229. doi: 10.1038/ng1460. [DOI] [PubMed] [Google Scholar]
- 19.Takamiya K., Yamamoto A., Furukawa K., Yamashiro S., Shin M., Okada M., Fukumoto S., Haraguchi M., Takeda N., Fujimura K. Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc. Natl. Acad. Sci. USA. 1996;93:10662–10667. doi: 10.1073/pnas.93.20.10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheikh K.A., Sun J., Liu Y., Kawai H., Crawford T.O., Proia R.L., Griffin J.W., Schnaar R.L. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc. Natl. Acad. Sci. USA. 1999;96:7532–7537. doi: 10.1073/pnas.96.13.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma Q., Kobayashi M., Sugiura M., Ozaki N., Nishio K., Shiraishi Y., Furukawa K., Furukawa K., Sugiura Y. Morphological study of disordered myelination and the degeneration of nerve fibers in the spinal cord of mice lacking complex gangliosides. Arch. Histol. Cytol. 2003;66:37–44. doi: 10.1679/aohc.66.37. [DOI] [PubMed] [Google Scholar]
- 22.Ohmi Y., Tajima O., Ohkawa Y., Yamauchi Y., Sugiura Y., Furukawa K., Furukawa K. Gangliosides are essential in the protection of inflammation and neurodegeneration via maintenance of lipid rafts: elucidation by a series of ganglioside-deficient mutant mice. J. Neurochem. 2011;116:926–935. doi: 10.1111/j.1471-4159.2010.07067.x. [DOI] [PubMed] [Google Scholar]
- 23.Ohmi Y., Ohkawa Y., Yamauchi Y., Tajima O., Furukawa K., Furukawa K. Essential roles of gangliosides in the formation and maintenance of membrane microdomains in brain tissues. Neurochem. Res. 2012;37:1185–1191. doi: 10.1007/s11064-012-0764-7. [DOI] [PubMed] [Google Scholar]
- 24.Wu G., Xie X., Lu Z.H., Ledeen R.W. Cerebellar neurons lacking complex gangliosides degenerate in the presence of depolarizing levels of potassium. Proc. Natl. Acad. Sci. USA. 2001;98:307–312. doi: 10.1073/pnas.011523698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takamiya K., Yamamoto A., Furukawa K., Zhao J., Fukumoto S., Yamashiro S., Okada M., Haraguchi M., Shin M., Kishikawa M. Complex gangliosides are essential in spermatogenesis of mice: possible roles in the transport of testosterone. Proc. Natl. Acad. Sci. USA. 1998;95:12147–12152. doi: 10.1073/pnas.95.21.12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Max S.R., Maclaren N.K., Brady R.O., Bradley R.M., Rennels M.B., Tanaka J., Garcia J.H., Cornblath M. GM3 (hematoside) sphingolipodystrophy. N. Engl. J. Med. 1974;291:929–931. doi: 10.1056/NEJM197410312911802. [DOI] [PubMed] [Google Scholar]
- 27.Fishman P.H., Max S.R., Tallman J.F., Brady R.O., Maclaren N.K., Cornblath M. Deficient Ganglioside Biosynthesis: a novel human sphingolipidosis. Science. 1975;187:68–70. doi: 10.1126/science.803227. [DOI] [PubMed] [Google Scholar]
- 28.Schüle R., Siddique T., Deng H.-X., Yang Y., Donkervoort S., Hansson M., Madrid R.E., Siddique N., Schöls L., Björkhem I. Marked accumulation of 27-hydroxycholesterol in SPG5 patients with hereditary spastic paresis. J. Lipid Res. 2010;51:819–823. doi: 10.1194/jlr.M002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.