

Abstract
SHORT syndrome is a rare, multisystem disease characterized by short stature, anterior-chamber eye anomalies, characteristic facial features, lipodystrophy, hernias, hyperextensibility, and delayed dentition. As part of the FORGE (Finding of Rare Disease Genes) Canada Consortium, we studied individuals with clinical features of SHORT syndrome to identify the genetic etiology of this rare disease. Whole-exome sequencing in a family trio of an affected child and unaffected parents identified a de novo frameshift insertion, c.1906_1907insC (p.Asn636Thrfs∗18), in exon 14 of PIK3R1. Heterozygous mutations in exon 14 of PIK3R1 were subsequently identified by Sanger sequencing in three additional affected individuals and two affected family members. One of these mutations, c.1945C>T (p.Arg649Trp), was confirmed to be a de novo mutation in one affected individual and was also identified and shown to segregate with the phenotype in an unrelated family. The other mutation, a de novo truncating mutation (c.1971T>G [p.Tyr657∗]), was identified in another affected individual. PIK3R1 is involved in the phosphatidylinositol 3 kinase (PI3K) signaling cascade and, as such, plays an important role in cell growth, proliferation, and survival. Functional studies on lymphoblastoid cells with the PIK3R1 c.1906_1907insC mutation showed decreased phosphorylation of the downstream S6 target of the PI3K-AKT-mTOR pathway. Our findings show that PIK3R1 mutations are the major cause of SHORT syndrome and suggest that the molecular mechanism of disease might involve downregulation of the PI3K-AKT-mTOR pathway.
Main Text
SHORT syndrome (MIM 269880) is a rare disorder characterized by short stature, hyperextensibility of joints and/or hernias, ocular depression, Rieger anomaly, and delays of tooth eruption.1 Although these features provide the condition’s acronym, they do not capture the full range of clinical features, which can include a recognizable facial gestalt (triangular facies, lack of facial fat, and hypoplastic nasal alae with overhanging columella), a near universal partial lipodystrophy, insulin resistance, nephrophrocalcinosis, and hearing deficits, among many others.2–7 Notably, both developmental milestones and cognition are normal for individuals with SHORT syndrome.3
The first description of SHORT syndrome was of a sibling pair whose parents displayed no obvious features.1 This has been followed by several reports of sporadic occurrences, suggesting an autosomal-recessive or de novo dominant mode of inheritance.6 However, there have also been several reports of parent-child transmissions, including male-to-male transmission, consistent with an autosomal-dominant inheritance pattern for SHORT syndrome.3,4,8,9 The majority of affected individuals still appear to be simplex cases,3 suggesting a significant contribution for de novo dominant mutations.
Specific genes have been suggested to play a role in the etiology of SHORT syndrome. PITX2 (MIM 601542) was highlighted in an individual with Rieger anomaly and syndromic features including lipoatrophy, hyperextensibility, a ventricular septal heart defect, and dysmorphic facial features.10 This individual had a familial chromosomal translocation involving PITX2.10 Another individual with eye anomalies and short stature was found by microarray to have a large deletion spanning PITX2 and several other genes.11 BMP4 (MIM 112262) is associated with microphthalmia and has been reported to be deleted, along with 14 other genes at 14q22.2, in a single individual diagnosed with SHORT syndrome.12 Although they appear to have a syndrome related to Axenfield-Rieger anomaly, affected individuals with PITX2 and BMP4 mutations10,12 do not have the characteristic facial gestalt shared by the individuals in the original and subsequent descriptions of SHORT syndrome.1–4,6,7,13
The FORGE (Finding of Rare Disease Genes) Canada Consortium is a collaborative project with the goal of identifying genetic mutations for rare childhood diseases.14–16 We ascertained individuals with SHORT syndrome by contacting the members of the FORGE Canada Consortium and selected international colleagues and asking whether the physicians were aware of any individual(s) diagnosed with SHORT syndrome. Affected individuals and family members were recruited from Medical Genetics Clinics in North America, Israel, and the United Kingdom. Approval of the study design was obtained from the institutional research ethics board at the Children’s Hospital of Eastern Ontario, and free and informed consent was obtained from each study subject (or parent, if appropriate) prior to enrollment. Each individual was assessed by a medical geneticist, ophthalmologist, and/or pediatrician. Affected individuals 2 and 5 were previously published as cases of SHORT syndrome with associated nephrocalcinosis.5 The clinical description of the affected individuals is presented in Table 1. DNA was extracted according to standard protocols. Paternity was confirmed by the genotyping of nine polymorphic simple-tandem-repeat markers.
Table 1.
Clinical Characteristics of Individuals with SHORT Syndrome
| Affected Individual 1 | Affected Individual 2 | Affected Individual 3 | Affected Individual 4 | Affected Individual 5 | Affected Individual 6 | Affected Individual 7 | |
|---|---|---|---|---|---|---|---|
| Age at assessment | 2 years | 10 years | 10.4 years | birth to 18 years | 4 years | 32 years | at birth |
| Gender | female | male | male | male | male | female | male |
| PIK3R1 mutation | c.1906_1907insC | - | c.1971T>G | c.1945C>T | c.1945C>T | c.1945C>T | c.1945C>T |
| Growth | |||||||
| Birth weight | 1.556 kg at 365 weeks | 0.760 kg at 32 weeks | 2.22 kg at 40 weeks | 2.52 kg at 38 weeks | 2.27 kg at term | 2.55 kg at term | 2.18 kg at term |
| Height (percentile) | 76.5 cm (<3rd) | 120 cm (<3rd) | 126 cm (3rd) | 132.5 cm at 11 years (5th); 160 cm at 18 years (<3rd) | 122.3 cm at 11 years (<3rd) | 148.5 cm (<3rd) | 45 cm |
| Weight (percentile) | 8 kg (<3rd) | not reported | 21.8 kg (<3rd) | 23.5 kg at 11 years (<3rd); 43 kg at 18 years (<3rd) | 17.9 kg at 11 years (<3rd) | 44 kg (3rd) | 2.18 kg |
| Head circumference (percentile) | 43 cm (<3rd) | not reported | 51.5 cm (25th) | 45.5 cm at 20 months (3rd) | 49 cm (25th) | 49.5 cm (<3rd) | 34 cm |
| Bone age | normal | delayed by 2.5 years | delayed by 24 months | delayed | not reported | not reported | not reported |
| Connective Tissue | |||||||
| Hyperextensibility | no | yes; especially small joints | yes; especially small joints | no | none reported | no | NA |
| Hernias | no | inguinal hernia | no | no | none reported | no | no |
| Facies | |||||||
| Triangular facies | yes | yes | yes | yes | yes | yes | yes |
| Ocular depression | yes | yes | yes | yes | yes | yes | yes |
| Aged appearance | yes | yes | yes | yes | yes | to hands | to hands |
| Dimple | yes | no | no | yes | yes | no | no |
| Hypoplastic ala nasi | yes | yes | yes | yes | yes | yes | yes |
| Eye Anomalies | |||||||
| Rieger anomaly | right optic-pit anomaly | right-sided; temporal; posterior; embryotoxin | not typical | possible | no | no | no |
| Glaucoma | no | no | no | no | no | no | no |
| Other anterior-chamber anomalies | no | no | atypical irises with hypoplastic appearance on transillumination; poorly dilating, constricting pupils | no | iridogoniodysgenesis | hypoplasia of iris stroma on transillumination | no |
| Dental | |||||||
| Teething delays or other | yes | yes; fusion of central and lateral incisors | delayed eruption of teeth; overcrowded teeth with micrognathia | yes | not reported | delays with primary dentition; crowding | NA |
| Other Common Features | |||||||
| Development and cognition | normal | normal range | speech delay | normal | normal | normal | normal |
| Insulin resistance or diabetes | no | no | no | no | no | no | no |
| Lipodystophy | yes | yes | yes | yes | no | hands | hands |
| Hearing loss | no | no | no | yes | no | no | unilateral |
| Radiological features | none reported | left acetabular dysplasia; right Perthes disease | slender, narrow iliac wings; slender long bones; “drumstick” distal phalanges; narrow upper thorax | none reported | none reported | none reported | none reported |
| Other | - | hypercalciuria; ASD; VSD; patent ductus arteriosus | vitiligo since age 7 years; poor response to growth hormone; behavioral problems | - | VSD; anorectal atresia; nephrocalcinosis; neuropathic bladder | - | - |
| Previously reported in the literature | no | yes5 | no | no | yes5 | yes8 | yes8 |
The following abbreviations are used: NA, not applicable; ASD, atrial septal defect; and VSD, ventricular septal defect.
We performed exome capture and high-throughput sequencing of samples for affected individual 1 and her two unaffected parents (trio design) and for affected individual 2 (simplex case). Target enrichment for the samples was performed with the Agilent SureSelect 50 Mb (V3) All Exon Kit. Sequencing (Illumina HiSeq) generated >14 Gbp of 100 bp paired-end reads per sample. Read alignment, variant calling, and annotation were done as for previous FORGE projects17 with a pipeline based on the Burrows-Wheeler Aligner (BWA), Picard, ANNOVAR, and custom annotation scripts. We removed adaptor sequences and quality trimmed reads by using the FASTX-Toolkit and then used a custom script to ensure that only read pairs with both mates present were subsequently used. Reads were aligned to UCSC Genome Browser hg19 with BWA 0.5.9,18 and indel realignment was done with the Genome Analysis Toolkit (GATK).19 Duplicate reads were then marked with Picard and excluded from downstream analyses. We assessed coverage of Consensus Coding Sequence (CCDS) bases by using the GATK, which showed that all samples had >92% of CCDS bases covered by at least 20 reads. For each sample, single-nucleotide variants (SNVs) and short insertions and deletions (indels) were called with SAMtools pileup20 with the extended base-alignment-quality adjustment (-E) and were then quality filtered so that at least 20% of reads supported the variant call. We annotated variants by using both ANNOVAR21 and custom scripts to identify whether they affect protein-coding sequence and whether they are present in dbSNP132, the 1000 Genomes data set (2012/04 release), the 6,500 exomes in the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server (data downloaded October 3, 2012; see Web Resources), or in approximately 800 exomes previously sequenced at the McGill University and Genome Quebec Innovation Centre.
First, we analyzed the data from affected individual 1 and her unaffected parents. Two de novo nonsynonymous variants were detected in the affected individual, and they were found to be absent in both parents. The first, SNV c.3656C>G (p.Ser1219Cys) (RefSeq accession number NM_001164496.1) in WDR52, is predicted to be a polymorphism by MutationTaster (see Web Resources). The second, frameshift insertion c.1906_1907insC (p.Asn636Thrfs∗18) (RefSeq NM_181523.2) in exon 14 of PIK3R1, generates a stop codon in the region encoding amino acid residue 654 and is expected to produce premature truncation of p85α, the longest protein isoform encoded by PIK3R1. No genes with two obvious deleterious mutations—one inherited from each parent—were detected.
Next, we assessed the sequencing results of affected individual 2 for any rare variants present in a heterozygous state. No variants were seen in PIK3R1 or WDR52 for affected individual 2; several variants were highlighted as rare and were confirmed by Sanger sequencing. All were found to be inherited and hence were unlikely candidate(s) for causing SHORT syndrome in this affected individual. There was excellent coverage for PIK3R1 (>50× across all exons) and no evidence of any structural variant. PIK3R1 was investigated by Sanger sequencing in affected individuals 3–5. In exon 14 of PIK3R1, affected individual 3 was found to have a nonsense mutation (c.1971T>G [p.Tyr657∗]) absent in both parents, and affected individual 4 was found to have a missense mutation (c.1945C>T [p.Arg649Trp]) absent in both parents and predicted to be damaging by PolyPhen. Affected individual 5,5 as well as his affected mother and brother,8 has been previously described. These three affected individuals were found to have the same missense mutation, c.1945C>T (p.Arg649Trp), as affected individual 4, and this recurrent mutation occurs within the context of a CpG motif. These mutations are not represented in dbSNP131, 1000 Genomes, or the NHLBI Exome Variant Server. The observation of rare mutations in the same exon of PIK3R1 in four of five affected individuals, the recurrence of the same mutation in two unrelated, affected individuals, and a consistent inheritance pattern in a family and de novo cases together provide strong genetic evidence that we have identified the molecular cause of SHORT syndrome.
The identification of a disease-causing mutation in a disorder such as SHORT syndrome can be a difficult task given the rarity of affected individuals and their varied clinical presentations. Despite the similar clinical presentation, we did not identify a PIK3R1 mutation in affected individual 2. All the affected individuals included in our study display features of SHORT syndrome (Table 1). Re-evaluation of the phenotype of affected individual 2 confirmed that he shows several of the core features of SHORT syndrome (short stature, inguinal hernias, delayed dentition, a lack of subcutaneous fat, and an anterior-chamber ocular defect; Table 1). However, his facial features appear distinct from those of the mutation-positive individuals (Figure 1G). The key facial features present in the mutation-positive individuals include a triangular face, deep-set eyes, hypoplasic alae nasae, low-hanging columella, and a downturned mouth (Figures 1A–1F). It appears that individual 2 does not have SHORT syndrome given the lack of a PIK3R1 mutation and the recognizable facial features. This suggests that SHORT syndrome is a highly specific diagnosis and that it relies heavily on the facial gestalt.
Figure 1.
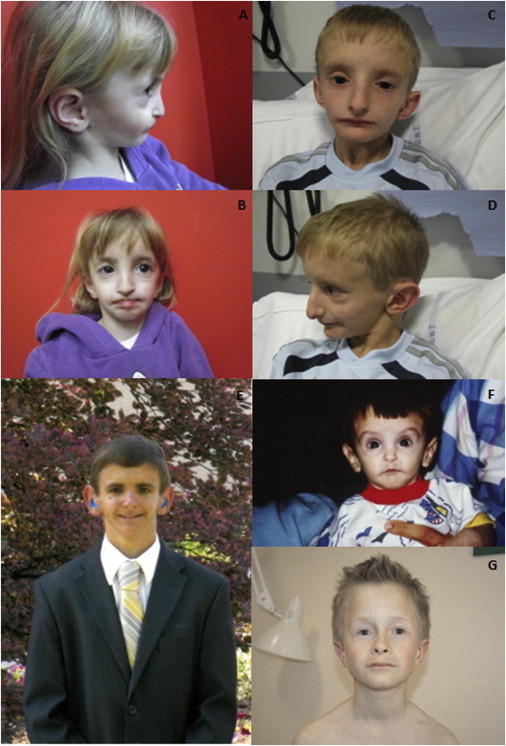
Facial Features of Individuals Clinically Diagnosed with SHORT Syndrome
(A–F) Photographs of affected individuals 1 (A and B), 3 (C and D), and 4 (E and F) capture the core facial gestalt, including the broad triangular facies, deep-set eyes, hypoplastic alae nasae, low-hanging columella, and downturned mouth.
(G) Photograph of individual 2, who has a diagnosis of SHORT syndrome.5 This individual has several features of SHORT syndrome; however, he appears to lack the facial features of the individuals in (A)–(F). Specifically, his face does not appear to be as triangular, and his eyes are not as deep set. His columella is not low hanging, and the ala nasi are not overly hypoplastic. His mouth is not downturned, and he does not have obvious micrognathia. This image was reproduced with permission from John Wiley & Sons (license number 3112610845148).
PIK3R1 encodes the regulatory subunit of PI3K, specifically the predominant regulatory subunit p85α, as well as the lesser isoforms p50α and p55α.22 The latter two isoforms are localized in the liver and skeletal muscle, respectively.23 p85α is ubiquitous and binds to PI3K’s catalytic subunit (p110α), negatively regulating its kinase activity. In order for p85α to perform its regulatory role, it first requires its Src homology 2 (SH2) domains to be phosphorylated. This can occur by its binding to insulin-responsive substrate (IRS) proteins, by its direct binding to a receptor tyrosine kinase, or by its phosphorylation via the RAS-GTP pathway.24 There are two SH2 domains in PI3K, and the amino acid alterations identified in the individuals with SHORT syndrome are located in the C-terminal SH2 domain (Figure 2). This SH2 domain is also present in the p50α and p55α isoforms. A lymphoblastoid cell line (LCL) was able to be established from affected individual 1 (with c.1906_1907insC [p.Asn636Thrfs∗18]) for functional analysis. LCLs were not available for affected individuals 3 or 4 or for individuals from the family of individual 5. Immunoblot analysis showed that the levels of the p85α-specific isoform were approximately 50% lower in extracts than in control samples; in addition, extracts from individuals with hemimegalencephaly caused by activating mutations in PIK3R2 and PIK3CA were available, and the findings were consistent with a heterozygous truncating PIK3R1 mutation (Figure 3). Further, immunoblot analysis using a pan-p85 antisera uniquely identified a smaller product in LCL extracts of affected individual 1; they were consistent in size with the expected truncation due to p.Asn636Thrfs∗18 (Figure 3). Levels of the p110α PI3K catalytic subunit appeared normal, in keeping with the costabilizing interaction between p110α and p85α (Figure 3). Interestingly, we found that levels of phospho-S6 (Ser240 and Ser244) were lower in extracts from the LCLs of affected individual 1 than in those from control LCLs, suggestive of suppressed signal transduction in the PI3K-AKT-mTOR pathway (Figure 3).
Figure 2.
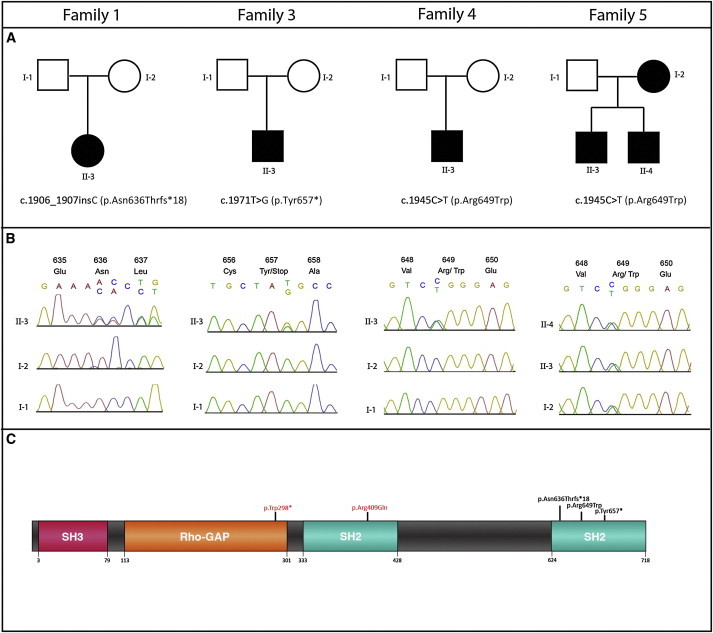
PIK3R1 Mutations in Families Affected by SHORT Syndrome
(A) Family pedigrees and the heterozygous mutations identified in PIK3R1 (RefSeq NM_181523.2).
(B) Electropherograms showing the mutation position in the father (top), mother (middle), and affected child (bottom). That of family 5 shows the mutation position in the mother, brother, and proband.
(C) A schematic of PI3K demonstrates the position of the substitutions and truncations in the SH2 domains of the protein. The alterations in red are those from the literature25,26 and are not associated with SHORT syndrome.
Figure 3.
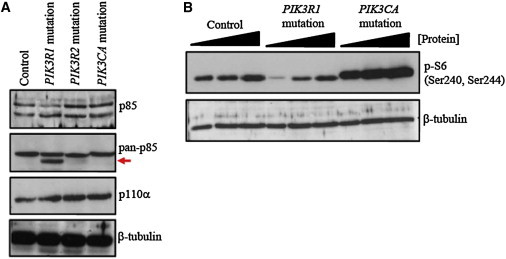
Immunoblot Analysis from LCL Extracts Derived from Individuals with SHORT Syndrome and Hemimegalencephaly
(A) The upper panel shows p85α-specific levels (top band) in extracts from the control, individual 1 (c.1906_1907insC [p.Asn636Thrfs∗18]), and two previously described individuals with hemimegalencephaly. The latter two individuals exhibit hyperactivating mutations in PIK3R2 (c.1117G>A [p.Gly373Arg]), encoding the p85β regulatory subunit of PI3K, and PIK3CA (c.1359_1361del [p.Glu453∗]), encoding the p110α catalytic subunit of PI3K.27 The middle panel shows the results of using a pan-p85 antibody capable of detecting the p85α isoform (encoded by PIK3R1), as well as p85β (encoded by PIK3R2) and p55γ (encoded by PIK3R3). A truncated product (red arrow) was detected in extracts from individual 1 only (p.Asn636Thrfs∗18). Both p85α and p110α directly interact and costabilize. The lower panel shows a band corresponding to p110α in LCLs from individual 1, potentially suggestive of stable interaction.
(B) Phosphorylation of S6 (by S6 kinase), which is directly activated by mTOR, is a terminal readout of PI3K-AKT-mTOR-pathway activity. Titration of whole-cell extract shows that compared to control extracts, those from individual 1 exhibit reduced S6 phosphorylation on serine 240 and 244. Cells from an individual with a PIK3CA mutation and hemimegalencephaly show elevated S6 phosphorylation, as previously demonstrated.27 These data are suggestive of diminished signaling through this pathway in the LCLs of individual 1.
Another alteration that has been described in PI3K is a substitution disrupting the N-terminal SH2 domain in an individual with acanthosis nigrecans and severe insulin resistance.25,28 Recently, there has been a report of an individual with agammaglobulinemia, absent B lineage cells, and a history of colitis and absent p85α resulting from a mutation in exon 6 of PIK3R1.26 However, isoforms p50α and p55α were still transcribed because the mutation was upstream of the p50α and p55α promoter site.26 These findings suggest phenotypic heterogeneity for mutations within this gene.
How mutations in PIK3R1 result in the multisystem phenotype of SHORT syndrome remains unknown. Although insulin resistance might be hypothesized to be an upstream consequence of impaired PI3K activation, it is not universal among individuals with SHORT syndrome and is not a feature of the four affected individuals reported here, although this feature might develop with time. Mice with a targeted deletion of Pik3r1 show a paradoxical increase in insulin sensitivity and episodes of hypoglycemia.29 Importantly, it was observed that the other isoforms (e.g., p50α) encoded by Pik3r1 were upregulated in these mice and most likely caused the disturbance in insulin homeostasis.29 Deletion of all isoforms (p85α, p55α, and p50α) also results in hypoglycemia, liver necrosis, and perinatal lethality in homozygous mice, suggesting that the p85α isoform is necessary for normal development.23
Upon activation of cell-surface receptors, PI3K catalyzes the phosphorylation of phosphatidylinositol (4,5) bis-phosphate (PtdIns(4,5)P2) and generates PtdIns (3,4,5)P3, an important lipid messenger that stimulates the localized surface membrane recruitment of important downstream kinases, including PDK1 and AKT, as part of the signal transduction cascade. PTEN phosphatase, a well-studied tumor suppressor, acts in opposition to PI3K by converting PtdIns (3,4,5)P3to PtdIns(4,5)P2 to finely regulate the amplitude and duration of signaling.30 Loss of PTEN function results in the inappropriate activation of the AKT-mTOR pathway.30 Constitutional mutations in PTEN (MIM 601728) are seen in disorders such as Cowden syndrome (MIM 158350) and Banayan-Riley-Ruvulcaba syndrome (MIM 153480), which are characterized by overgrowth and a predisposition to malignancy. More recently mosaic or germline mutations resulting in hyperactivation of this pathway have been implicated in Proteus syndrome (MIM 176920), megalencephaly-capillary malformation syndrome31 (MIM 602501), megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome31 (MIM 603387), and isolated hemimegalencephaly.27,31 Described here, the preliminary analysis of a single case-derived LCL with a heterozygous truncating mutation in PIK3R1 suggests that the mechanism underlying SHORT syndrome operates in an opposite manner to the above examples, given that the spontaneously reduced phosphorylation of S6 seems to indicate that there is diminished function of the AKT-mTOR pathway (Figure 3). Interestingly, constitutionally reduced signaling by the PI3K-AKT-mTOR pathway, as demonstrated in conditions such as Donohue syndrome (MIM 246200), and various knockout mouse models of this pathway are associated with impaired cell division and growth retardation. Although further analysis of cell lines from other individuals with SHORT syndrome will be required for fully ascertaining the impact of PIK3R1 mutations on the function of the PI3K-AKT-mTOR pathway, our analysis here of a single cell line provides a working paradigm.
In summary, we have shown that dominant mutations in PIK3R1 cause SHORT syndrome, a recognizable malformation syndrome. The findings also suggest that the molecular mechanism of the disease might involve downregulation of the PI3K-AKT-mTOR pathway, which is important for cellular proliferation and growth. It is possible that dysregulation of other members of this complex pathway will result in similar, overlapping phenotypes.
Acknowledgments
The authors would firstly like to thank the study participants and their families—without their participation, this work would not have been possible. This work was funded by the Government of Canada through Genome Canada, the Canadian Institutes of Health Research (CIHR), and the Ontario Genomics Institute (OGI-049). Additional funding was provided by Genome Quebec and Genome British Columbia. M.O.D. is a Senior Cancer Research UK (CR-UK) Fellow whose lab is supported by CR-UK, the Medical Research Council (UK), and Leukaemia Lymphoma Research (UK). A.C.S. is supported by a CIHR postdoctoral fellowship. D.A.D and K.M.B are the recipients of a CIHR Clinical Investigator award from the Institute of Genetics. This work was selected for study by the FORGE Canada Steering Committee, consisting of K.M.B., J. Friedman, J. Michaud, F. Bernier, M. Brudno, B. Fernandez, B. Knoppers, M. Samuels, and S. Scherer.
Contributor Information
David A. Dyment, Email: ddyment@cheo.on.ca.
A. Micheil Innes, Email: micheil.innes@alberthealthservices.ca.
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants (DGV), http://projects.tcag.ca/variation/
FASTX-Toolkit, http://hannonlab.cshl.edu/fastx_toolkit/
MutationTaster, http://www.mutationtaster.org
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Picard, http://picard.sourceforge.net/
SAMtools, http://samtools.sourceforge.net/
References
- 1.Gorlin R.J., Cervenka J., Moller K., Horrobin M., Witkop C.J., Jr. Malformation syndromes. A selected miscellany. Birth Defects Orig. Artic. Ser. 1975;11:39–50. [PubMed] [Google Scholar]
- 2.Lipson A.H., Cowell C., Gorlin R.J. The SHORT syndrome: further delineation and natural history. J. Med. Genet. 1989;26:473–475. doi: 10.1136/jmg.26.7.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koenig R., Brendel L., Fuchs S. SHORT syndrome. Clin. Dysmorphol. 2003;12:45–49. doi: 10.1097/00019605-200301000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Aarskog D., Ose L., Pande H., Eide N. Autosomal dominant partial lipodystrophy associated with Rieger anomaly, short stature, and insulinopenic diabetes. Am. J. Med. Genet. 1983;15:29–38. doi: 10.1002/ajmg.1320150104. [DOI] [PubMed] [Google Scholar]
- 5.Reardon W., Temple I.K. Nephrocalcinosis and disordered calcium metabolism in two children with SHORT syndrome. Am. J. Med. Genet. A. 2008;146A:1296–1298. doi: 10.1002/ajmg.a.32250. [DOI] [PubMed] [Google Scholar]
- 6.Brodsky M.C., Whiteside-Michel J., Merin L.M. Rieger anomaly and congenital glaucoma in the SHORT syndrome. Arch. Ophthalmol. 1996;114:1146–1147. doi: 10.1001/archopht.1996.01100140348022. [DOI] [PubMed] [Google Scholar]
- 7.Schwingshandl J., Mache C.J., Rath K., Borkenstein M.H. SHORT syndrome and insulin resistance. Am. J. Med. Genet. 1993;47:907–909. doi: 10.1002/ajmg.1320470619. [DOI] [PubMed] [Google Scholar]
- 8.Bankier A., Keith C.G., Temple I.K. Absent iris stroma, narrow body build and small facial bones: a new association or variant of SHORT syndrome? Clin. Dysmorphol. 1995;4:304–312. doi: 10.1097/00019605-199510000-00005. [DOI] [PubMed] [Google Scholar]
- 9.Sorge G., Ruggieri M., Polizzi A., Scuderi A., Di Pietro M. SHORT syndrome: a new case with probable autosomal dominant inheritance. Am. J. Med. Genet. 1996;61:178–181. doi: 10.1002/(SICI)1096-8628(19960111)61:2<178::AID-AJMG16>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 10.Karadeniz N.N., Kocak-Midillioglu I., Erdogan D., Bökesoy I. Is SHORT syndrome another phenotypic variation of PITX2? Am. J. Med. Genet. A. 2004;130A:406–409. doi: 10.1002/ajmg.a.30206. [DOI] [PubMed] [Google Scholar]
- 11.Lines M.A., Kozlowski K., Kulak S.C., Allingham R.R., Héon E., Ritch R., Levin A.V., Shields M.B., Damji K.F., Newlin A., Walter M.A. Characterization and prevalence of PITX2 microdeletions and mutations in Axenfeld-Rieger malformations. Invest. Ophthalmol. Vis. Sci. 2004;45:828–833. doi: 10.1167/iovs.03-0309. [DOI] [PubMed] [Google Scholar]
- 12.Reis L.M., Tyler R.C., Schilter K.F., Abdul-Rahman O., Innis J.W., Kozel B.A., Schneider A.S., Bardakjian T.M., Lose E.J., Martin D.M. BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum. Genet. 2011;130:495–504. doi: 10.1007/s00439-011-0968-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haan E., Morris L. SHORT syndrome: distinctive radiographic features. Clin. Dysmorphol. 1998;7:103–107. doi: 10.1097/00019605-199804000-00004. [DOI] [PubMed] [Google Scholar]
- 14.Majewski J., Schwartzentruber J.A., Caqueret A., Patry L., Marcadier J., Fryns J.P., Boycott K.M., Ste-Marie L.G., McKiernan F.E., Marik I., FORGE Canada Consortium Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum. Mutat. 2011;32:1114–1117. doi: 10.1002/humu.21546. [DOI] [PubMed] [Google Scholar]
- 15.Bernier F.P., Caluseriu O., Ng S., Schwartzentruber J., Buckingham K.J., Innes A.M., Jabs E.W., Innis J.W., Schuette J.L., Gorski J.L., FORGE Canada Consortium Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am. J. Hum. Genet. 2012;90:925–933. doi: 10.1016/j.ajhg.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hood R.L., Lines M.A., Nikkel S.M., Schwartzentruber J., Beaulieu C., Nowaczyk M.J., Allanson J., Kim C.A., Wieczorek D., Moilanen J.S., FORGE Canada Consortium Mutations in SRCAP, encoding SNF2-related CREBBP activator protein, cause Floating-Harbor syndrome. Am. J. Hum. Genet. 2012;90:308–313. doi: 10.1016/j.ajhg.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srour M., Hamdan F.F., Schwartzentruber J.A., Patry L., Ospina L.H., Shevell M.I., Désilets V., Dobrzeniecka S., Mathonnet G., Lemyre E., FORGE Canada Consortium Mutations in TMEM231 cause Joubert syndrome in French Canadians. J. Med. Genet. 2012;49:636–641. doi: 10.1136/jmedgenet-2012-101132. [DOI] [PubMed] [Google Scholar]
- 18.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanhaesebroeck B., Leevers S.J., Panayotou G., Waterfield M.D. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem. Sci. 1997;22:267–272. doi: 10.1016/s0968-0004(97)01061-x. [DOI] [PubMed] [Google Scholar]
- 23.Fruman D.A., Mauvais-Jarvis F., Pollard D.A., Yballe C.M., Brazil D., Bronson R.T., Kahn C.R., Cantley L.C. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85 alpha. Nat. Genet. 2000;26:379–382. doi: 10.1038/81715. [DOI] [PubMed] [Google Scholar]
- 24.Taniguchi C.M., Emanuelli B., Kahn C.R. Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 25.Baynes K.C., Beeton C.A., Panayotou G., Stein R., Soos M., Hansen T., Simpson H., O’Rahilly S., Shepherd P.R., Whitehead J.P. Natural variants of human p85 alpha phosphoinositide 3-kinase in severe insulin resistance: a novel variant with impaired insulin-stimulated lipid kinase activity. Diabetologia. 2000;43:321–331. doi: 10.1007/s001250050050. [DOI] [PubMed] [Google Scholar]
- 26.Conley M.E., Dobbs A.K., Quintana A.M., Bosompem A., Wang Y.D., Coustan-Smith E., Smith A.M., Perez E.E., Murray P.J. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. J. Exp. Med. 2012;209:463–470. doi: 10.1084/jem.20112533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J.H., Huynh M., Silhavy J.L., Kim S., Dixon-Salazar T., Heiberg A., Scott E., Bafna V., Hill K.J., Collazo A. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet. 2012;44:941–945. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lappalainen I., Thusberg J., Shen B., Vihinen M. Genome wide analysis of pathogenic SH2 domain mutations. Proteins. 2008;72:779–792. doi: 10.1002/prot.21970. [DOI] [PubMed] [Google Scholar]
- 29.Terauchi Y., Tsuji Y., Satoh S., Minoura H., Murakami K., Okuno A., Inukai K., Asano T., Kaburagi Y., Ueki K. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85 alpha subunit of phosphoinositide 3-kinase. Nat. Genet. 1999;21:230–235. doi: 10.1038/6023. [DOI] [PubMed] [Google Scholar]
- 30.Shi Y., Paluch B.E., Wang X., Jiang X. PTEN at a glance. J. Cell Sci. 2012;125:4687–4692. doi: 10.1242/jcs.093765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rivière J.B., Mirzaa G.M., O’Roak B.J., Beddaoui M., Alcantara D., Conway R.L., St-Onge J., Schwartzentruber J.A., Gripp K.W., Nikkel S.M., Finding of Rare Disease Genes (FORGE) Canada Consortium De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]