

Abstract
Myopathies are a clinically and etiologically heterogeneous group of disorders that can range from limb girdle muscular dystrophy (LGMD) to syndromic forms with associated features including intellectual disability. Here, we report the identification of mutations in transport protein particle complex 11 (TRAPPC11) in three individuals of a consanguineous Syrian family presenting with LGMD and in five individuals of Hutterite descent presenting with myopathy, infantile hyperkinetic movements, ataxia, and intellectual disability. By using a combination of whole-exome or genome sequencing with homozygosity mapping, we identified the homozygous c.2938G>A (p.Gly980Arg) missense mutation within the gryzun domain of TRAPPC11 in the Syrian LGMD family and the homozygous c.1287+5G>A splice-site mutation resulting in a 58 amino acid in-frame deletion (p.Ala372_Ser429del) in the foie gras domain of TRAPPC11 in the Hutterite families. TRAPPC11 encodes a component of the multiprotein TRAPP complex involved in membrane trafficking. We demonstrate that both mutations impair the binding ability of TRAPPC11 to other TRAPP complex components and disrupt the Golgi apparatus architecture. Marker trafficking experiments for the p.Ala372_Ser429del deletion indicated normal ER-to-Golgi trafficking but dramatically delayed exit from the Golgi to the cell surface. Moreover, we observed alterations of the lysosomal membrane glycoproteins lysosome-associated membrane protein 1 (LAMP1) and LAMP2 as a consequence of TRAPPC11 dysfunction supporting a defect in the transport of secretory proteins as the underlying pathomechanism.
Main Text
Limb girdle muscular dystrophies (LGMDs) are a heterogeneous group of genetic myopathies leading primarily to proximal muscle weakness, with relative sparing of heart and bulbar muscles, except for some subtypes.1,2 So far mutations at over 50 loci with either autosomal-dominant (LGMD1) or autosomal-recessive inheritance (LGMD2) have been described.3 The major forms of LGMD result from mutations in genes encoding constituents of the sarcolemmal dystrophin complex, e.g., laminin (LGMD1B), sarcoglycan (LGMD2C-F), and dysferlin (LGMD2B). Other forms, however, result from mutations in genes affecting muscle function via different pathomechanisms involving membrane trafficking,4 muscle remodeling,5 and posttranslational modification of sarcolemmal proteins.6 The age of onset, severity, and rate of progression vary considerably between LGMD subtypes, ranging from early childhood myopathy to adult onset with long-time preserved ambulation. The spectrum of dystroglycanopathies even ranges from mild LGMD to severe congenital muscular dystrophy with brain and eye involvement and severe intellectual disability (ID).6 ID is defined as an intelligence quotient <70 and significant limitations in two or more adaptive skills identified in childhood7 and is found in 1%–3% of the general population.8 ID is etiologically heterogeneous with genetic and nongenetic causes and can be found as the sole clinical feature in nonsyndromic ID or as part of a syndrome with other clinical manifestations. Disruption of a number of cellular and embryological processes have been linked to ID such as transcriptional control of neuronal genes, neuronal development and synapse formation, and intracellular signaling, as well as intracellular trafficking.8,9 Here, we report on the clinical, molecular, and cellular phenotype of an autosomal-recessively inherited disease spectrum that ranges from LGMD to a syndrome characterized by myopathy, ID, hyperkinetic movements, and ataxia, resulting from altered vesicle trafficking.
The study was conducted on a consanguineous Syrian family with an uncharacterized form of LGMD (family 1; Figure 1A) and two families of Hutterite ancestry10,11 (families 2 and 3; Figure 1A) with affected individuals presenting with a myopathic syndrome associated with moderate ID, infantile hyperkinetic movements, and ataxia. All subjects or their legal representatives gave written informed consent to the study and the research protocols were approved by the respective institutional review board. The study was performed in accordance with the Declaration of Helsinki protocols. Fibroblasts were cultivated from a skin biopsy from individual III-8 of family 1 and the two siblings (II-6 and II-8) from family 2 following standard protocols after written informed consent had been given.
Figure 1.

Pedigrees and Molecular Characterization of the TRAPPC11 Mutations
(A) Pedigree structure of families 1, 2, and 3.
(B) Representative wild-type (WT) and mutant electropherograms from family 1. The c.2938G>A mutation is indicated by a black arrow.
(C) Representative WT and mutant electropherograms from families 2 and 3 demonstrating the homozygous c.1287+5G>A mutation (indicated by a black arrow) in relation to the exon 12 and intron 12 boundary.
(D) RT-PCR analysis confirmed the presence of altered C11 splicing in lymphocytes obtained from one carrier parent, two affected individuals, and two controls. Numbers 1, 2, 3, and 4 correspond to different transcripts illustrated in (E).
(E) Schematic representation of these RT-PCR products after gel isolation and Sanger sequencing. In control cells, the full-length transcript (1) predominates over an alternatively spliced minor transcript lacking exon 11 (2). In cells from affected individuals, the minor transcript lacks only exon 12 (3), whereas the predominant transcript (4) lacks both exons 11 and 12, resulting in an in-frame deletion of 58 amino acids (p.Ala372_Ser429del).
(F) Schematic representation of TRAPPC11 and the location of the foie gras and gryzun domains. The two mutations identified in this study are indicated by black arrows.
The three affected individuals from family 1 were born to healthy first-cousin parents (Figures S1A–S1C). They suffer from a progressive proximal muscle weakness, with onset by early school age and 9- to 16-fold increased serum creatine kinase (CK) levels, that led to different degrees of impaired ambulation. The younger cousins are ambulatory with moderate limitations due to fatigue and muscle pain, whereas the eldest cousin (III-3), at age 26, has severely limited mobility (unable to climb stairs, walks short distances with much difficulty) reflecting the progressive nature of the disease. In all affected individuals, the shoulder girdle muscles are less severely involved than the hip girdle musculature. Skeletal findings, present in all affected individuals, include hip dysplasia and scoliosis. In addition, individual III-3 presented with a mild bilateral cataract and individual III-8 showed strabismus convergens. Except for a slight enlargement of the right cardiac ventricle in individual III-9, there was no obvious cardiac or bulbar muscle involvement (Table 1). Pulmonary function testing revealed a moderate restrictive respiratory disorder in individual III-3, while clinical signs of sleep disordered breathing could not be objectified by overnight oxymetry and capnography. The younger cousins do not yet show signs of respiratory involvement. Individual III-3 displayed mild ID.
Table 1.
Clinical Features
| Individual |
Family 1 |
Family 2 |
Family 3 |
|||||
|---|---|---|---|---|---|---|---|---|
| III:8 | III:9 | III:3 | II:2 | II:6 | II:8 | II:2 | II:3 | |
| TRAPPC11 mutation | c.2938G>A p.Gly980Arg | c.2938G>A p.Gly980Arg | c.2938G>A p.Gly980Arg | c.1287+5G>A | c.1287+5G>A | c.1287+5G>A | c.1287+5G>A | c.1287+5G>A |
| Gender | female | female | female | male | male | male | female | male |
| Age (years) | 20 | 16 | 26 | 25 | 22 | 20 | 20 | 18 |
| Height (centile) | 10th | 25–50th | 25th | <5th | 3rd | <3rd | 50th | 5–10th |
| OFC (centile) | 25th | 50–75th | 10–25th | 50th | <2nd | <<<3rd | <3rd | <3rd |
| Muscular symptoms | prox. weakness, muscle pain, muscle cramps | prox. weakness, muscle pain, muscle cramps | prox. weakness, muscle pain | none | mild muscle weakness | none | none | Hypotonia in early childhood |
| Muscle biopsy | n/a | n/a | myopathic changes | myopathic changes | myopathic changes | n/a | n/a | n/a |
| CK levels (u/l) | ∼2,700 | 1,300–2,800 | 600–1,600 | 490–1,215 | 300–600 | 700–1,000 | 1,009 | 728 |
| DTR | UE+/ LE− | UE+/ LE+ | UE−/ LE− | UE+/ LE+ | UE+/ LE+ | UE+/ LE+ | UE+/ LE+ | UE+/ LE+ |
| Heart | not involved | enlarged RV | not involved | not involved | not involved | not involved | not involved | not involved |
| Skeletal symptoms | hip dysplasia, scoliosis, no contractures | hip dysplasia, mild scoliosis, no contractures | hip dysplasia, scoliosis, no contractures | limb asymmetry | none | none | none | none |
| Intellectual disability | no | no | mild | mild to moderate | moderate | moderate | mild, IQ 60 (WISC-III) | mild, IQ 60 (TONI-3) |
| Development | motor delay | normal | unknown | global delay | global delay | global delay | global delay | global delay |
| Seizures | none | none | none | none | abnormal EEG | primary generalized | abnormal EEG | none |
| Ataxia | none | none | none | truncal | truncal | truncal | truncal | truncal |
| Choreiform movements | none | none | none | truncal and limb | truncal and limb | generalized | generalized | limb and facial |
| Neuroimaging (MRI) | n/a | n/a | n/a | normal | mild cerebral atrophy | mild cerebral atrophy | normal | normal |
| Ocular | esotropia, myopia | none | bilateral cataract, myopia | none | none | none | exophoria anisometropia, amblyopia | none |
CK, creatine kinase; DTR, deep tendon reflexes; EEG, electroencephalogram; ID, intellectual disability; LE, lower extremity; MRI, magnetic resonance imaging; n/a, not applicable; OFC, occipital frontal circumference; Prox., proximal; RV, right ventricle; TONI-3, Test of Nonverbal Intelligence, Third Edition v3; UE, upper extremity; u/l, units/liter; WISC, Wechsler Intelligence Scale for Children; +, normal; −, absent.
We performed whole-exome sequencing on DNA extracted from blood lymphocytes of individual III-8. After enrichment of exonic sequences with the SureSelect Human All Exon 50 Mb kit (Agilent Technologies, Santa Clara, CA, USA), the exome was sequenced on an Illumina Genome Analyzer IIx Sequencer (Illumina, San Diego, CA, USA) with two lanes of a single-end 150 basepair protocol. Bioinformatic analysis of all exome variants was used to determine stretches of homozygosity. To refine the homozygous regions, we additionally performed genome-wide linkage analysis in the three affected individuals and their parents by using the Affymetrix GeneChip® Human Mapping 250K Array (Affymetrix, Santa Clara, CA, USA). Genotypes were called by the GeneChip® DNA Analysis Software (GDAS v2.0, Affymetrix) and analyzed as previously decribed.12–16 Linkage analysis was performed assuming autosomal-recessive inheritance, full penetrance, consanguinity, and a causative variant frequency of 0.0001. A single linked homozygous region of 1.03 Mb on chromosome 4q35.1 (defined by SNPs rs6823077 and rs12502711; chr4: 183833940–184866656, hg19; Figures S1D and S1E) containing nine annotated genes was determined. The only unannotated variant in the exome data set within this critical region was the homozygous missense variant c.2938G>A (p.Gly980Arg; Figure 1B) in TRAPPC11 (Table S1; RefSeq accession number NM_021942.5). TRAPPC11 codes for a 1,133 amino acid protein containing a so-called foie gras and a gryzun domain (Figure 1F). The variant affects a highly conserved amino acid residue within the gryzun domain (Figure 1F; Figure S1F) that is predicted to affect protein structure by PolyPhen-2. Cosegregation of the c.2938G>A variant within the family was confirmed by Sanger sequencing (data not shown). The mutation was not found in 100 healthy Turkish controls tested either by restriction enzyme digestion with the enzyme Mval or direct Sanger sequencing and is not annotated in any current database of human variation including >12,000 alleles of the Exome Variant Server (Exome Variant Server, NHLBI GO Exome Sequencing Project [ESP], Seattle, WA). Furthermore, screening of all coding exons of TRAPPC11 by Sanger sequencing in 32 German single cases of unclassified recessive LGMD did not identify an additional TRAPPC11 mutation. Screening of the exome data of genes in which recessive mutations are known to cause LGMD did not identify a pathogenic mutation.
The Hutterites are an endogamous Anabaptist group that arose during the Protestant Reformation in South Tyrol and, upon arrival in North America during the 1870s, established three groups, the Dariusleut, Schmiedeleut, and Lehrerleut that have remained mostly genetically isolated from one another.17 The Hutterite families in this study consisted of two affected brothers and their affected first cousin from the Dariusleut Hutterites of Alberta (family 2; Figure 1A; Figures S2A and S2B) and two affected siblings (one male, one female) from the Schmiedeleut Hutterites of South Dakota (family 3; Figure 1A; Figures S2C and S2D). An eight-generation pedigree connects the affected individuals to their most recent common Hutterite ancestors, a couple born in the 1790s (Figure S3), prior to the founding of the three leuts. All affected individuals from families 2 and 3 have a history of early onset developmental delay and as young adults now have mild to moderate ID. There is no history of regression, and neuroimaging was unremarkable; in particular, no abnormalities in the cerebellum or basal ganglia were observed. As young children, they all had significant evidence of a hyperkinetic movement disorder mainly characterized by choreiform movements of trunk, limbs, and head although athetoid movements, tremors, and dystonic posturing were also noted. In addition, all affected individuals had a truncal ataxia resulting in further gait instability (Table 1). Metabolic investigations including plasma, urine, and cerebrospinal fluid failed to identify any specific anomalies. Mild muscle weakness with persistently elevated CK levels suggested an associated myopathy. The two limb girdle muscular dystrophies known to be present in the Hutterite population, LGMD2H (MIM 254110; TRIM32, NM_012210.3, c.1459G>A; p.Asp487Asn)18 and LGMD2I (MIM 607155; FKRP, NM_024301.4, c.826C>A; p.Leu276Ile)19 were excluded in all five individuals by Sanger sequencing. Although the two families were not closely related, a founder mutation with an autosomal recessive mode of inheritance was suspected because of the Hutterites’ history of genetic isolation.
Homozygosity mapping in the two affected siblings from family 2 and two of their unaffected siblings was performed by using a 10K SNP genotyping microarray (Affymetrix), and two relevant blocks of homozygosity on chromosomes 1p31.3 and 4q34.3-q35.1 were identified (Figure S2E). Haplotype analysis across the two regions including all three affected individuals from family 2 ruled out the region on chromosome 1p31.3 (data not shown). The remaining region on chromosome 4q34.3-q35.1 (flanked by the SNPs rs721684 and rs726466; chr4: 177850523–186546056, hg19) was 8.7 Mb in size and contained 47 predicted or known genes and 10 predicted or known noncoding RNAs (Table S2). Whole-exome sequencing was undertaken on a single affected individual from family 2 by using the SureSelect Human All Exon v2 kit (Agilent Technologies) and paired-end sequencing on an Illumina GAII (Illumina) performed commercially by Perkin Elmer (Branford, CT, USA). Reads were mapped to the reference sequence and variants were called by using the CLC genomics workbench (CLCbio, Cambridge, MA, USA). Within the single region of shared identity by descent, only three variants were not in dbSNP with a population frequency of less than 0.02. Only the c.1287+5G>A splice-site variant at the donor site of exon 12 of TRAPPC11 (NM_021942.5; Figure 1C) was considered to be pathogenic. The variant was predicted to abolish the exon 12 splice donor site by in silico analysis by using the Alamut software (Interactive Software, San Diego, CA).
Family 3 was investigated by whole-genome sequencing (Complete Genomics, Mountain View, CA, USA) of both affected siblings and both parents. A total of 2,714 variants were heterozygous in both parents, homozygous in both affected siblings, and absent in the homozygous state in a database of 94 other Hutterite genomes. However, only eight of these were annotated as altering the protein-coding sequence or splicing of a gene. With the exception of TRAPPC11 c.1287+5G>A, the same mutation identified in family 2, these remaining variants were common (minor allele frequency ≥10%) in the other Hutterite genomes or in dbSNP. TRAPPC11 is located within a 1.69 Mb homozygous haplotype on chromosome 4q35.1 (chr4: 184562444–186254248, hg19) shared by both affected siblings in family 3. Segregation studies via Sanger sequencing of TRAPPC11 c.1287+5G>A in both family 2 and 3 verified that all five affected individuals were homozygous for the variant, all parents were heterozygous, and all unaffected siblings carried one or no copies of the variant (data not shown).
The same c.1287+5G>A mutation identified in two separate Hutterite leuts in individuals with similar phenotypes substantiated the suspicion of a founder mutation present prior to the leut subdivision. Thus, we screened for this mutation in a normal control population of 1,827 individuals from all three Hutterite leuts by using a custom TaqMan genotyping assay (Applied Biosystems, Foster City, CA, USA) and did not identify any individuals who were homozygous for the mutant allele, adding support for the pathogenicity of this mutation. We observed carrier frequencies of approximately 7% in both the Dariusleut and Schmiedeleut. The mutation was not detected in the Lehrerleut (Table S3). Given the carrier frequencies in the two leuts, we would expect approximately 1 in 750 individuals to be homozygous for this mutation, suggesting that this mutation might account for some of the unexplained syndromic forms of ID in the Hutterite population.
Because the TRAPPC11 c.1287+5G>A mutation was predicted to alter splicing of the exon 12 donor site, we determined the effect on the mature TRAPPC11 transcript by using RT-PCR followed by sequencing of the resulting fragments. In control lymphocytes, two splice isoforms were observed: the predominant splice isoform consists of the full-length transcript, whereas the minor splice isoform lacks exon 11 and is predicted to result in premature protein truncation (Figures 1D and 1E). In lymphocytes from affected individuals, the predominant splice isoform lacks both exons 11 and 12 resulting in a 58 amino acid in-frame deletion in the foie gras domain (p.Ala372_Ser429del), whereas the minor splice isoform lacks only exon 12 and is predicted to result in a prematurely truncated protein (Figures 1D and 1E). Multiple species alignment of TRAPPC11 using Multalin17 demonstrated numerous amino acids within the deleted segment that are highly conserved (Figure S2F), suggesting that this region is important in protein function.
TRAPP (transport protein particle) is a multiprotein complex involved in ER-to-Golgi trafficking.20 It was initially identified in Saccharomyces cervisiae21 and is conserved across species.22 The subunit TRAPPC11 (C11) is a TRAPP component that is important for complex integrity and anterograde membrane transport from the endoplasmic reticulum (ER) to the ER-to-Golgi intermediate compartment (ERGIC) in mammals.22 It is ubiquitously expressed in humans, reflecting its important role in basic cellular functions. C11 participates in numerous interactions with other TRAPP complex components, most notably TRAPPC2 (C2), TRAPPC2L (C2L), TRAPPC6, TRAPPC10, and TRAPPC12.22 Loss-of-function mutations in the zebrafish ortholog of C11 have been found in the foie gras mutant, characterized by steatohepatitis and eye development defects, respectively.23,24 In Drosophila S2 cells, RNAi knockdown of C11 blocks Golgi exit.25 In HeLa cells, small interfering RNA (siRNA) knockdown of C11 leads to impaired binding of other TRAPP components, functional impairment of the complex, retention of secretory proteins in the ERGIC, and fragmentation of the Golgi apparatus.22,25
Immunostaining of the Golgi apparatus with the marker protein GM130 (BD Biosciences, San Jose, CA, USA) was performed in primary fibroblasts from individual III-8 of family 1 and the two siblings from family 2, as well as on control cells. Punctate Golgi dispersal was observed in cells from affected individuals similar to that seen after TRAPPC11 knockdown in HeLa cells22 (Figures 2A–2E). Quantitation of the fragmented phenotype indeed shows a higher percentage of cells with a fragmented Golgi in cells from affected individuals compared to controls (Figure 2F). The Golgi phenotype was confirmed by immunostaining for a second Golgi marker mannosidase II (manII; courtesy of Dr. Kelley Moremon) in family 2 (data not shown). Confocal microscopy of the Golgi in individual III-8 from family 1 further illustrated the scattered Golgi network (Figure 2G) and three-dimensional (3D) reconstruction of confocal microscopy images from family 2 confirmed that the fragmentation is due to fewer and/or thinner connections between the GM130-positive structures (Figures 2H and 2I).
Figure 2.
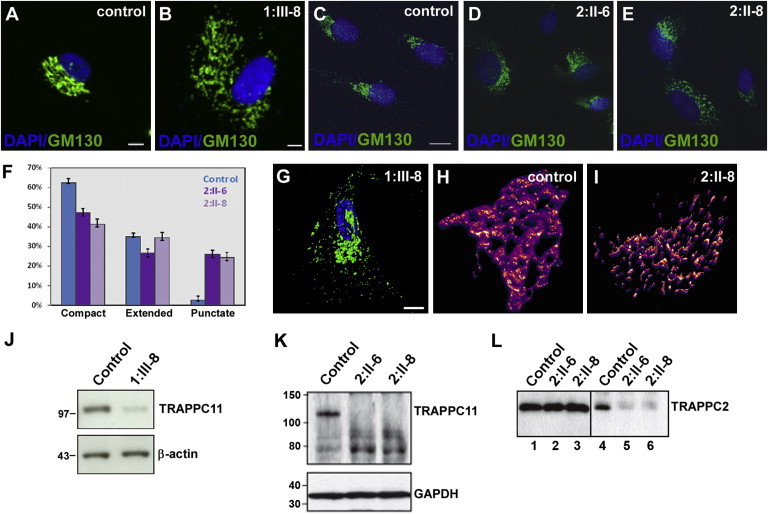
TRAPPC11 Mutations Alter Golgi Morphology, Protein Stability and TRAPP Assembly
(A–E) Immunostaining of fibroblasts from affected individuals and controls with GM130 antibody and DAPI show that the affected individuals have disrupted Golgi morphology compared to healthy controls. Scale bars represent 10 μm.
(F) Quantitation of the Golgi phenotype seen in the cells from (C)–(E). A minimum of 300 cells for each sample were quantitated over three independent experiments.
(G) High-resolution confocal image of a fibroblast from individual III-8 of family 1 illustrating the scattered Golgi structure. The scale bar represents 10 μm.
(H and I) 3D reconstruction of a z stack of the Golgi from a control cell and from individual II-8 of family 2.
(J and K) Immunoblot analysis of cell lysates from individual 1:III-8 (J) or 2:II-6 and 2:II-8 (K) fibroblasts show a reduction of full-length C11 in comparison to control cells. Different mobility of C11 results from different types of gel and buffer used for the experiments shown in (J) and (K). Note the presence of possible C11 fragments in (K) suggesting a destabilization of the protein as a result of the c.1287+5G>A mutation.
(L) Lysates from fibroblasts from individuals II-6 and II-8 of family 2 (lanes 1–3) and control were prepared, and equal amounts (0.5 mg) were incubated with anti-C11 IgG. The immune complexes were collected onto beads (lanes 4–6), eluted, and probed for the TRAPP protein C2. Lanes 1–3 show equal amounts of C2 in the starting material. The C2 and C11 antibodies are noncommercial, generated by the group of M.S. in rabbits against full-length His-tagged C2 and a peptide derived from the carboxy-terminal region of C11. Antibodies were used as described.22
Consistent with the punctate Golgi seen upon C11 knockdown, immunoblot analysis demonstrated a quantitative reduction of full-length mutant C11 protein in fibroblasts from families 1 and 2 (Figures 2J and 2K). In family 2, there is an accumulation of what might possibly be TRAPPC11 fragments due to their cross-reactivity with anti-TRAPPC11. If these fragments are indeed truncated polypeptides of TRAPPC11, they are incapable of interacting with TRAPPC2 (Figure 2L), suggesting that the instability of TRAPPC11 in these individuals might either cause or result from an inability to interact with TRAPP, a distinction that we cannot presently address. Similarly, although C11 p.Gly980Arg was unstable in cells derived from affected individuals, we exploited the stability of this protein in a yeast system to demonstrate that the protein loses its ability to interact with several TRAPP proteins by yeast two-hybrid analysis (Figure S4). An unrelated substitution in proximity to C11 p.Gly980Arg (C11 p.Trp986_Arg988delinsAlaGluAsp) also showed the same inability to interact with TRAPP proteins (Figure S4; Table S4) indicating that this region of C11 might be important for its interactions and stability.
In order to determine whether cells from affected individuals also displayed a block in membrane traffic between the endoplasmic reticulum (ER) and the Golgi, we used VSV-G-ts045-GFP (VSV-G) as a marker for transport along the secretory pathway. This commonly used marker protein misfolds at elevated temperatures (40°C) and is retained in the ER. Upon shifting to lower temperature (32°C) and in the presence of the protein synthesis inhibitor cycloheximide, the ER-restricted material exits and is transported to the Golgi and ultimately to the cell surface.26 The kinetics of this transport is well documented with appearance of the protein in the Golgi within minutes of release, where it can reside for up to 40 minutes before transport to the cell surface.27 We assessed VSV-G localization in fibroblasts from affected individuals from family 2 and control fibroblasts, in conjunction with the Golgi marker manII, at fixed intervals. The trafficking experiments were performed in cell lines from both affected individuals from family 2; however, data is shown only for individual II-8 for simplicity. After incubation at 40°C, VSV-G is found exclusively in the ER in both control cells and cells from affected individuals, displaying the typical diffuse, reticular pattern (Figure 3A). Colocalization of VSV-G with the Golgi marker manII is clearly seen after 30 minutes following release from the ER at 32°C in both control cells and cells from affected individuals (Figure 3A). After 120 minutes, however, virtually all of the VSV-G is found on the cell surface of control cells while a significant amount of VSV-G remains in the Golgi of cells from affected individuals (Figure 3A). Live-cell imaging confirmed delayed VSV-G exit from the Golgi in cells from affected individuals, pronounced from approximately 90 minutes, consistent with the findings of the timed intervals above (Movie S1). The delay of VSV-G exit from the Golgi in cells from the affected individuals suggests that generalized trafficking of molecules through the Golgi might be delayed in these individuals as a result of the in-frame deletion of TRAPPC11.
Figure 3.
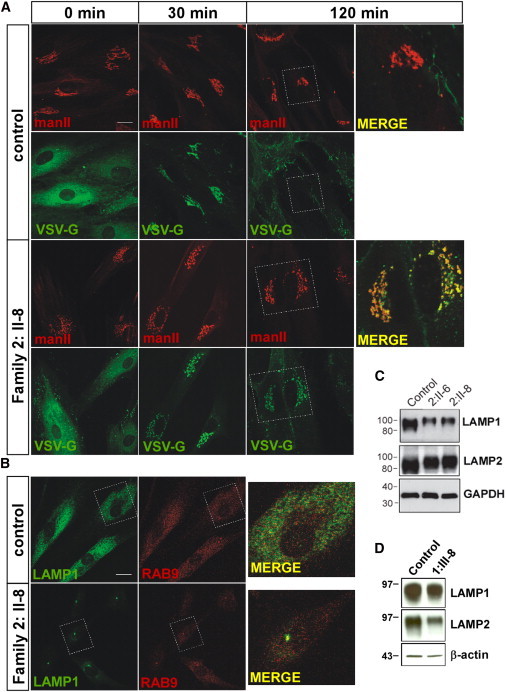
Altered VSV-G Trafficking and LAMP1 Localization in Cells from Affected Individuals
(A) Fibroblasts from a control and individual II:8 of family 2 were infected with virus expressing VSV-G-ts045-GFP (VSV-G) for 1 hr and then incubated overnight at 37°C. The cells were then shifted to 40°C for 6 hours at which point cyclohexamide was added and the samples were transferred to 32°C. Samples were removed at 0, 30, and 120 min following transfer to 32°C and stained with the Golgi marker mannosidase II (manII). The scale bar represents 10 μm.
(B) Immunostaining with the late endosome/lysosome markers Rab9 and LAMP1 (Abcam, Cambridge, UK) demonstrated a normal diffuse pattern of both proteins in control fibroblasts but strong perinuclear localization in cells from affected individuals. The scale bar represents 10 μm.
(C) Equal amounts of lysate prepared from control, individual II:6 and individual II:8 of family 2 were probed for LAMP1, LAMP2 (Abcam), and GAPDH as a loading control.
(D) Equal amounts of cell lysate from fibroblasts of individual III:8 of family 1 and control fibroblasts were incubated with monoclonal antibodies against LAMP1, LAMP2 (H4A3 and H4B4, respectively; Santa Cruz Biotechnology, Santa Cruz, USA), and β-actin as a loading control.
Delayed exit from the Golgi could arise from a defect in anterograde traffic from the Golgi to the cell surface or indirectly from defects in the endocytic pathway that would fail to retrieve material from the Golgi needed for anterograde traffic. To determine whether the trafficking defect observed in cells from affected individuals was due to defects in the endocytic pathway, we examined the uptake of fluorescently tagged transferrin from the cell medium but did not observe any uptake defects (data not shown). However, immunostaining of cells from affected individuals from family 2 for the late endosomal/lysosomal component lysosome-associated membrane protein 1 (LAMP1) identified a striking difference in the localization pattern compared to control cells (Figure 3B). LAMP1 in control cells was seen in puncta throughout the cell with the occasional deposition in the perinuclear region (24% ± 1% of control cells [n = 189]), similar to other studies reporting LAMP1 localization.28 Conversely, fibroblasts from affected individuals displayed prominent perinuclear staining for LAMP1 in a high proportion of cells (78% ± 2% and 87%±3% of cells in individual II-6 and individual II-8, respectively [n = 189 for all cases]). Immunostaining of cells from affected individuals from family 2, as well as immunoblotting of the corresponding cell lysates, showed a reduced level of LAMP1 compared to control (Figures 3B and 3C) and was concentrated in a higher molecular-size region than in the control, suggesting that it might be more highly glycosylated. Levels of lysosome associated membrane protein 2 (LAMP2) did not appear to be significantly reduced in cells from affected individuals from family 2 but, like LAMP1, the protein appeared to be more highly glycosylated (Figure 3C). Moreover, immunoblot analysis of fibroblasts from individual III-8 of family 1 also showed LAMP1 and LAMP2 alterations (Figure 3D).
Retention of the human growth-hormone marker protein GFP-FM4-hGH in the Golgi of HeLa cells following knockdown of C11 was previously reported.25 In addition, ablation of C11 by siRNA was reported to result in accumulation of VSV-G in a brefeldin A-resistant compartment, interpreted to be related to the ERGIC (ER-Golgi intermediate compartment).22 Our finding that partial deletion of the foie gras domain of C11 leads to defects in a post-Golgi compartment might be explained by a milder functional effect of the p.Ala372_Ser429del deletion compared to knockdown of the entire protein. It is noteworthy that several recent studies implicated the TRAPP complex in retrograde traffic originating at the cell surface; however, the precise location where TRAPP acts in this pathway was not addressed.29,30 Our present study suggests that a C11-containing TRAPP complex is involved in the formation and/or movement of late endosomes/lysosomes. A prominent perinuclear staining of LAMP1 has been seen in cells depleted of Rab9, leading to the suggestion that effector molecules required for late endosomal transport are not recruited in the absence of this GTPase.31 In addition, Rab7 and the lipid composition of membranes has been implicated in the motility of late endosomes/lysosomes.32 It will be of interest in the future to examine the relationship between a C11-containing TRAPP complex and various GTPases involved in late endosome dynamics.
Mutations in TRAPPC2 and TRAPPC9, encoding TRAPP complex components, cause X-linked spondyloepiphyseal dysplasia tarda33 and postnatal microcephaly with ID,34,35 respectively. It has been suggested that the different effects of mutations in TRAPPC2 and TRAPPC9 on human disease are linked to the specific function and binding capacities of each subunit.36 The similar Golgi phenotype observed in fibroblasts from affected individuals from families 1 and 2 suggests similar functional effects of both TRAPPC11 mutations on TRAPP composition and function. Phenotypic differences between our families could be explained by different functional consequences of each alteration, residing in different protein domains.
Our finding of LAMP1/LAMP2 alterations suggest a similarity with the pathomechanism of Danon disease (MIM 300257), a myopathy caused by LAMP2 deficiency,37 characterized by dilatative cardiomyopathy, proximal skeletal muscle weakness, and ID.38 Danon disease is a rare X-linked dominant disorder with early childhood onset and an aggressive disease course in male affected individuals, leading to death from cardiac complications in the 2nd to 3rd decade, if not prevented by heart transplantation. Female affected individuals show a milder disease course, but many suffer from mild ID and muscle weakness in addition to cardiac manifestations. Both sexes might develop retinopathy, hepatic, and pulmonary disease.38,39 The molecular pathogenesis of Danon disease exemplifies that defects in intracellular trafficking components can cause a skeletal muscle phenotype with associated ID.
In summary, we present a form of autosomal-recessive, slowly progressive LGMD with childhood onset and high CK, as well as a syndrome consisting of myopathy, ID, hyperkinetic movements, and ataxia, caused by homozygous mutations in the membrane trafficking component TRAPPC11. Thus, the presence of myopathy and high CK with or without additional symptoms in a person should alert clinicians for TRAPPC11 deficiency. Both mutations lead to a range of molecular defects including altered TRAPP complex composition, impaired Golgi morphology, and altered protein transport along the secretory pathway. These results suggest that altered membrane trafficking is the underlying molecular mechanism of this disease spectrum.
Acknowledgements
We are grateful to all family members that participated in this study and to Karin Boss and Bob Argiropolous for critically reading the manuscript, Leo Dimnik for assistance with NGS data, and Kelley Moremon for the manII antibody. This work was supported by the German Federal Ministry of Education and Research (BMBF) by grant number 01GM1211A (E-RARE network CRANIRARE-2) to B.W., by the Canadian Institutes of Health Research and the Natural Sciences and Engineering Council of Canada to M.S., research grant #5-FY09-529 from the March of Dimes Foundation to K.M.B., and the National Institutes of Health grants R01HD21244 and R01HL085197 to C.O. R.E.L. was supported by a Remax clinical fellowship from the Alberta Children’s Hospital Foundation and K.M.B. is supported by a Clinical Investigatorship Award from the Canadian Institutes of Health Research, Institute of Genetics. M.S. is a member of the Groupe de Recherche Axé sur la Structure des Protéines (GRASP) network.
Contributor Information
Bernd Wollnik, Email: bwollnik@uni-koeln.de.
Michael Sacher, Email: michael.sacher@concordia.ca.
Supplemental Data
Cells were infected with VSV-G-ts045-GFP for 1 hr and then incubated at 37°C for ∼20 hr. The cells were then shifted to 40°C for 6 hr. Immediately prior to imaging, cycloheximide was added and the cells were placed on a heated stage (32°C) in a 5% CO2 environment. Imaging was performed every 30 s over a 120 m period on a Nikon Livescan sweptfield microscope. The first 90 min are shown.
Web Resources
The URLs for the data presented herein are as follows:
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
GeneCards: http://www.genecards.org
National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
UCSC Genome Browser, http://genome.ucsc.edu
UniProt, http://www.uniprot.org/
References
- 1.Rosales X.Q., Tsao C.Y. Childhood onset of limb-girdle muscular dystrophy. Pediatr. Neurol. 2012;46:13–23. doi: 10.1016/j.pediatrneurol.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 2.Bushby K. Diagnosis and management of the limb girdle muscular dystrophies. Pract. Neurol. 2009;9:314–323. doi: 10.1136/jnnp.2009.193938. [DOI] [PubMed] [Google Scholar]
- 3.Pegoraro, E., and Hoffman, E. (2000, updated 2012). Limb-Girdle Muscular Dystrophy Overview. In GeneReviews™ [Internet], Pagon RA, Bird TD, Dolan CR, et al., ed. (University of Washington, Seattle, WA, USA), 1993-. http://www.ncbi.nlm.nih.gov/books/NBK1408/ [PubMed]
- 4.Minetti C., Sotgia F., Bruno C., Scartezzini P., Broda P., Bado M., Masetti E., Mazzocco M., Egeo A., Donati M.A. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat. Genet. 1998;18:365–368. doi: 10.1038/ng0498-365. [DOI] [PubMed] [Google Scholar]
- 5.Richard I., Broux O., Allamand V., Fougerousse F., Chiannilkulchai N., Bourg N., Brenguier L., Devaud C., Pasturaud P., Roudaut C. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell. 1995;81:27–40. doi: 10.1016/0092-8674(95)90368-2. [DOI] [PubMed] [Google Scholar]
- 6.Mercuri E., Muntoni F. The ever-expanding spectrum of congenital muscular dystrophies. Ann. Neurol. 2012;72:9–17. doi: 10.1002/ana.23548. [DOI] [PubMed] [Google Scholar]
- 7.American Association on Intellectual and Developmental Disabilities . American Association on Intellectual and Developmental Disabilities; Washington, DC: 2010. Intellectual disability: definition, classification, and systems of supports. [DOI] [PubMed] [Google Scholar]
- 8.Kaufman L., Ayub M., Vincent J.B. The genetic basis of non-syndromic intellectual disability: a review. J Neurodev Disord. 2010;2:182–209. doi: 10.1007/s11689-010-9055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 2011;45:81–104. doi: 10.1146/annurev-genet-110410-132512. [DOI] [PubMed] [Google Scholar]
- 10.Boycott K.M., Parboosingh J.S., Chodirker B.N., Lowry R.B., McLeod D.R., Morris J., Greenberg C.R., Chudley A.E., Bernier F.P., Midgley J. Clinical genetics and the Hutterite population: a review of Mendelian disorders. Am. J. Med. Genet. A. 2008;146A:1088–1098. doi: 10.1002/ajmg.a.32245. [DOI] [PubMed] [Google Scholar]
- 11.Hostetler J.A. History and relevance of the Hutterite population for genetic studies. Am. J. Med. Genet. 1985;22:453–462. doi: 10.1002/ajmg.1320220303. [DOI] [PubMed] [Google Scholar]
- 12.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 13.O’Connell J.R., Weeks D.E. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 15.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 16.Thiele H., Nürnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 17.Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frosk P., Weiler T., Nylen E., Sudha T., Greenberg C.R., Morgan K., Fujiwara T.M., Wrogemann K. Limb-girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am. J. Hum. Genet. 2002;70:663–672. doi: 10.1086/339083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frosk P., Greenberg C.R., Tennese A.A., Lamont R., Nylen E., Hirst C., Frappier D., Roslin N.M., Zaik M., Bushby K. The most common mutation in FKRP causing limb girdle muscular dystrophy type 2I (LGMD2I) may have occurred only once and is present in Hutterites and other populations. Hum. Mutat. 2005;25:38–44. doi: 10.1002/humu.20110. [DOI] [PubMed] [Google Scholar]
- 20.Kim Y.G., Raunser S., Munger C., Wagner J., Song Y.L., Cygler M., Walz T., Oh B.H., Sacher M. The architecture of the multisubunit TRAPP I complex suggests a model for vesicle tethering. Cell. 2006;127:817–830. doi: 10.1016/j.cell.2006.09.029. [DOI] [PubMed] [Google Scholar]
- 21.Sacher M., Jiang Y., Barrowman J., Scarpa A., Burston J., Zhang L., Schieltz D., Yates J.R., 3rd, Abeliovich H., Ferro-Novick S. TRAPP, a highly conserved novel complex on the cis-Golgi that mediates vesicle docking and fusion. EMBO J. 1998;17:2494–2503. doi: 10.1093/emboj/17.9.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scrivens P.J., Noueihed B., Shahrzad N., Hul S., Brunet S., Sacher M. C4orf41 and TTC-15 are mammalian TRAPP components with a role at an early stage in ER-to-Golgi trafficking. Mol. Biol. Cell. 2011;22:2083–2093. doi: 10.1091/mbc.E10-11-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadler K.C., Amsterdam A., Soroka C., Boyer J., Hopkins N. A genetic screen in zebrafish identifies the mutants vps18, nf2 and foie gras as models of liver disease. Development. 2005;132:3561–3572. doi: 10.1242/dev.01918. [DOI] [PubMed] [Google Scholar]
- 24.Gross J.M., Perkins B.D., Amsterdam A., Egaña A., Darland T., Matsui J.I., Sciascia S., Hopkins N., Dowling J.E. Identification of zebrafish insertional mutants with defects in visual system development and function. Genetics. 2005;170:245–261. doi: 10.1534/genetics.104.039727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wendler F., Gillingham A.K., Sinka R., Rosa-Ferreira C., Gordon D.E., Franch-Marro X., Peden A.A., Vincent J.P., Munro S. A genome-wide RNA interference screen identifies two novel components of the metazoan secretory pathway. EMBO J. 2010;29:304–314. doi: 10.1038/emboj.2009.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergmann J.E., Singer S.J. Immunoelectron microscopic studies of the intracellular transport of the membrane glycoprotein (G) of vesicular stomatitis virus in infected Chinese hamster ovary cells. J. Cell Biol. 1983;97:1777–1787. doi: 10.1083/jcb.97.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirschberg K., Miller C.M., Ellenberg J., Presley J.F., Siggia E.D., Phair R.D., Lippincott-Schwartz J. Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J. Cell Biol. 1998;143:1485–1503. doi: 10.1083/jcb.143.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Humphries W.H., 4th, Szymanski C.J., Payne C.K. Endo-lysosomal vesicles positive for Rab7 and LAMP1 are terminal vesicles for the transport of dextran. PLoS ONE. 2011;6:e26626. doi: 10.1371/journal.pone.0026626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bassik M.C., Kampmann M., Lebbink R.J., Wang S., Hein M.Y., Poser I., Weibezahn J., Horlbeck M.A., Chen S., Mann M. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell. 2013;152:909–922. doi: 10.1016/j.cell.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moreau D., Kumar P., Wang S.C., Chaumet A., Chew S.Y., Chevalley H., Bard F. Genome-wide RNAi screens identify genes required for Ricin and PE intoxications. Dev. Cell. 2011;21:231–244. doi: 10.1016/j.devcel.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 31.Ganley I.G., Carroll K., Bittova L., Pfeffer S. Rab9 GTPase regulates late endosome size and requires effector interaction for its stability. Mol. Biol. Cell. 2004;15:5420–5430. doi: 10.1091/mbc.E04-08-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lebrand C., Corti M., Goodson H., Cosson P., Cavalli V., Mayran N., Fauré J., Gruenberg J. Late endosome motility depends on lipids via the small GTPase Rab7. EMBO J. 2002;21:1289–1300. doi: 10.1093/emboj/21.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gedeon A.K., Colley A., Jamieson R., Thompson E.M., Rogers J., Sillence D., Tiller G.E., Mulley J.C., Gécz J. Identification of the gene (SEDL) causing X-linked spondyloepiphyseal dysplasia tarda. Nat. Genet. 1999;22:400–404. doi: 10.1038/11976. [DOI] [PubMed] [Google Scholar]
- 34.Mochida G.H., Mahajnah M., Hill A.D., Basel-Vanagaite L., Gleason D., Hill R.S., Bodell A., Crosier M., Straussberg R., Walsh C.A. A truncating mutation of TRAPPC9 is associated with autosomal-recessive intellectual disability and postnatal microcephaly. Am. J. Hum. Genet. 2009;85:897–902. doi: 10.1016/j.ajhg.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mir A., Kaufman L., Noor A., Motazacker M.M., Jamil T., Azam M., Kahrizi K., Rafiq M.A., Weksberg R., Nasr T. Identification of mutations in TRAPPC9, which encodes the NIK- and IKK-beta-binding protein, in nonsyndromic autosomal-recessive mental retardation. Am. J. Hum. Genet. 2009;85:909–915. doi: 10.1016/j.ajhg.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zong M., Wu X.G., Chan C.W., Choi M.Y., Chan H.C., Tanner J.A., Yu S. The adaptor function of TRAPPC2 in mammalian TRAPPs explains TRAPPC2-associated SEDT and TRAPPC9-associated congenital intellectual disability. PLoS ONE. 2011;6:e23350. doi: 10.1371/journal.pone.0023350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishino I., Fu J., Tanji K., Yamada T., Shimojo S., Koori T., Mora M., Riggs J.E., Oh S.J., Koga Y. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 38.Boucek D., Jirikowic J., Taylor M. Natural history of Danon disease. Genet. Med. 2011;13:563–568. doi: 10.1097/GIM.0b013e31820ad795. [DOI] [PubMed] [Google Scholar]
- 39.Prall F.R., Drack A., Taylor M., Ku L., Olson J.L., Gregory D., Mestroni L., Mandava N. Ophthalmic manifestations of Danon disease. Ophthalmology. 2006;113:1010–1013. doi: 10.1016/j.ophtha.2006.02.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cells were infected with VSV-G-ts045-GFP for 1 hr and then incubated at 37°C for ∼20 hr. The cells were then shifted to 40°C for 6 hr. Immediately prior to imaging, cycloheximide was added and the cells were placed on a heated stage (32°C) in a 5% CO2 environment. Imaging was performed every 30 s over a 120 m period on a Nikon Livescan sweptfield microscope. The first 90 min are shown.