

Abstract
Short stature, hyperextensibility of joints and/or inguinal hernia, ocular depression, Rieger anomaly, and teething delay (SHORT) syndrome is a developmental disorder with an unknown genetic cause and hallmarks that include insulin resistance and lack of subcutaneous fat. We ascertained two unrelated individuals with SHORT syndrome, hypothesized that the observed phenotype was most likely due to de novo mutations in the same gene, and performed whole-exome sequencing in the two probands and their unaffected parents. We then confirmed our initial observations in four other subjects with SHORT syndrome from three families, as well as 14 unrelated subjects presenting with syndromic insulin resistance and/or generalized lipoatrophy associated with dysmorphic features and growth retardation. Overall, we identified in nine affected individuals from eight families de novo or inherited PIK3R1 mutations, including a mutational hotspot (c.1945C>T [p.Arg649Trp]) present in four families. PIK3R1 encodes the p85α, p55α, and p50α regulatory subunits of class IA phosphatidylinositol 3 kinases (PI3Ks), which are known to play a key role in insulin signaling. Functional data from fibroblasts derived from individuals with PIK3R1 mutations showed severe insulin resistance for both proximal and distal PI3K-dependent signaling. Our findings extend the genetic causes of severe insulin-resistance syndromes and provide important information with respect to the function of PIK3R1 in normal development and its role in human diseases, including growth delay, Rieger anomaly and other ocular affections, insulin resistance, diabetes, paucity of fat, and ovarian cysts.
Main Text
Insulin resistance is closely associated with many common, complex, and multifactorial conditions, including obesity, type 2 diabetes, atherosclerosis, nonalcoholic fatty-liver disease, and polycystic-ovary syndrome. Rare monogenic forms of insulin resistance also exist, and identifying the genetic basis of such phenotypes provides unique opportunities for deciphering the multiple molecular mechanisms leading to insulin resistance.1 SHORT syndrome (MIM 269880)—whose acronym stands for short stature, hyperextensibility of joints and/or inguinal hernia, ocular depression, Rieger anomaly, and teething delay—is a developmental disorder of unknown genetic cause and is associated with generalized thin habitus despite normal food intake. Onset of insulin resistance and/or diabetes mellitus typically occurs in adolescence.2 Although affected individuals frequently present with short stature and relative microcephaly when compared to unaffected family members, height is in the low-normal range in one-third of cases. Other features include characteristic facial dysmorphism, delayed bone age and gracile long bones, variable ocular anomalies, normal intellect or mild impairment with speech delay, frequent illnesses, and lack of subcutaneous fat with wrinkled skin and readily visible veins enhancing the progeroid appearance. Vertical and male-to-male transmission have been described, indicative of autosomal-dominant inheritance.2 Mutations in PITX2 (MIM 601542) and BMP4 (MIM 112262) have been discussed as putatively causative, but no mutations were identified in subjects with typical SHORT syndrome.3,4
We ascertained two unrelated individuals with a strikingly similar clinical presentation consistent with SHORT syndrome (Table 1 and Figures 1A–1D). Given the absence of recurrence of the syndrome in both families, we hypothesized that the observed phenotype was most likely due to de novo mutations in the same gene and performed whole-exome sequencing in the two probands and their unaffected parents. We then confirmed our initial observations in four other subjects with SHORT syndrome from three families, as well as 14 unrelated subjects presenting with syndromic insulin resistance and/or generalized lipoatrophy associated with dysmorphic features and growth retardation. Written, informed consent was obtained from all subjects, legal representatives, and relatives before enrollment in the study. The research protocol was approved by the ethics committees at participating institutions. Individuals were diagnosed with SHORT syndrome on the basis of previously reported clinical features.2
Table 1.
Clinical Features of Individuals with Mutations in PIK3R1
|
Individual |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P3a | P4 | P5 | P6 | P7 | P8 | P9 | |
| Sex | male | male | female | male | female | male | female | female | male |
| Features of SHORT Syndrome Acronym | |||||||||
| Measurements at birth | |||||||||
| Gestational age | 34 weeks of gestation + 4 days | 34 weeks of gestation | 35.5 weeks of gestation | 35 weeks of gestation | term | 27 weeks of gestation | term | ND | prematurity |
| Length (cm) | 42 (<3rd per) | 40 (<3rd per) | 43.5 (<3rd per) | 47 (25th per) | 52 (20th per) | 40 (50th per) | NA | NA | NA |
| Weight (g) | 1,460 (<3rd per) | 1,380 (<3rd per) | 1,830 (<3rd per) | 2,000 (3rd per) | 2,260 (<3rd per) | 1,400 (50th per) | 2,700 (3rd per) | ND | 950 |
| OFC (cm) | 29.5 (3rd per) | 28.5 (<3rd per) | 32 (20th per) | 33 (40th per) | unknown | 25 (10th per) | ND | ND | ND |
| Measurements at last follow-up examination | |||||||||
| Age | 7 years and 7 months | 7 years and 8 months | 19 years | 20 months | 40 years | ND | 28 years | 60 years | 28 years |
| Length (cm) | 109.7 (<3rd per) | 111.8 (<3rd per) | 146.5 (<3rd per) | 80.5 (40th per) | 160 (25th per) | ND | 145 (<3rd per) | 143 (<3rd per) | 155 (<3rd per) |
| Weight (kg) | 15 (<3rd per) | 15.5 (<3rd per) | 36 (<3rd per) | 8.76 (<3rd per) | 50 (<3rd per) | ND | 44.6 (<3rd per) | 35 (<3rd per) | 42 (<3rd per) |
| OFC (cm) | 47 (<3rd per) | 47 (<3rd per) | unknown | 47 (20th per) | 51 (<3rd per) | ND | ND | ND | ND |
| BMI | 12.5 (<3rd per) | 12.4 (<3rd per) | 16.8 (<3rd per) | 13.5 (<3rd per) | 19.5 (35th per) | ND | 21.2 (25th per) | 17.1 (3rd per) | 17.5 (3rd per) |
| Hyperextensibility of joints | − | − | + | − | + | − | ND | ND | ND |
| Inguinal hernia | − | − | − | − | − | ND | − | − | − |
| Ocular depression | + | + | + | + | + | + | ND | + | − |
| Rieger anomaly | − | − | + | − | + | ND | − | − | − |
| Teeth delay | + | + | + | +b | + | ND | + | ND | ND |
| Number of features of acronym | 3/5 | 3/5 | 5/5 | 2/5 | 4/5 | 1/3 | 2/3 | 2/3 | 1/3 |
| Other Findings | |||||||||
| Ophthalmological findings | |||||||||
| Hyperopia | + | + | + | + | − | ND | ND | ND | ND |
| Astigmatism | + | − | + | − | − | ND | + | ND | ND |
| Myopia | − | − | − | − | + | ND | ND | ND | ND |
| Esotropia | − | − | + | − | − | ND | ND | ND | ND |
| Lack of subcutaneous fat | + | + | + | + | + | ND | + | + | + |
| Insulin resistance | + | + | + | ND | + | ND | + | + | + |
| Diabetes | − | − | + | − | + | ND | + | + | + |
| Facial dysmorphim | |||||||||
| Triangular shape | + | + | + | + | + | + | ND | + | + |
| Prominent forehead | + | + | + | + | + | + | + | + | − |
| Hypoplastic or thin alae nasi | + | + | + | + | + | + | + | + | + |
| Small chin | + | + | + | + | + | + | + | + | + |
| Large low-set ears | + | + | + | + | + | + | + | + | + |
| Mild midface hypoplasia | + | + | + | + | + | + | ND | + | − |
| Micrognathia | + | + | + | + | + | + | + | + | + |
| Thin, wrinkled skin | + | + | − | + | − | ND | + | + | ND |
| Readily visible veins | + | + | − | + | − | ND | + | + | ND |
| Frequent illnesses | − | − | − | − | + | ND | ND | + | ND |
| Ovarian cysts | NA | NA | + | NA | + | NA | + | ND | NA |
| Mental development | |||||||||
| Normal | − | − | − | + | + | ND | + | + | − |
| Mild impairment and/or speech delay | + | + | + | − | − | ND | − | − | + |
| Radiographic findings | |||||||||
| Delayed bone age | + | + | + | ND | ND | ND | ND | ND | ND |
| Gracile long bones | − | − | + | ND | ND | ND | ND | ND | ND |
Abbreviations are as follows: per, percentile; +, presence of a feature; −, absence of a feature; OFC, occipitofrontal circumference; BMI, body mass index; NA, not applicable; and ND, no data.
Subject previously reported.
No teeth at last follow-up examination.
Figure 1.
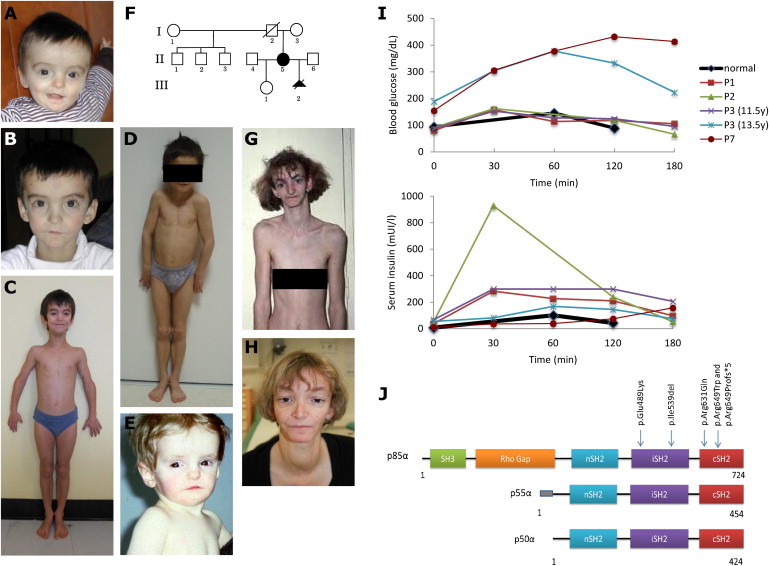
Appearance of Individuals with SHORT Syndrome, Test Results for Oral Glucose Tolerance, and Distribution of PIK3R1 Mutations
(A–H) Photos of subjects P1 (A–C), P2 (D), and P3 (E) and familial pedigree and photos of subject P5 (F–H). Subjects presented with a typical triangular facial shape, deep-set eyes, thin and hypoplastic alae nasi, a small chin, mild midfacial hypoplasia, and lipoatrophy. We obtained written consent to publish photographs of the subjects.
(I) Test results for oral glucose tolerance show hyperglycemia in subjects P3 (at 13.5 years of age) and P7 and increased serum insulin levels in all affected children (subjects P1, P2, and P3).
(J) Schematic representation of the three isoforms encoded by PIK3R1, their functional domains, and the alterations identified in individuals with SHORT syndrome.
For all study subjects, genomic DNA was extracted from blood according to standard procedures. Whole-exome capture and sequencing were performed from 5 μg of genomic DNA at the PerkinElmer DNA Sequencing Service. Exome capture was performed with the 51 Mb SureSelect Human All Exon V4 kit (Agilent). Generated libraries were sequenced on a HiSeq 2000 (Illumina) according to the manufacturer’s recommendations for paired-end 100 bp reads. We generated over 4 Gb of mappable sequence per individual, resulting in a depth of coverage of at least ten reads for more than 85% of the targeted exome (Table S1, available online). Sequencing data were processed as previously described.5,6 Raw single-nucleotide variants (SNVs) and indels were identified with the Genome Analysis Toolkit (GATK v.2.1-10)7 Unified Genotyper tool with a variant quality score ≥ 30. Variants were then filtered with the GATK Variant Filtration tool and the following hard-filtering expressions: “QD < 2.0,” “ReadPosRankSum < −8.0,” and “FS > 200.0.” Filtered variants were annotated with SeattleSeq SNP Annotation (see Web Resources). As previously described,6 we systematically identified candidate de novo events by focusing on protein-altering and splice-site DNA changes (1) absent in the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server (EVS; see Web Resources), (2) supported by at least two reads and ≥20% of total reads in the proband, and (3) absent in both parents (two variant reads or fewer and ≤5% of total reads) at base-pair positions covered by at least four reads in the entire trio. Candidate de novo changes were then manually inspected with the Integrative Genomics Viewer.8 This analysis identified de novo mutations in PIK3R1 (MIM 171833, RefSeq accession number NM_181523.2) in both affected individuals, P1 (c.1615_1617delATT [p.Ile539del]) and P2 (c.1465G>A [p.Glu489Lys]) ( Table S2). Both mutations were confirmed as de novo by standard PCR protocols with the use of custom intronic primers combined with capillary sequencing. PIK3R1 encodes the p85α, p55α, and p50α regulatory subunits of class IA phosphatidylinositol 3 kinases (PI3Ks), which are dimers composed of a p110 catalytic subunit and a p85 regulatory subunit. PI3K signaling is involved in multiple cellular functions, such as cell growth, proliferation, migration, metabolism, angiogenesis, survival, and apoptosis, and mutations in several of the core components of the PI3K pathway have been associated with some of the most prevalent human diseases, including diabetes and cancer.9
We screened PIK3R1 for mutations in four individuals with SHORT syndrome from three additional families. The coding and flanking intronic regions of PIK3R1 (RefSeq NM_181523.2, NM_181524.1, and NM_181504.3) were amplified by standard PCR protocols (primer sequences are available upon request). Sequencing was performed with the ABI BigDye Terminator Cycle Sequencing kit (v.3.1) and an ABI 3130 Genetic Analyzer (Applied Biosystems) according to the manufacturer’s instructions. Sequence traces were analyzed with Mutation Surveyor (v.4.0.7, SoftGenetics). This experiment identified a recurrent substitution (c.1945C>T [p.Arg649Trp]) in all four subjects (Table 2), and it proved to be de novo in the only simplex case for which parental DNA was available (P3, who was previously reported;10 Figure 1E). The remaining three individuals with PIK3R1 mutations included a simplex case (P4) and an affected woman and her terminated fetus (P5 [II-5 in Figure 1F] and P6 [III-2 in Figure 1F], Figures 1F–1H). The c.1945C>T mutation occurs within the context of a CpG dinucleotide, which might explain its recurrence. We then sequenced PIK3R1 in a heterogeneous clinical group of 14 additional unrelated subjects presenting with severe insulin resistance and/or generalized lipoatrophy associated with dysmorphic features and growth retardation (Table S3). None of these cases had been previously diagnosed with SHORT syndrome, and none had mutations in AGPAT2 (MIM 603100), BSCL2 (MIM 606158), LMNA (MIM 150330), PPARG (MIM 601487), or INSR (MIM 147670). Capillary sequencing identified mutations in three of these subjects: the previously identified recurrent substitution (c.1945C>T [p.Arg649Trp]) in P7, a single-nucleotide insertion predicted to result in a prematurely truncated protein (c.1943dupT [p.Arg649Profs∗5]) in P8, and a substitution (c.1892G>A [p.Arg631Gln]) in P9 (Table 2). Although all three individuals had facial dysmorphism retrospectively consistent with SHORT syndrome, the limited available clinical data did not allow confirmation of the clinical diagnosis of SHORT syndrome (Table 1). Nucleotide-level conservation and the impact of amino acid substitutions were assessed with Genomic Evolutionary Rate Profiling (GERP),11 Grantham matrix,12 and PolyPhen-2 (with the HumVar-trained model)13 scores. In addition to the fact that three of the identified missense PIK3R1 mutation sites (encoding p.Ile539del, p.Glu489Lys, and p.Arg649Trp) were found to be de novo in one subject each, all mutation sites affect highly conserved nucleotides and are predicted to be possibly or probably damaging (Table 2). None of the five mutation sites were reported in the NHLBI EVS (Table S4), which consists of whole-exome-sequencing data from ∼6,500 individuals (the EVS was accessed March 2013), dbSNP137 (see Web Resources), or the 1000 Genomes Project (see Web Resources). A single truncating mutation (c.901C>T [p.Arg301∗]), which was present in a heterozygous state in a single individual, was reported in the EVS data set. Contrary to the truncating mutation identified in the present study and predicted to affect all three subunits encoded by PIK3R1 (i.e., p85α, p55α, and p50α), the mutation encoding p.Arg301∗ is predicted to affect only the p85α subunit. We can speculate that disruption of all three PIK3R1 subunits is required for the development of SHORT syndrome, which is consistent with a recent report of a complete loss of p85α (but normal p55α and p50α expression) in an individual with absent B cells but normal growth, development, and fasting glucose and insulin levels.14
Table 2.
Summary of PIK3R1 Mutations Identified in Nine Affected Individuals from Eight Families
| Individual | Mutation Coordinates (hg19) | Alleles (Ref/Alt) | cDNA Change | Amino Acid Change | Grantham Score | GERP Score | PolyPhen-2 | Inheritance |
|---|---|---|---|---|---|---|---|---|
| P1 | chr5: 67,591,018 | AATT/A | c.1615_1617delATT | p.Ile539del | - | - | - | de novo |
| P2 | chr5: 67,590,403 | G/A | c.1465G>A | p.Glu489Lys | 56 | 5.07 | possibly damaging (0.67) | de novo |
| P3 | chr5: 67,592,129 | C/T | c.1945C>T | p.Arg649Trp | 101 | 5.15 | probably damaging (1.00) | de novo |
| P4 | chr5: 67,592,129 | C/T | c.1945C>T | p.Arg649Trp | 101 | 5.15 | probably damaging (1.00) | absent in mother, father not available |
| P5 | chr5: 67,592,129 | C/T | c.1945C>T | p.Arg649Trp | 101 | 5.15 | probably damaging (1.00) | absent in mother, father not available |
| P6 | chr5: 67,592,129 | C/T | c.1945C>T | p.Arg649Trp | 101 | 5.15 | probably damaging (1.00) | inherited from mother (p5) |
| P7 | chr5: 67,592,129 | C/T | c.1945C>T | p.Arg649Trp | 101 | 5.15 | probably damaging (1.00) | parents not available |
| P8 | chr5: 67,592,127 | T/TT | c.1943dupT | p.Arg649Profs∗5 | - | - | - | parents not available |
| P9 | chr5: 67,592,076 | G/A | c.1892G>A | p.Arg631Gln | 43 | 5.15 | probably damaging (0.94) | parents not available |
Mutation nomenclature is based on RefSeq accession number NM_181523.2. Abbreviations are as follows: Ref, reference allele; alt, alternative allele; and GERP, Genomic Evolutionary Rate Profiling.
Class IA PI3Ks play a key role in insulin signaling through binding of phosphorylated insulin-receptor substrates, production of phosphatidylinositol-4,5-trisphosphate (PIP3), and subsequent activation of AKT serine-threonine kinase.15 A test for oral glucose tolerance was performed according to the recommended procedures16 and showed increased insulin levels in the three affected children tested (subjects P1, P2, and P3 [Figure 1I and Table S5]). Subjects P3 and P7 developed diabetes at 13.5 and 12 years of age, respectively, which explains their hyperglycemia and why their levels of serum insulin were lower than those of other affected individuals. Interestingly, in contrast to other monogenic forms of insulin resistance and lipodystrophy, SHORT syndrome resulting from mutations in PIK3R1 is not associated with severe hypertriglyceridemia (Table S5). Multiple lines of evidence indicate that the PIK3R1 mutations identified here should result in decreased PI3K activity. In humans, loss of insulin receptor (INSR) causes Donohue (MIM 246200) and Rabson-Mendenhall (MIM 262190) syndromes, both of which share many features with SHORT syndrome, including growth restriction, extreme insulin resistance, paucity of body fat, dysmorphic features, inguinal hernia, wrinkled skin, ovarian cysts, and/or dysplastic teeth.17 Mice carrying a heterozygous mutation abrogating p110α kinase activity display small body size, insulin resistance, and glucose intolerance.18 Interestingly, loss of AKT2 function is associated with severe insulin resistance, diabetes, and lipoatrophy in humans and mice,19,20 but unlike in SHORT syndrome, hypertriglyceridemia and increased de novo lipogenesis are part of the phenotype.21 Conversely, mutations in several PI3K-AKT-pathway-related genes result in increased PI3K signaling and cause severe brain and/or body overgrowth,5 and an activating mutation in AKT2 in humans was shown to cause severe hypoglycemia, weight gain, and somatic overgrowth,22 which is the opposite phenotype to SHORT syndrome. To assess the impact of PIK3R1 mutations, we evaluated the effects of short-term insulin stimulation on proximal (i.e., AKT, also known as protein kinase B) and distal (i.e., glycogen synthesis and glucose transport) PI3K-dependent insulin signaling in fibroblast cells derived from two individuals (P1 and P2) with PIK3R1 mutations and two unaffected males. Skin fibroblasts were prepared according to standard procedures, as described previously.23 Protein levels of PIK3R1 were evaluated by immunoblot on whole-cell lysates with antibodies to the p85α subunit (sc-423 [Z-8], Santa Cruz Biotechnology). Serum-depleted cells were incubated for 10 (for AKT detection) or 30 min (for the glycogen synthesis and the glucose-transport assays) with or without insulin (100 nMole/l). Insulin stimulation was evaluated by immunoblot analysis on whole-cell lysates with antibodies to AKT and phospho-AKT-ser473 (SC 8312 and SC 7985, Santa Cruz Biotechnology). Glycogen synthesis was evaluated by the addition of 14C-glucose (75 kBq/ml, PerkinElmer) to fibroblasts for 2 hr. Radiolabeled glycogen was then extracted and counted, as described previously.24 Similarly, glucose uptake was evaluated with 3H-2-deoxy-D-glucose (PerkinElmer). As shown in Figure 2, the studied PIK3R1 mutations did not affect the protein levels of PIK3R1 (Figure 2A). However, cells from both affected individuals displayed a markedly decreased effect of insulin on AKT activation, glycogen synthesis, and glucose uptake (by 70%–90%; Figures 2B and 2C), thus indicating severe insulin resistance for both proximal and distal PI3K-dependent signaling. These results might partly be explained by the increased levels of basal activity in cells from affected individuals (3- to 4-fold, 1.5- to 2-fold, and 1- to 2-fold increase for AKT activity, glycogen synthesis, and glucose uptake, respectively). This increased basal activity with further desensitization to insulin action has been described previously in other states of severe insulin resistance caused by mutations in INSR.24–26 Although further metabolic investigations will be needed for precisely deciphering the underlying signaling defects, our observations are consistent with impaired insulin-stimulated pathways, which lead to a generalized insulin-resistance syndrome affecting glucose homeostasis, growth, adipogenesis, and lipogenesis (Figure 1, Table 1, and Table S5).
Figure 2.
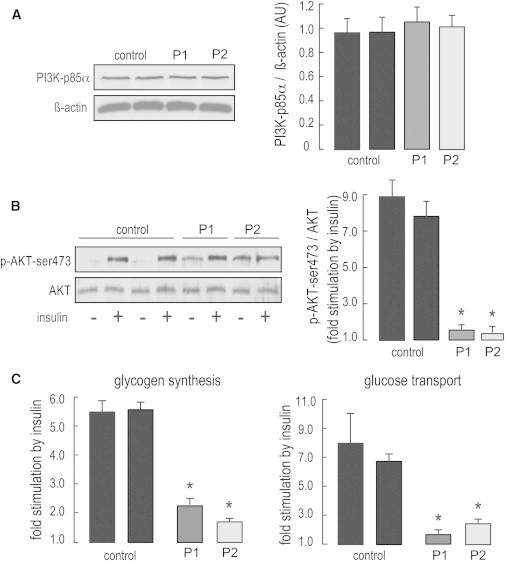
Cellular Insulin Resistance in Subjects with PIK3R1 Mutations
(A and B) PIK3R1 levels and short-term effects of insulin on proximal PI3K-dependent insulin signaling in fibroblasts from affected individuals (P1 and P2) and controls. PIK3R1 levels (A) and the effect of insulin on AKT activation (B) were evaluated by immunoblotting on whole-cell lysates. Intensities were quantified by densitometry scanning. PIK3R1 levels and AKT phosphorylation levels were normalized to β-actin and total AKT, respectively. Representative immunoblots are shown. The following abbreviation is used: p-AKT-ser473, phospho-AKT-ser473.
(C) Short-term effects of insulin on distal PI3K-dependent signaling. The effects of insulin on glycogen synthesis and glucose transport were evaluated by the incorporation of 14C-glucose into glycogen and the cellular uptake of 3H-2-deoxy-D-glucose, respectively. In fibroblasts from control subjects and individuals P1 and P2, basal glycogen synthesis was 22.3 ± 1.6, 54.6 ± 6.0, and 52.5 ± 0.4 pmoles of glucose incorporated into glycogen per mg protein per hour, respectively, and basal glucose transport was 10.5 ± 1.1, 23.2 ± 2.4, and 11.4 ± 1.5 pmole of cellular 2-deoxyglucose per mg protein per min, respectively.
Results are expressed as means ± SEM. Asterisks indicate a statistically significant difference compared to control cells (p < 0.05, Student’s t test).
Mice lacking Pik3r1 display enhanced insulin sensitivity and glucose tolerance,27 whereas p85 overexpression is associated with insulin resistance in skeletal muscle,28 thus indicating that p85 subunits act as negative regulators of PI3K signaling downstream of the insulin receptor. However, the mechanisms underlying negative regulation of insulin signaling by the PI3K regulatory subunits remain unclear.15 Two of the mutations identified here are located in PIK3R1’s inter-Src homology 2 (iSH2) domain (Figure 1J), which is a major interacting domain with p110 catalytic subunits. This interaction regulates PI3K activity through the formation of active and inactive dimers.29 Although further functional studies will be necessary for elucidating the exact mechanisms leading to SHORT syndrome, we can speculate that these mutations might affect the interaction between p85 regulatory subunits and p110 catalytic subunits and thus lead to decreased PI3K activity.
Other clinical SHORT syndrome features, such as bone demineralization, ovarian cysts, Rieger anomaly, and teething delay, are consistent with decreased PI3K-AKT signaling. Indeed, the PI3K-AKT pathway is critical for bone development, growth, and integrity and is a key regulator of osteoclastic bone resorption.30 Mice lacking both Akt1 and Akt2 display a severe bone-deficiency phenotype, and Akt1–/– mice exhibit growth deficiency, along with impaired bone growth early in life and mild osteopenia in adulthood.31 Besides severe diabetes and lipoatrophy, loss of Akt2 was shown to predispose mice to the formation of ovarian cysts.32 Finally, haploinsufficiency of PITX2, which is a downstream phosphorylation target of AKT2,33 causes Axenfeld-Rieger syndrome (MIM 602482), a condition that has significant clinical overlap with SHORT syndrome and is characterized by anterior-segment eye defects and umbilical and teeth anomalies.34
Because individuals with SHORT syndrome are usually born small for gestational age (SGA), which predisposes them to permanent short stature,35 two of the subjects reported here (P1 and P2) were being treated by growth hormone (GH). This treatment was approved in 2001 and 2003 in the United States and Europe, respectively, for SGA children without catch-up growth after the age of 4 years. However, GH treatment is known to induce hyperinsulinemia and to decrease insulin sensitivity; this phenomenon might aggravate insulin resistance and accelerate the onset of diabetes in subjects with SHORT syndrome, particularly during puberty, when insulin resistance is observed in both normal and diabetic children.36 Given the presence of diabetes in all adult PIK3R1-mutation-positive individuals presented here (Table 1), the well-known association between insulin resistance and PI3K-pathway dysfunction, and the fact that GH-induced insulin resistance is rapidly reversible upon treatment cessation,37 GH treatment was ceased in both subjects. This well illustrates the fact that genetic findings can have direct implications for care practice by enabling a more personalized clinical management.
In conclusion, our findings provide important information with respect to the function of PIK3R1 in normal development and its role in human diseases, including growth delay, Rieger anomaly and other ocular affections, insulin resistance, diabetes, paucity of fat, and ovarian cysts. The relatively mild clinical presentation of several of the subjects reported here suggests that SHORT syndrome might be underdiagnosed and that mutations in PIK3R1 might be a rare cause of apparently nonsyndromic insulin resistance in lean cases.
Acknowledgments
We wish to thank the subjects and families involved in the study, PerkinElmer DNA Sequencing Service for the whole-exome-sequencing experiments, the University of Burgundy Centre de Calcul (see Web Resources) for technical support and management of the informatics platform, and the Dyscerne network (see Web Resources) for their help with this project. This work was supported by the Regional Council of Burgundy, a French government grant (ANR-10-IAHU) managed by the Agence Nationale de la Recherche (program “Investments for the Future,” Institute of Cardiometabolism and Nutrition), the Institut de la Santé et de la Recherche Médicale, and the Université Pierre et Marie Curie. We also thank the National Heart, Lung, and Blood Institute Grand Opportunity (GO) Exome Sequencing Project (see Web Resources) and its ongoing studies, which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the Women’s Health Initiative Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010).
Contributor Information
Christel Thauvin-Robinet, Email: christel.thauvin@chu-dijon.fr.
Jean-Baptiste Rivière, Email: jean-baptiste.riviere@u-bourgogne.fr.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
Dyscerne, http://www.dyscerne.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
SeattleSeq Annotation 137, http://snp.gs.washington.edu/SeattleSeqAnnotation137/
University of Burgundy Centre de Calcul, https://haydn2005.u-bourgogne.fr/dsi-ccub/
References
- 1.Semple R.K., Savage D.B., Cochran E.K., Gorden P., O’Rahilly S. Genetic syndromes of severe insulin resistance. Endocr. Rev. 2011;32:498–514. doi: 10.1210/er.2010-0020. [DOI] [PubMed] [Google Scholar]
- 2.Koenig R., Brendel L., Fuchs S. SHORT syndrome. Clin. Dysmorphol. 2003;12:45–49. doi: 10.1097/00019605-200301000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Karadeniz N.N., Kocak-Midillioglu I., Erdogan D., Bökesoy I. Is SHORT syndrome another phenotypic variation of PITX2? Am. J. Med. Genet. A. 2004;130A:406–409. doi: 10.1002/ajmg.a.30206. [DOI] [PubMed] [Google Scholar]
- 4.Reis L.M., Tyler R.C., Schilter K.F., Abdul-Rahman O., Innis J.W., Kozel B.A., Schneider A.S., Bardakjian T.M., Lose E.J., Martin D.M. BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum. Genet. 2011;130:495–504. doi: 10.1007/s00439-011-0968-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rivière J.B., Mirzaa G.M., O’Roak B.J., Beddaoui M., Alcantara D., Conway R.L., St-Onge J., Schwartzentruber J.A., Gripp K.W., Nikkel S.M., Finding of Rare Disease Genes (FORGE) Canada Consortium De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rivière J.B., van Bon B.W., Hoischen A., Kholmanskikh S.S., O’Roak B.J., Gilissen C., Gijsen S., Sullivan C.T., Christian S.L., Abdul-Rahman O.A. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat. Genet. 2012;44:440–444. doi: 10.1038/ng.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelman J.A., Luo J., Cantley L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 10.Bonnel S., Dureau P., LeMerrer M., Dufier J.L. SHORT syndrome: a case with high hyperopia and astigmatism. Ophthalmic Genet. 2000;21:235–238. [PubMed] [Google Scholar]
- 11.Cooper G.M., Goode D.L., Ng S.B., Sidow A., Bamshad M.J., Shendure J., Nickerson D.A. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat. Methods. 2010;7:250–251. doi: 10.1038/nmeth0410-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 13.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conley M.E., Dobbs A.K., Quintana A.M., Bosompem A., Wang Y.D., Coustan-Smith E., Smith A.M., Perez E.E., Murray P.J. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. J. Exp. Med. 2012;209:463–470. doi: 10.1084/jem.20112533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taniguchi C.M., Emanuelli B., Kahn C.R. Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 16.Bartoli E., Fra G.P., Carnevale Schianca G.P. The oral glucose tolerance test (OGTT) revisited. Eur. J. Intern. Med. 2011;22:8–12. doi: 10.1016/j.ejim.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Longo N., Wang Y., Smith S.A., Langley S.D., DiMeglio L.A., Giannella-Neto D. Genotype-phenotype correlation in inherited severe insulin resistance. Hum. Mol. Genet. 2002;11:1465–1475. doi: 10.1093/hmg/11.12.1465. [DOI] [PubMed] [Google Scholar]
- 18.Foukas L.C., Claret M., Pearce W., Okkenhaug K., Meek S., Peskett E., Sancho S., Smith A.J., Withers D.J., Vanhaesebroeck B. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature. 2006;441:366–370. doi: 10.1038/nature04694. [DOI] [PubMed] [Google Scholar]
- 19.Garofalo R.S., Orena S.J., Rafidi K., Torchia A.J., Stock J.L., Hildebrandt A.L., Coskran T., Black S.C., Brees D.J., Wicks J.R. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J. Clin. Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.George S., Rochford J.J., Wolfrum C., Gray S.L., Schinner S., Wilson J.C., Soos M.A., Murgatroyd P.R., Williams R.M., Acerini C.L. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–1328. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Semple R.K., Sleigh A., Murgatroyd P.R., Adams C.A., Bluck L., Jackson S., Vottero A., Kanabar D., Charlton-Menys V., Durrington P. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J. Clin. Invest. 2009;119:315–322. doi: 10.1172/JCI37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hussain K., Challis B., Rocha N., Payne F., Minic M., Thompson A., Daly A., Scott C., Harris J., Smillie B.J. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334:474. doi: 10.1126/science.1210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Villegas J., McPhaul M. Establishment and culture of human skin fibroblasts. Curr. Protoc. Mol. Biol. 2005;Chapter 28 doi: 10.1002/0471142727.mb2803s71. 28, 3. [DOI] [PubMed] [Google Scholar]
- 24.Desbois-Mouthon C., Danan C., Amselem S., Blivet-Van Eggelpoel M.J., Sert-Langeron C., Goossens M., Besmond C., Capeau J., Caron M. Severe resistance to insulin and insulin-like growth factor-I in cells from a patient with leprechaunism as a result of two mutations in the tyrosine kinase domain of the insulin receptor. Metabolism. 1996;45:1493–1500. doi: 10.1016/s0026-0495(96)90178-x. [DOI] [PubMed] [Google Scholar]
- 25.Quon M.J., Cama A., Taylor S.I. Postbinding characterization of five naturally occurring mutations in the human insulin receptor gene: impaired insulin-stimulated c-jun expression and thymidine incorporation despite normal receptor autophosphorylation. Biochemistry. 1992;31:9947–9954. doi: 10.1021/bi00156a013. [DOI] [PubMed] [Google Scholar]
- 26.Formisano P., Sohn K.J., Miele C., Di Finizio B., Petruzziello A., Riccardi G., Beguinot L., Beguinot F. Mutation in a conserved motif next to the insulin receptor key autophosphorylation sites de-regulates kinase activity and impairs insulin action. J. Biol. Chem. 1993;268:5241–5248. [PubMed] [Google Scholar]
- 27.Fruman D.A., Mauvais-Jarvis F., Pollard D.A., Yballe C.M., Brazil D., Bronson R.T., Kahn C.R., Cantley L.C. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85 alpha. Nat. Genet. 2000;26:379–382. doi: 10.1038/81715. [DOI] [PubMed] [Google Scholar]
- 28.Barbour L.A., Shao J., Qiao L., Leitner W., Anderson M., Friedman J.E., Draznin B. Human placental growth hormone increases expression of the p85 regulatory unit of phosphatidylinositol 3-kinase and triggers severe insulin resistance in skeletal muscle. Endocrinology. 2004;145:1144–1150. doi: 10.1210/en.2003-1297. [DOI] [PubMed] [Google Scholar]
- 29.Miled N., Yan Y., Hon W.C., Perisic O., Zvelebil M., Inbar Y., Schneidman-Duhovny D., Wolfson H.J., Backer J.M., Williams R.L. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317:239–242. doi: 10.1126/science.1135394. [DOI] [PubMed] [Google Scholar]
- 30.Mukherjee A., Rotwein P. Selective signaling by Akt1 controls osteoblast differentiation and osteoblast-mediated osteoclast development. Mol. Cell. Biol. 2012;32:490–500. doi: 10.1128/MCB.06361-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng X.D., Xu P.Z., Chen M.L., Hahn-Windgassen A., Skeen J., Jacobs J., Sundararajan D., Chen W.S., Crawford S.E., Coleman K.G., Hay N. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17:1352–1365. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Restuccia D.F., Hynx D., Hemmings B.A. Loss of PKBβ/Akt2 predisposes mice to ovarian cyst formation and increases the severity of polycystic ovary formation in vivo. Dis Model Mech. 2012;5:403–411. doi: 10.1242/dmm.008136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gherzi R., Trabucchi M., Ponassi M., Gallouzi I.E., Rosenfeld M.G., Briata P. Akt2-mediated phosphorylation of Pitx2 controls Ccnd1 mRNA decay during muscle cell differentiation. Cell Death Differ. 2010;17:975–983. doi: 10.1038/cdd.2009.194. [DOI] [PubMed] [Google Scholar]
- 34.Semina E.V., Reiter R., Leysens N.J., Alward W.L., Small K.W., Datson N.A., Siegel-Bartelt J., Bierke-Nelson D., Bitoun P., Zabel B.U. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat. Genet. 1996;14:392–399. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- 35.Karlberg J., Albertsson-Wikland K. Growth in full-term small-for-gestational-age infants: from birth to final height. Pediatr. Res. 1995;38:733–739. doi: 10.1203/00006450-199511000-00017. [DOI] [PubMed] [Google Scholar]
- 36.Amiel S.A., Sherwin R.S., Simonson D.C., Lauritano A.A., Tamborlane W.V. Impaired insulin action in puberty. A contributing factor to poor glycemic control in adolescents with diabetes. N. Engl. J. Med. 1986;315:215–219. doi: 10.1056/NEJM198607243150402. [DOI] [PubMed] [Google Scholar]
- 37.Krusenstjerna-Hafstrøm T., Clasen B.F., Møller N., Jessen N., Pedersen S.B., Christiansen J.S., Jørgensen J.O. Growth hormone (GH)-induced insulin resistance is rapidly reversible: an experimental study in GH-deficient adults. J. Clin. Endocrinol. Metab. 2011;96:2548–2557. doi: 10.1210/jc.2011-0273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.