

Abstract
Intellectual disability (ID), often attributed to autosomal-recessive mutations, occurs in 40% of autism spectrum disorders (ASDs). For this reason, we conducted a genome-wide analysis of runs of homozygosity (ROH) in simplex ASD-affected families consisting of a proband diagnosed with ASD and at least one unaffected sibling. In these families, probands with an IQ ≤ 70 show more ROH than their unaffected siblings, whereas probands with an IQ > 70 do not show this excess. Although ASD is far more common in males than in females, the proportion of females increases with decreasing IQ. Our data do support an association between ROH burden and autism diagnosis in girls; however, we are not able to show that this effect is independent of low IQ. We have also discovered several autism candidate genes on the basis of finding (1) a single gene that is within an ROH interval and that is recurrent in autism or (2) a gene that is within an autism ROH block and that harbors a homozygous, rare deleterious variant upon analysis of exome-sequencing data. In summary, our data suggest a distinct genetic architecture for participants with autism and co-occurring intellectual disability and that this architecture could involve a role for recessively inherited loci for this autism subgroup.
Main Text
Autism spectrum disorders (ASDs [MIM 209850]) represent a heterogeneous group of neurodevelopmental disorders with a wide range of cognitive function. Intellectual disability (ID) (defined as an IQ ≤ 70 and diminished adaptive function) occurs in 40% of autism cases.1 Many more males than females are affected by autism, but the proportion of affected females is increased for affected individuals with a lower IQ.1,2 Although there has been substantial progress regarding de novo mutations, identification of inherited loci in autism has been challenging. Many related neurodevelopmental conditions such as ID are associated with X-linked recessive and/or autosomal-recessive inherited loci. To date, there have been few genome-wide studies designed to examine excess homozygosity in autism, particularly in pedigrees without recent shared ancestry,3–7 which could indicate the effect of autosomal-recessive loci.
Because of the important role played by autosomal-recessively inherited loci in ID and related neurodevelopmental disorders,8,9 in this study, we investigated a possible role for autosomal-recessive loci through the study of autosomal runs of homozygosity (ROH) with the use of SNP arrays. We analyzed the Simons Simplex Collection (SSC), composed of 2,108 North American simplex ASD-affected families.10 These families were recruited for the presence of a single affected offspring and were phenotyped for ensuring that siblings and parents were unaffected (i.e., had no ASDs, learning disorders, broader autism phenotypes, or related psychopathologies). We were able to capitalize on the recruitment of the unaffected sibling as a matched control to compare the burden of ROH. We tested the hypothesis that ROH is associated with affected status in those children with autism and co-occurring ID. Our data indicate that ROH is associated with affected status in individuals with a full-scale IQ ≤ 70 and also that low IQ is a predictor of increased ROH. Although girls with autism appear to have a higher burden of ROH than do their same-sex unaffected siblings, our data cannot discern whether this association between female gender in autism and increased ROH is independent of low IQ. Overall, our analysis supports significant differences in the genetic architecture for children with autism and low IQ. Although other causes are possible, the simplest explanation for increased ROH is that autosomal-recessive loci play an important part in the genetic susceptibility to autism with co-occurring ID.
At the time of writing this manuscript, the SSC data set included 2,108 genotyped families. All research was approved by the appropriate institutional review boards, and informed consent was obtained. The pedigree structure involves one affected child per pedigree (i.e., simplex) for a total of 3,941 children. Of these pedigrees, 1,656 had a quartet structure with unaffected parents, one proband, and one unaffected sibling. The remaining 452 families were trios without an unaffected sibling and were not analyzed in the case-control study. The overall male-to-female ratio for probands was 6.4:1, and for probands with an IQ ≤ 70, the male-to-female ratio was 4.7:1. Phenotype and ancestry information can be found in recent publications.10,11 Girls with autism appear to be lower functioning than boys in this SSC sample, as has been described previously for other data sets.12 The average full-scale IQ was 82.9 for boys and 75.3 for girls (p < 0.001). Composite scores on the Vineland Adaptive Behavior Scale were 73.4 for boys and 71.4 for girls (p < 0.001). Affected boys and girls had equivalent calibrated Autism Diagnostic Observation Schedule (ADOS) severity scores: 7.4 for boys and 7.4 for girls (p = 0.68).
Noting that the parents do not have known shared ancestry, we evaluated autosomal homozygosity as a potential marker of autosomal-recessive loci in this SSC cohort. Individuals in the SSC were genotyped on one of three different array versions—Illumina 1Mv1 (327 families), Illumina 1Mv3 Duo (1,159 families), or Illumina HumanOMNI 2.5 (622 families)—at the Keck Foundation Yale Center for Genomic Analysis. Members of each family were analyzed on the same array version.11 Illumina 1Mv1 and 1Mv3 Duo have 1,040,853 probes in common (97% of probes in the 1Mv1 and 87% of probes in the 1Mv3).11 The HumanOMNI 2.5 array has 2,450,000 probes, including single-nucleotide variants (SNVs) with a minor allele frequency down to 1%. For the HumanOMNI 2.5 arrays, rare SNVs with an allele frequency below 2.5% were removed for the elimination of spurious calls of homozygosity due to the largely monomorphic major allele of these variants. In the Illumina 1Mv3 Duo array, 53,405 CNVi probes were excluded in ROH analysis.
We used PLINK version 1.0713 to identify autosomal ROH. To prevent the program from underestimating ROHs as a result of an occasional genotyping error or missing genotype, we allowed one heterozygous and five missing calls per window. We analyzed a range of minimum segment lengths, including 1,000 kb, 1,750 kb, 2,000 kb, 2,500 kb, 3,000 kb, and 3,500 kb, as the lower thresholds for calling a ROH (Figure 1 and Table S1, available online). Data from the X and Y chromosomes were not analyzed.
Figure 1.
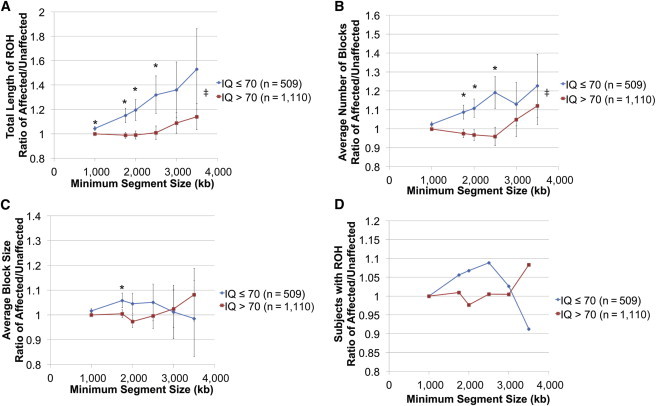
The Burden of Autosomal ROH Is Higher in Autism-Affected Individuals with IQ ≤ 70 than in Unaffected Siblings in Simplex Pedigrees
Autosomal ROH burden was higher in probands with an IQ ≤ 70 (blue) than in unaffected siblings in the SSC data set. This effect was not observed in probands with an IQ > 70 (red). Each ratio is plotted for different minimum segment sizes used for determining a block of ROH. Asterisks represent the statistical test if the point is different from a ratio of 1, i.e., the proband and unaffected sibling have equal values (p value < 0.05). Specific p values are presented in Table S1. Conditional-logistic-regression analysis in STATA version 11.1 was performed for comparing probands to their designated siblings. Error bars represent the SEM. Ratios were computed in STATA version 11.1 with standard errors. ‡ symbols represent the statistical test if the IQ ≤ 70 curve is different from the IQ > 70 curve (p value < 0.05). A one-way ANOVA was performed for comparing all data points for IQ ≤ 70 and > 70. The ANOVA p value was 0.01 for (A) and (B). The number of discordant sibling pairs, indicated by n, contributed to the data. Further information regarding this association analysis can be found in Table S1.
(A) The ratio of total length of ROH per individual for probands compared to designated unaffected siblings is plotted.
(B) The ratio of the average number of blocks per individual for probands compared to designated unaffected siblings is plotted.
(C) The ratio of the average block size per individual for probands compared to designated unaffected siblings is plotted.
(D) The ratio between the proportion of probands with at least one ROH block and the proportion of designated unaffected siblings with at least one ROH block is plotted.
We ensured that measurements of ROH were not confounded by large copy-number variants (CNVs) (i.e., genomic deletions that appear as a segment of homozygosity but actually reflect hemizygosity). First, we discovered 59 children for whom a ROH call (minimum segment size of 1,000 kb or larger) was actually a large CNV. These segments of hemizygosity were removed from the ROH data set. Second, we examined the logR data regarding probe intensity for the probes contained within all blocks of ROH for skewness toward nonzero values (Figure S1). By this method, we found little evidence to suggest that analysis of ROH was confounded by overlapping CNVs. Third and finally, the size of ROH argued against false-negative CNV calls. For example, 40% of study participants had at least one ROH with a minimum segment size greater than 2,500 kb. CNV calling methods are excellent at predicting CNVs of this large size (positive predictive value of 96%11). Notably, the proportion of autism probands with CNVs of this size was far lower than 40% at <2.3%.11 Finally, we examined the logR data directly for the most recurrent ROH intervals in autism for each relevant family, and these data did not support the presence of large CNVs within these ROH intervals (Figures S2–S5).
From the outset, we chose for our major analyses to involve strictly within-family studies. The family-based discordant sibling-pair design of the SSC study allows for proband-sibling comparisons that mitigate a wide range of technical and methodological confounders, such as those introduced by differences in ancestry.14 When we compared total burden of autosomal homozygosity between affected children and unaffected children across the study without regard to family, we did not observe differences (data not shown). As our major analysis, we compared ROH measurements between affected participants and their matched unaffected siblings. In addition, we stratified the discordant sibling pairs into two groups: those with probands with a full-scale IQ ≤ 70 and those with probands with an IQ > 70. We varied the minimum size limit for the definition of ROH between 1,000 and 3,500 kb (Figure 1 and Table S1). As the lower limit of the final segment for ROH is increased, the expectation is that the blocks are derived from a more recent shared ancestor. At a minimum final segment size of 1,000 kb or greater, every participant in the study had at least one ROH (Table S1). At an increased minimum segment size of 1,750 kb, 2,500 kb, and 3,500 kb, the proportion of controls with at least one ROH decreased to 86.2%, 37.5%, and 10%, respectively (Table S1). Conditional logistic regression contrasted probands with their designated siblings for the number of ROH blocks, total length of ROH, and average block size. (Similar results were obtained from the Wilcoxon matched-pairs signed-rank test.)
We discovered that affected status is associated with an increased burden of ROH in probands with an IQ ≤ 70. As the minimum block size was increased, we observed that the ROH burden was higher in affected probands with an IQ ≤ 70 than in their unaffected matched siblings, but no such excess was seen for an IQ > 70 (Figure 1 and Table S1). For example, at a minimum segment size ≥ 2,500 kb, the total burden of ROH in probands with an IQ ≤ 70 was 1.32× greater than that of their unaffected siblings (p = 0.03) (Figure 1A). By contrast, the ratio of the ROH burden in affected versus unaffected siblings in the cohort with a proband IQ > 70 was 1.01 (p = 0.89). The average number of ROH seems to be the major factor associated with the increased burden (Figure 1B) because the average length of ROH and proportion of subjects carrying ROH segments were only negligibly higher than those of the controls (i.e., ratios were generally less than 1.1) (Figures 1C and 1D). We also studied IQ < 55 in autism. Relative to their unaffected siblings, probands with an IQ < 55 were observed to have rising ROH burden with increasing minimum segment sizes. We ran conditional-regression analysis to test the statistical significance, and when the minimum segment sizes were between 1,000 and 1,750 kb, statistical significance was obtained (p < 0.05). Compared to that of controls, the ROH burden for probands with an IQ from 55 to 70 increased substantially between the minimum segment sizes of 1,000 and 3,500 kb. However, because of the low sample size (n = 208), statistical significance was not obtained (Table S2). The same trend was observed for the average number of blocks (Table S2).
As has been true for other forms of genetic variation, such as CNVs, ROH segments are widely distributed across the entire genome (Figure 2). There exist genomic regions that contain more ROH in affected probands than in controls. Despite the large sample size, the statistical significance for any given single locus failed to survive conservative genome-wide correction (Figure S6). The most highly recurrent locus reached p values between 0.015 and 0.0383. The complete set of loci and candidate genes recurrent in autism (with a p value less than 0.2) is shown in Table S3. Of note, several regions contain only a single gene or very few genes. The genes implicated in this way include CADM2 (MIM 609938) on chromosome 3, PLCXD3 and FGF10 (MIM 602115) on chromosome 5, and UNC5D and RIPK2 (MIM 603455) on chromosome 8. Interestingly, both CADM2 and FGF10 have been previously implicated as candidate genes by a homozygosity approach in autism.4 Also of note, one region on chromosome 8 contains TRAPPC9 (MIM 611966), a gene known to be mutated in autosomal-recessive ID.15–17 Overlapping blocks in four examples of recurrent regions and relative logR and B allele frequency (BAF) values are shown in Figures S2–S5. According to relative logR and BAF data for all relevant families, we were unable to detect any CNVs or heterozygous regions for the probands for whom the ROH blocks were called.
Figure 2.
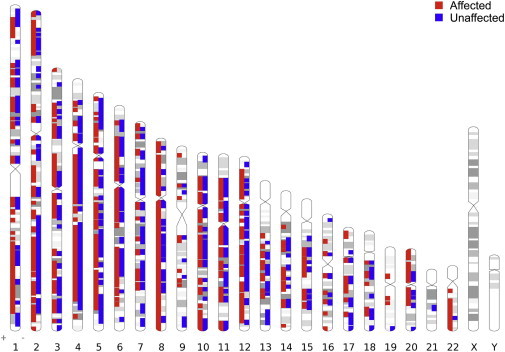
Genome-wide Distribution of ROH Blocks for the Minimum Final Segment Size of 2,500 kb According to Disease Status
Genome-wide distribution of ROH blocks for the minimum final segment size of 2,500 kb is shown and was created with Idiographica (see Web Resources). Chromosomes X and Y were excluded from the analysis. All ROH blocks, which occur at least once in a subject, are shown.
Further, we investigated whether there are homozygous, rare deleterious coding variants in any of the genes within intervals in which ROH was more common in affected probands than in their control siblings (Table S4) by using available exome-sequencing data for the SSC data set.18,19,20 Although sequence data were only available for a subset of the participants in this study, we were able to find several candidate mutations. We identified putative deleterious, homozygous missense variants in affected individuals in two distinct dynein-encoding genes. For example, one affected individual (with an IQ of 28) exhibited a homozygous missense variant (a highly conserved uncharged glutamine to positively charged histidine in dynein, axonemal, heavy chain 7 [DNAH7 (MIM 610061)]) that transmitted in the pedigree, i.e., was homozygous in the proband. We also identified putative deleterious, homozygous variants in BMP3 (MIM 112263) and BMP5 (MIM 112265) in two different affected individuals (Table S4). Determining a potential role in disease for each of these variants will require further investigation.
We next set out to investigate genotype-phenotype correlations for ROH in the group of affected individuals in the SSC. Across four groupings of participants (females with and without ROH and males with and without ROH), we examined the association between ROH (defined as a segment ≥ 2,500 kb) and IQ measures, Vineland Adaptive Behavior Scale scores, the presence of epilepsy, and autism measures such as the Autism Diagnostic Interview, Revised (ADI-R), the ADOS, and the calibrated ADOS severity score. We conducted bivariate analyses to determine the association between group (ROH or non-ROH) and phenotypic characteristics. For continuous variables (cognition, adaptive functioning, and autism symptoms), the mean and SD were calculated in each group. We used the generalized linear model to compute least-squares means for each group. We calculated the p values for differences of the least-squares means to compare groups to each other. For categorical variables (epilepsy), we calculated the number and percentage in each group and computed a chi-square p value.
Within the autism cohort, we noticed a strong statistical association between ROH and measures of intellectual (IQ) and adaptive (Vineland) function, but not measures of autism symptoms or severity (Figure 3). With a minimum ROH ≥ 2,500 kb, girls with ASD and carrying at least one ROH consistently demonstrated lower IQ and Vineland scores than did other groups. For example, the mean full-scale IQ for probands was 81.9 for the entire sample and 74.0 for female probands with ROH (Figure 3 and Table S5). Relative to ASD males without ROH, ASD boys with ROH also showed statistically significant impairment in cognition and functioning (Figure 3 and Table S5). Males with autism and ROH had IQ levels approximately five points lower than did autism boys without ROH (IQ of 80.2 with ROH versus IQ of 84.4 without ROH, p = 0.002). Based on ADI-R and ADOS scores, autism symptoms did not show distinct differences across these groups (Figure 3 and Table S5). Of note, the highest rate of epilepsy was also observed in girls carrying at least one ROH (6.8%) as compared to affected females without ROH (1.2%) (p < 0.02, chi-square = 6.232, 1 degree of freedom).
Figure 3.
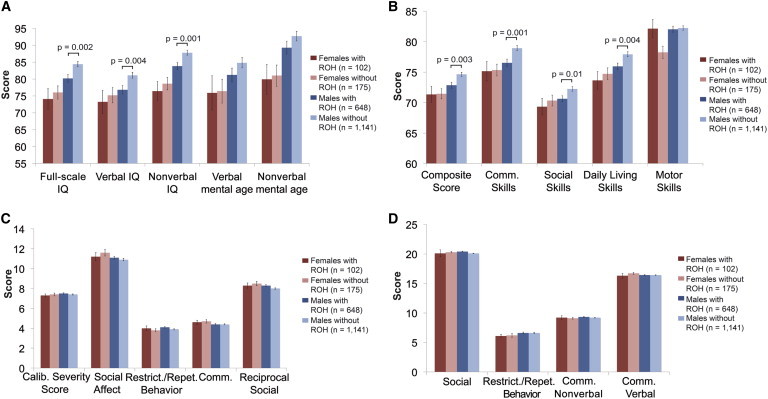
Children with Autism and Autosomal ROH Have Lower Cognitive Function and Adaptive Function
(A) Comparison of IQ tests for autism probands with and without autosomal ROH (minimum final segment size of 2,500 kb). The y axis indicates the mean score, and error bars represent the SEM.
(B) Comparison of adaptive functioning based on Vineland scores for autism probands with and without ROH (minimum final segment size of 2,500 kb). The y axis indicates mean composite scores for each group, and error bars represent the SEM.
(C) Comparison of autistic symptoms based on ADOS scores for autism probands with and without ROH (minimum final segment size of 2,500 kb). The y axis indicates mean ADOS scores for each group, and error bars represent the SEM.
(D) Comparison of autistic symptoms based on ADI-R scores for autism probands with and without ROH (minimum final segment size of 2,500 kb). The y axis indicates mean ADI-R scores for each group, and error bars represent the SEM.
All tests were conducted in SAS version 9.2. Statistically significant p values (<0.05) are shown in the graphs. Further information regarding phenotype-genotype analysis can be found in Table S5.
Given the increase in proportion of girls with autism in the IQ ≤ 70 group, we tested the hypothesis that girls with autism have an increased burden of ROH regardless of IQ level, but this contrast was not significant (data not shown). Given the possibility of reduced penetrance of autism susceptibility loci in females compared to males, we then strictly tested same-sex-discordant sibling pairs. As shown in Figure S7, compared to their same-sex siblings, girls with autism revealed a statistically significant burden of ROH (p < 0.01), whereas relative to their male unaffected siblings, boys with autism did not show this pattern (Table S6). However, when we conducted statistical models for predictors of ROH by using logistic-regression analysis, only ID was independently associated with ROH burden (Table S7). Although female gender might be associated with increased ROH in autism, we were not able to disentangle female gender from low IQ with regard to this association with increased ROH in autism (Table S7).
Many genome-wide studies have focused on de novo genetic variants that confer risk in autism; however, in the current study, we have potentially identified an inherited component of the genetic architecture. The methods of Kong et al.21 estimate that a block of homozygosity that is 2,500 kb dates to a common ancestor approximately 40 generations back in the pedigree, or approximately 1,000 years ago (Figure S8A). We also estimated the generational age of ROH blocks with a minimum segment size of 2,500 kb for probands and siblings (Figures S8B and S8C). The effect size of the genome-wide burden measures appears to decrease with the addition of older and smaller loci, suggesting that as a group, more recent ROH blocks have a greater effect size (Figure 1). On the basis of these estimates, it is evident that these inherited ROH risk loci are distinct from the de novo variation in the genetic architecture.
In summary, we conducted a genome-wide analysis of ROH in the SSC data set and present two principal findings. First, autism involving ID is associated with an increased burden of ROH in affected probands compared to unaffected siblings in these simplex pedigrees. Second, among affected probands, we observed a strong association between autosomal homozygosity and the level of intellectual and adaptive function. Individuals with lower functioning appear more likely to harbor at least one large block of homozygosity. Relative to prior studies of homozygosity in autism or other neuropsychiatric disease, our study has several strengths. Our data set capitalizes on the internal sibling control that circumvents the challenging confounds regarding population stratification, a luxury other studies did not have. Our study argues that autosomal-recessive loci might contribute to an important part of the genetic architecture of autism in low-IQ probands at least in simplex autism. In addition, we present data that support a model involving a distinct genetic architecture in girls and autism-affected individuals with low IQ. Our data are consistent with the possibility that the effect seen in girls is principally driven by level of IQ.
The mechanisms through which ROH loci confer susceptibility to autism involving ID remain to be proven. Much of the excess ROH might map onto a heterogeneous group of recessive loci, that is, a series of loci wherein loss-of-function or hypomorphic alleles are homozygous and thereby confer disease susceptibility. In addition to loss-of-function alleles, homozygous hypermorphic alleles might also confer susceptibility to disease. Further, as opposed to coding variants alone, variants that alter gene-expression levels might be particularly pertinent. In several ways, ROH loci appear comparable to large genomic CNVs that are well known to be associated with autism and ID.22 Similar to ROH, CNVs are heterogeneously distributed across the genome and can confer reduced dosage (deletions) or increased dosage (duplications) mechanisms. Another possible interpretation regarding ROH loci is that, like CNVs, some ROH loci can have features of a contiguous gene disorder. Instead of a single variant's conferring disease susceptibility, there might be additive or interactive effects of contiguous variants within an ROH interval. Further analysis of DNA sequence within ROH loci will be required for identifying and/or distinguishing the mechanisms by which ROH might confer susceptibility to low-IQ autism as observed here.
Acknowledgments
This work was supported by Simons Foundation Autism Research Initiative (SFARI) grant 124827EM to E.M.M. We are grateful to all of the families at the participating Simons Simplex Collection (SSC) sites, as well as the principal investigators (A.L. Beaudet, R. Bernier, J. Constantino, E.H.C., Jr., E. Fombonne, D.H.G., E. Hanson, D.E. Grice, A. Klin, R. Kochel, D. Ledbetter, C. Lord, C. Martin, D.M. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M.W.S., W. Stone, J.S. Sutcliffe, C.A. Walsh, Z. Warren, and E. Wijsman). We appreciate obtaining access to phenotypic data in SFARI Base. Approved researchers can obtain the SSC population data set described in this study by applying at https://base.sfari.org. E.M.M. was also supported by the Career Award in Medical Science from the Burroughs Wellcome Fund and National Institutes of Health grant 5K23MH080954-05. E.D.G. is the first Brown University Alpert Medical School Translational Neuroscience Postdoctoral Fellow jointly sponsored by the Lifespan Research Institute, the Lifespan Division of Psychiatry, the Brown Institute for Brain Science, and the Norman Prince Neurosciences Institute. E.W.V. receives partial support from the Brown Institute for Brain Science. We would like to acknowledge the strong collaborations of the SSC Genetics Consortium, led by M.W.S. and also including B.D., J.S. Sutcliffe, C.A. Walsh, D.H.G., E.H.C., Jr, R.M. Cantor, D.E. Grice, A.L. Beaudet, D.M. Martin, and E.M.M.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Idiographica, http://www.ncrna.org/idiographica
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SFARI Base, https://sfari.org/resources/sfari-base
References
- 1.Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators. Centers for Disease Control and Prevention Prevalence of autism spectrum disorders—Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill. Summ. 2012;61:1–19. [PubMed] [Google Scholar]
- 2.Fombonne E. Epidemiology of pervasive developmental disorders. Pediatr. Res. 2009;65:591–598. doi: 10.1203/PDR.0b013e31819e7203. [DOI] [PubMed] [Google Scholar]
- 3.Chahrour M.H., Yu T.W., Lim E.T., Ataman B., Coulter M.E., Hill R.S., Stevens C.R., Schubert C.R., Greenberg M.E., Gabriel S.B., Walsh C.A., ARRA Autism Sequencing Collaboration Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012;8:e1002635. doi: 10.1371/journal.pgen.1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casey J.P., Magalhaes T., Conroy J.M., Regan R., Shah N., Anney R., Shields D.C., Abrahams B.S., Almeida J., Bacchelli E. A novel approach of homozygous haplotype sharing identifies candidate genes in autism spectrum disorder. Hum. Genet. 2012;131:565–579. doi: 10.1007/s00439-011-1094-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novarino G., El-Fishawy P., Kayserili H., Meguid N.A., Scott E.M., Schroth J., Silhavy J.L., Kara M., Khalil R.O., Ben-Omran T. Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science. 2012;338:394–397. doi: 10.1126/science.1224631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrow E.M., Yoo S.Y., Flavell S.W., Kim T.K., Lin Y., Hill R.S., Mukaddes N.M., Balkhy S., Gascon G., Hashmi A. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L.S., Hranilovic D., Wang K., Lindquist I.E., Yurcaba L., Petkovic Z.B., Gidaya N., Jernej B., Hakonarson H., Bucan M. Population-based study of genetic variation in individuals with autism spectrum disorders from Croatia. BMC Med. Genet. 2010;11:134. doi: 10.1186/1471-2350-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ropers H.H. Genetics of intellectual disability. Curr. Opin. Genet. Dev. 2008;18:241–250. doi: 10.1016/j.gde.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 9.Basel-Vanagaite L. Genetics of autosomal recessive non-syndromic mental retardation: recent advances. Clin. Genet. 2007;72:167–174. doi: 10.1111/j.1399-0004.2007.00881.x. [DOI] [PubMed] [Google Scholar]
- 10.Fischbach G.D., Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–195. doi: 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amiet C., Gourfinkel-An I., Bouzamondo A., Tordjman S., Baulac M., Lechat P., Mottron L., Cohen D. Epilepsy in autism is associated with intellectual disability and gender: evidence from a meta-analysis. Biol. Psychiatry. 2008;64:577–582. doi: 10.1016/j.biopsych.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 13.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altshuler D., Daly M.J., Lander E.S. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mir A., Kaufman L., Noor A., Motazacker M.M., Jamil T., Azam M., Kahrizi K., Rafiq M.A., Weksberg R., Nasr T. Identification of mutations in TRAPPC9, which encodes the NIK- and IKK-beta-binding protein, in nonsyndromic autosomal-recessive mental retardation. Am. J. Hum. Genet. 2009;85:909–915. doi: 10.1016/j.ajhg.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Philippe O., Rio M., Carioux A., Plaza J.M., Guigue P., Molinari F., Boddaert N., Bole-Feysot C., Nitschke P., Smahi A. Combination of linkage mapping and microarray-expression analysis identifies NF-kappaB signaling defect as a cause of autosomal-recessive mental retardation. Am. J. Hum. Genet. 2009;85:903–908. doi: 10.1016/j.ajhg.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mochida G.H., Mahajnah M., Hill A.D., Basel-Vanagaite L., Gleason D., Hill R.S., Bodell A., Crosier M., Straussberg R., Walsh C.A. A truncating mutation of TRAPPC9 is associated with autosomal-recessive intellectual disability and postnatal microcephaly. Am. J. Hum. Genet. 2009;85:897–902. doi: 10.1016/j.ajhg.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iossifov I., Ronemus M., Levy D., Wang Z.H., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kong A., Masson G., Frigge M.L., Gylfason A., Zusmanovich P., Thorleifsson G., Olason P.I., Ingason A., Steinberg S., Rafnar T. Detection of sharing by descent, long-range phasing and haplotype imputation. Nat. Genet. 2008;40:1068–1075. doi: 10.1038/ng.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrow E.M. Genomic copy number variation in disorders of cognitive development. J. Am. Acad. Child Adolesc. Psychiatry. 2010;49:1091–1104. doi: 10.1016/j.jaac.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.