

Abstract
RAS GTPases mediate a wide variety of cellular functions, including cell proliferation, survival, and differentiation. Recent studies have revealed that germline mutations and mosaicism for classical RAS mutations, including those in HRAS, KRAS, and NRAS, cause a wide spectrum of genetic disorders. These include Noonan syndrome and related disorders (RAS/mitogen-activated protein kinase [RAS/MAPK] pathway syndromes, or RASopathies), nevus sebaceous, and Schimmelpenning syndrome. In the present study, we identified a total of nine missense, nonsynonymous mutations in RIT1, encoding a member of the RAS subfamily, in 17 of 180 individuals (9%) with Noonan syndrome or a related condition but with no detectable mutations in known Noonan-related genes. Clinical manifestations in the RIT1-mutation-positive individuals are consistent with those of Noonan syndrome, which is characterized by distinctive facial features, short stature, and congenital heart defects. Seventy percent of mutation-positive individuals presented with hypertrophic cardiomyopathy; this frequency is high relative to the overall 20% incidence in individuals with Noonan syndrome. Luciferase assays in NIH 3T3 cells showed that five RIT1 alterations identified in children with Noonan syndrome enhanced ELK1 transactivation. The introduction of mRNAs of mutant RIT1 into 1-cell-stage zebrafish embryos was found to result in a significant increase of embryos with craniofacial abnormalities, incomplete looping, a hypoplastic chamber in the heart, and an elongated yolk sac. These results demonstrate that gain-of-function mutations in RIT1 cause Noonan syndrome and show a similar biological effect to mutations in other RASopathy-related genes.
Main Text
RAS GTPases are monomeric G proteins with a molecular mass of 20–40 kDa and cycle between a GTP-bound active and a GDP-bound inactive state. The members of the RAS superfamily are structurally classified into at least five subfamilies: RAS, Rho, Rab, Sar1/Arf, and Ran families.1,2 The Ras subfamily consists of classical RAS proteins (HRAS, KRAS, and NRAS), RRAS, RRAS2 (TC21), RRAS3 (MRAS), RAPs, RAEB, RALs, RIT1, and RIT2 (RIN). RAS proteins interact with multiple effectors, including RAF kinases, phosphatidylinositol 3-kinase (PI-3 kinase), RalGDS, p120GAP, MEKK1, RIN1, AF-6, phospholipase C epsilon, and the Nore-MST1 complex, and activate multiple downstream signaling cascades.1,2 Of these signaling pathways, the RAS/mitogen-activated protein kinase (RAS/MAPK) signaling pathway plays a central role in cellular proliferation and differentiation.
Noonan syndrome (MIM 163950) is an autosomal-dominant disorder characterized by short stature, distinctive facial features, and congenital heart defects.3,4 The distinctive facial features include hypertelorism, downslanting palpebral fissures, ptosis, a webbed or short neck, and low-set, posteriorly rotated ears. Congenital heart defects, including pulmonary valve stenosis and atrial septal defects, occur in 50%–80% of individuals. Hypertrophic cardiomyopathy is observed in 20% of affected individuals. Other clinical manifestations include cryptorchidism, mild intellectual disability, bleeding tendency, and hydrops fetalis. The incidence of this syndrome is estimated to be between 1 in 1,000 to 1 in 2,500 live births. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia (JMML), a myeloproliferative disorder characterized by excessive production of myelomonocytic cells.4 Noonan syndrome exhibits phenotypic overlap with Costello syndrome (MIM 218040) and cardiofaciocutaneous (CFC) syndrome (MIM 115150).
In 2001, Tartaglia et al. identified missense mutations in protein-tyrosine phosphatase, nonreceptor type 11 (PTPN11 [MIM 176876]), which encodes the tyrosine phosphatase SHP-2 in 50% of individuals with Noonan syndrome.5 In contrast, loss-of-function or dominant-negative mutations in PTPN11 have been reported in individuals with Noonan syndrome with multiple lentigines6 (formerly referred to as LEOPARD [multiple lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonic stenosis, abnormal genitalia, retardation of growth, and sensorineural deafness] syndrome [MIM 151100]). To date, germline mutations in PTPN11, KRAS (MIM 190070), SOS1 (MIM 182530), RAF1 (MIM 164760), and NRAS (MIM 164790) have been identified in individuals with Noonan syndrome7–12 (NS1 [MIM 163950], NS3 [MIM 609942], NS4 [MIM 610733], NS5 [MIM 611553], and NS6 [MIM 613224]), and mutations in SHOC2 (MIM 602775) and CBL (MIM 165360) have been identified in two Noonan-syndrome-like syndromes13–16 (NSLH [MIM 607721] and NSLL [MIM 613563], respectively) (Figure S1, available online). Moreover, we and another group have identified germline mutations in HRAS (MIM 190020) in individuals with Costello syndrome17 and germline mutations in KRAS, BRAF (MIM 164757), MAP2K1 (MIM 176872), and MAP2K2 (MIM 601263) in individuals with CFC syndrome.18,19 Mutations in BRAF have been also identified in a small percentage of individuals with Noonan syndrome (NS7 [MIM 613706]). A line of studies have shown that a group of the above genetic disorders result from dysregulation of the RAS and downstream signaling cascade (RAS/MAPK pathway syndromes, or RASopathies).20,21 Recently, mosaicism for KRAS and HRAS mutations has been reported in nevus sebaceous and Schimmelpenning syndrome,22 further extending a spectrum of diseases with a dysregulated RAS/MAPK pathway.
To identify genetic causes of Noonan syndrome, we recruited 180 individuals with Noonan syndrome or a related phenotype; they were negative for all coding exons in PTPN11, KRAS, HRAS, and SOS1; exons 6 and 11–16 in BRAF; exons 7, 14, and 17 in RAF1; exons 2 and 3 in MAP2K1 and MAP2K2; and exon 1 in SHOC2. Further genetic analysis has been conducted according to their first diagnoses.17,23–29 This study was approved by the ethics committee of Tohoku University School of Medicine. We obtained informed consent from all subjects involved in the study. We sequenced the exomes of 14 individuals whose clinical manifestations had been confirmed to be consistent with Noonan syndrome by trained dysmorphologists. Targeted enrichment was performed with the Agilent SureSelect Human All Exon v.1 Kit for four individuals and with the SureSelect Human All Exon 50Mb kit for ten individuals. Exon-enriched DNA libraries from these 14 individuals were sequenced on the Illumina Hiseq 2000 for 91 bp (v.1 kit) or 101 bp (50Mb kit). The Burrows-Wheeler Aligner (BWA) was used to align the sequence reads to the human genome (UCSC Genome Browser hg19);30 all BWA parameters were kept at the default settings. After the removal of duplicates from the alignments, realignment around known indels, recalibration, and SNP and indel calling were performed with the Genome Analysis Toolkit (v.1.5).31 ANNOVAR was used for annotation against the RefSeq database and dbSNP.32 We identified approximately 10,000 nonsynonymous, nonsense, and splice-site variations and coding indels per individual (Table S1). Filtering steps using variant databases (dbSNP132 and the 1000 Genome Project database) and in-house exome data were carried out, resulting in the identification of 122–282 variants per individual. By visual inspection of the generated data, four heterozygous RIT1 (MIM 609591; RefSeq accession number NM_006912.5) variants (c.246T>G [p.Phe82Leu], c.265T>C [p.Tyr89His], c.270G>T [p.Met90Ile], and c.284G>C [p.Gly95Ala]) were found in four individuals. Sanger sequencing validated the heterozygous state of the four variants. We did not find any other strong candidate genes in the results of exome sequencing.
RIT1 shares approximately 50% sequence identity with RAS, has an additional N-terminal extension, and does not possess a C-terminal CAAX motif, a specific motif for posttranslational modification.33,34 RIT1 is located in chromosomal region 1q22 and consists of six exons. We analyzed an additional 166 individuals diagnosed with Noonan syndrome or a related disorder but without mutations in known genes.17,23–29 Sanger sequencing of all coding exons in RIT1 in the 166 individuals showed that 13 in 166 individuals had changes. Combining with the 4 in 14 individuals from exome sequencing, a total of nine missense, nonsynonymous mutations were identified in 17 of 180 (9%) individuals who were suspected to have Noonan syndrome or a related disorder (Table 1 and Figures 1A–1L). The identified germline RIT1 mutations encode alterations located in the G1 domain (c.104G>C [p.Ser35Thr]); the switch I region, involving the G2 domain (c.170C>G [p.Ala57Gly]); and the switch II region, corresponding to RAS (c.242A>G [p.Glu81Gly], c.244T>G [p.Phe82Val], c.246T>G [p.Phe82Leu], c.247A>C [p.Thr83Pro], c.265T>C [p.Tyr89His], c.270G>T [p.Met90Ile], and c.284G>C [p.Gly95Ala]) (Figure S2). Amino acids where alterations are located are conserved among species (Figure S3). The RIT1 mutations encode alterations clustered in the switch II region. In contrast, HRAS germline mutations identified in Costello syndrome are clustered at codon 12 and 13 in the region encoding the G1 domain17 (Figure 1M). Mutations in parents were not identified in seven families. These mutations are apparently de novo, but biologic confirmation of parentage was not performed. One mutation, c.104G>C, was inherited from a mother with a Noonan syndrome phenotype (Table 1). None of these mutations were identified in 480 controls.
Table 1.
Mutations in RIT1, Family Status, and Heart Defects of Mutation-Positive Individuals
| Subject | Exon | Nucleotide Changea | Amino Acid Changeb | Father | Mother | HCMc | PSc | Other Heart Defectsc |
|---|---|---|---|---|---|---|---|---|
| NS414 | 2 | c.104G>C | p.Ser35Thr | WT | p.Ser35Thr | + | – | MVP, MR |
| KCC27 | 2 | c.104G>C | p.Ser35Thr | NA | NA | + | + | – |
| NS43 | 4 | c.170C>G | p.Ala57Gly | NA | NA | + | – | MR, TR |
| NS185 | 4 | c.170C>G | p.Ala57Gly | NA | NA | + | + | ASD, PDA |
| NS216 | 4 | c.170C>G | p.Ala57Gly | NA | NA | + | – | – |
| NS402 | 4 | c.170C>G | p.Ala57Gly | WT | WT | + | + | – |
| NS168 | 5 | c.242A>G | p.Glu81Gly | NA | NA | – | + | VSD |
| NS410 | 5 | c.244T>G | p.Phe82Val | WT | WT | + | – | – |
| NS358 | 5 | c.246T>G | p.Phe82Leu | WT | WT | – | + | ASD |
| NS465 | 5 | c.246T>G | p.Phe82Leu | NA | NA | – | + | VSD |
| NS276 | 5 | c.247A>C | p.Thr83Pro | WT | WT | + | + | PVC |
| KCC8 | 5 | c.265T>C | p.Tyr89His | NA | NA | + | + | – |
| KCC38 | 5 | c.270G>T | p.Met90Ile | WT | WT | + | + | ASD, VSD, PDA |
| NS234 | 5 | c.284G>C | p.Gly95Ala | WT | WT | – | – | ASD |
| NS265 | 5 | c.284G>C | p.Gly95Ala | WT | WT | + | + | – |
| Og22 | 5 | c.284G>C | p.Gly95Ala | NA | NA | – | – | – |
| Og45 | 5 | c.284G>C | p.Gly95Ala | NA | NA | + | + | ASD |
PCR primers used for sequencing are shown in Table S3. Nucleotide changes are not located in CpG dinucleotides, suggesting that they exhibit baseline mutation rates with a phenotypic filtering effect and that only these mutations lead to this phenotype. Abbreviations are as follows: WT, wild-type; HCM, hypertrophic cardiomyopathy; PS, pulmonic stenosis; MVP, mitral valve prolapse; MR, mitral regurgitation; TR, tricuspid regurgitation; ASD, atrial septal defect; PDA, patent ductus arteriosus; VSD, ventricular septal defect; PVC, premature ventricular contraction; and NA, not available.
RefSeq NM_006912.5.
RefSeq NP_008843.1.
HCM and heart anomalies were diagnosed by echocardiography.
Figure 1.
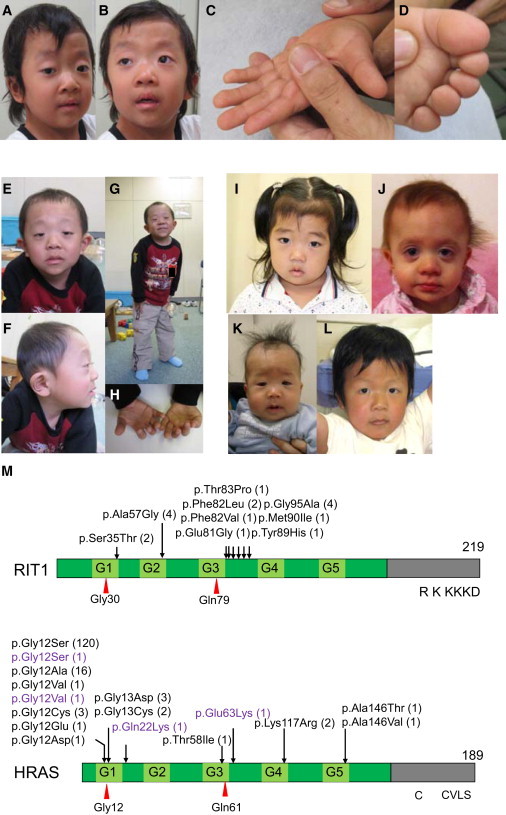
Photographs of Six Individuals in whom RIT1 Mutations Were Identified
(A–D) KCC38 at 3 years of age. Broad forehead, sparse eyebrows, ptosis, hypertelorism, and hyperpigmentation were observed (A and B). Prominent finger pads were observed (C and D).
(E–H) NS358 at 4 years of age. Hypertelorism, epicanthus, sparse eyebrows, and low-set ears were observed.
(I) NS414 at 3 years of age.
(J) NS465 at 1 year of age.
(K) NS276 at 5 months.
(L) NS265 at 5 years of age.
(M) Structure and identified germline alterations in RIT1 and HRAS. HRAS alterations identified in individuals with Costello syndrome were described before20 or shown in The RAS/MAPK Syndromes Homepage (see Web Resources). HRAS alterations identified in individuals with congenital myopathy with excess of muscle spindles35 are indicated in purple.
We obtained specific consent for photographs from six individuals.
To assess the functional consequences of RIT1 mutations identified in affected individuals, we introduced a single-base substitution (p.Ser35Thr, p.Ala57Gly, p.Glu81Gly, p.Phe82Leu, or p.Gly95Ala) identified in individuals with Noonan syndrome into a pCAGGS expression vector36 harboring RIT1 cDNA. As an experimental control, cDNAs harboring RIT1 c.89G>T (p.Gly30Val), c.104G>C (p.Ser35Asn), and c.236A>T (p.Gln79Leu) and Braf c.1910T>A (p.Val637Glu) (RefSeq NM_139294), which corresponds to oncogenic p.Val600Glu in humans, were also generated. RIT1 p.Gly30Val and p.Gln79Leu correspond to oncogenic RAS alterations p.Gly12Val and p.Gln61Leu, respectively. We introduced pFR-luc, pFA2-Elk1, phRLnull-luc, and wild-type (WT) or mutant expression constructs of RIT1 into NIH 3T3 cells to examine the transcriptional activation by ELK1,18,33 a transcription factor that is activated by MAPK. The results revealed that compared with the WT cDNA, all RIT1 mutations exhibited significant activation. RIT1 p.Gln79Leu, followed by p.Gly95Ala, p.Ala57Gly, p.Phe82Leu, and p.Glu81Gly, showed the highest ELK1 transactivation, as also shown in a past study37 (Figure 2A). The c.104G>C (p.Ser35Thr) substitution was identified in two affected individuals. RIT1 p.Ser35Asn, which corresponds to dominant-negative alteration p.Ser17Asn in RAS, has been used as a dominant-negative substitution in cell experiments.38 To examine the functional consequence of p.Ser35Thr, identified in affected individuals, we compared the ELK1 transactivation in cells expressing p.Ser35Thr and those expressing p.Ser35Asn. Enhanced ELK1 transactivation was observed in cells expressing p.Ser35Thr, but not in cells expressing p.Ser35Asn (Figure 2B). These results suggest that RIT1 mutations identified in affected individuals were gain-of-function mutations.
Figure 2.
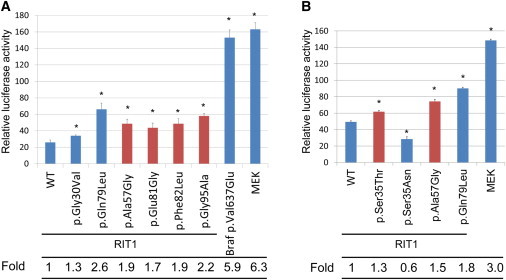
Stimulation of ELK Transcription in NIH 3T3 Cells Expressing RIT1 Germline Mutations
(A) The ELK1-GAL4 vector and the GAL4 luciferase trans-reporter vector were transiently transfected with various RIT1 germline mutations and activating mutations in BRAF and MAP2K1 in NIH 3T3 cells. c.1910T>A (p.Val637Glu) in mouse Braf corresponds to oncogenic c.1799T>A (p.Val600Glu) in human BRAF. Relative luciferase activity was calculated by normalization to the activity of a cotransfected control vector, phRLnull-luc, containing distinguishable R. reniformis luciferase.
(B) ELK1 transactivation in cells expressing p.Ser35Thr, identified in individuals with Noonan syndrome, and p.Ser35Asn, were examined. p.Ser35Asn corresponds to dominant-negative alteration p.Ser17Asn in RAS.
Results are expressed as the means of quadruplicate (A) and triplicate (B) samples. Error bars represent the SDs of mean values. Red bars indicate germline RIT1 mutations identified in Noonan syndrome. The following abbreviation is used: WT, wild-type. ∗p < 0.01 by t test.
RIT1 is expressed ubiquitously in embryonic and adult tissues.33,34 Rit1-null mice have been shown to grow to adulthood without any apparent abnormalities;39 hence, physiological roles of RIT1 in development remain unknown. To examine the developmental effect of identified mutations, we introduced mRNA of the WT and three RIT1 mutations (c.236A>T [p.Gln79Leu], c.242A>G [p.Glu81Gly], and c.284G>C [p.Gly95Ala]) into 1-cell-stage zebrafish embryos and observed the phenotype at 11 hr postfertilization (hpf). An oval-shaped egg sack, a typical manifestation of the gastrulation defect, was observed in embryos expressing RIT1 alterations (Figure 3A). This characteristic shape change was also observed in zebrafish expressing gain-of-function mutations of human NRAS40. Next, we observed the phenotype at later stages (48–52 hpf) (Figure 3B and Figure S4). The introduction of the WT mRNA did not interfere with the normal development, resulting in generally normal morphology in 125/132 (94.7%) embryos; however, 7/132 (5.3%) embryos had limited mild craniofacial and heart abnormalities (Table 2). In contrast, a combined manifestation of craniofacial abnormalities, pericardial edema, and an elongated yolk sac was observed in 66.1%, 52.4%, and 40.5% of embryos expressing p.Gln79Leu, p.Glu81Gly, and p.Gly95Ala, respectively. Development was severely retarded in approximately 7% of embryos expressing RIT1 alterations; these embryos displayed the formation of a disorganized round body shape with a dysmorphic head and body trunk. In the head region, a hypoplastic brain, especially in the telencephalic area, was observed and resulted in misshapen morphology. In the ventral part of the head, the jaw structure was also hypoplastic, and the eyes were translocated medially. These morphological changes gave a cyclopia-like appearance. The ventral sides of the eyes were small, and coloboma along with a loss of pigment was evident (Figure 3B). These phenotypic changes are compatible with the gastrulation defect observed at 11 hpf (Figure 3A). Because the Fgf/Ras/MAPK signaling cascade plays an essential role in the convergent and extension cell movement during gastrulation,41 perturbation by the RIT1 alterations could cause abnormal cell movement in the axial portions and thus lead to an elongated shape of the egg and the hypoplastic ventral side of the head. Detailed inspection of the morphology in mutant-injected embryos revealed abnormal cardiogenesis, namely, incomplete looping, hypoplastic chambers, and stagnation of blood flow in the yolk sac (Figure 3B). Although the atrium of these hearts beat regularly, the ventricle seemed to twitch passively by the contraction of the atrium (Movies S1, S2, S3, S4, S5, and S6). These results indicate that activating mutations in RIT1 induce abnormal craniofacial and heart defects in zebrafish.
Figure 3.
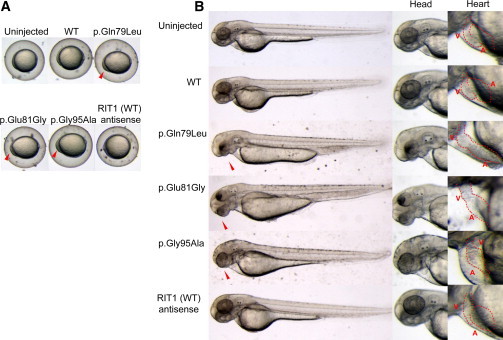
Morphology of Embryos Injected with the WT or Mutant RIT1 mRNA
In vitro transcription of each mRNA was performed with the mMESSAGE mMACHINE kit (Applied Biosystems) according to the manufacturer’s instructions. Synthesized mRNAs were purified with G-50 Micro Columns (GE Healthcare) and subsequently adjusted to a 300 ng/μl concentration for microinjection. Approximately 1 nl (300 pg) of RNA in water with 0.2% phenol red was injected into the cytoplasm of 1-cell-stage zebrafish embryos. Injected embryos were incubated at 28°C until observation.
(A) At 11 hpf, the shapes of the embryos injected with the WT sense or antisense mRNA were round, a normal morphology as observed in the uninjected embryos. In contrast, embryos expressing mutations (c.236A>T [p.Gln79Leu], c.242A>G [p.Glu81Gly], and c.284G>C [p.Gly95Ala]) are oval and compressed along the dorsal-ventral axis, indicative of a gastrulation defect. Note that cells have a hump in the head region at the anterior end of the body axis, the earliest manifestation of a craniofacial defect.
(B) Lateral views at 48 hpf are shown. Embryos expressing mutations (c.236A>T [p.Gln79Leu], c.242A>G [p.Glu81Gly], and c.284G>C [p.Gly95Ala]) formed swollen yolk sacs equally along the anterior posterior axis but did not show narrowing in the caudal half, which was clearly visible in the uninjected embryos and in those injected with the WT sense or antisense mRNA. In the craniofacial area, misshapen head and jaw structures and small eyes with hypoplasia on the ventral side were observed (middle panel); these phenotypes are consistent with the gastrulation defect. Shapes of the hearts (highlighted by red dotted lines) are shown in the right panel at a higher magnification. Normal looping of the heart tube and correct formation of two distinct chambers are observed in embryos injected with the WT sense or antisense mRNA. When mutations (c.236A>T [p.Gln79Leu], c.242A>G [p.Glu81Gly], and c.284G>C [p.Gly95Ala]) were expressed, looping was incomplete, resulting in stretched straight heart tubes. Constrictions at the atrial-ventricular canal are obscure, and the heart chambers are hypoplastic. Abbreviations are as follows: A, atrium; and V, ventricle.
Table 2.
Morphologic Abnormality at 48–52 hpf of Zebrafish Embryos Injected with WT or Mutant RNA at the 1-Cell Stage
| No Abnormalities | Heart and Facial Abnormalitiesa | Severely Disorganizedb | Total Number of Embryos | |
|---|---|---|---|---|
| WT | 125 | 7 (5.3%) | 0 (0%) | 132 |
| p.Gln79Leu | 31 | 78 (66.1%) | 9 (7.6%) | 118 |
| p.Glu81Gly | 42 | 55 (52.4%) | 8 (7.6%) | 105 |
| p.Gly95Ala | 44 | 34 (40.5%) | 6 (7.1%) | 84 |
Craniofacial abnormalities, pericardial heart edema, and an elongated yolk sac were observed.
Disorganized round body shape with a dysmorphic head and body trunk as shown in Figure S4.
RIT1-mutation-positive individuals showed a distinct facial appearance, congenital heart defects, and skeletal abnormalities and were diagnosed with Noonan syndrome by diagnostic criteria developed by van der Burgt (Figures 1A–1L and Table 1).4 Two individuals (NS358 and KCC38) were suspected to have CFC syndrome in the infantile period because of curly, sparse hair, a high cranial vault, and hypoplasia of the supraorbital ridges. Nine individuals showed perinatal abnormality, including polyhydramnios, nuchal translucency, and chylothorax (Table S2). It is of note that one individual (Og45) showing severe pleural effusion, hypertrophic cardiomyopathy, and hepatomegaly that ended in severe body edema and compromised circulation died 53 days after birth. Seven individuals showed high birth weight, probably as a result of subcutaneous edema, which is a typical manifestation observed in individuals with Noonan syndrome.4 Out of 17 affected individuals, 16 (94%) had heart defects (Table 1): hypertrophic cardiomyopathy (HCM) in 12 (71%) individuals, pulmonary stenosis in 11 (65%) individuals, and atrial septal defects in 5 (29%) individuals. The incidence of pulmonic stenosis and mild cognitive defects is close to the overall incidence of these features in Noonan syndrome cohorts. By contrast, the incidence of HCM is far greater than in individuals with Noonan syndrome overall (25/118 in Noonan syndrome42 versus 12/17 in individuals with RIT1 mutations; p < 0.0001 by Fisher’s exact test). It is of note that a high frequency of HCM (70%) was also reported in individuals with RAF1 mutations.10,11,24 It is possible that RIT1 interacts with RAF1 and that gain-of-function mutations in RIT1 and RAF1 exert similar effects in heart development.
Somatic alterations in classical RAS have been identified in approximately 30% of tumors.43 Noonan syndrome and related disorders confer an increased risk of developing malignant tumors.20,44 In a summary of the literature, it has been reported that 45 of 1,151 (3.9%) individuals with Noonan syndrome (but with an unknown mutation status) developed malignant tumors.44 Since molecular analysis became available, gene-specific association with malignant tumors has been revealed. The association with JMML, a myeloproliferative disorder characterized by the excessive production of myelomonocytic cells, has been reported in individuals with PTPN11, CBL, and KRAS mutations. Recent reports showed that two individuals with SOS1 mutations developed embryonal rhabdomyosarcoma.45,46 A somatic RIT1 variant, c.270G>A (p.Met90Ile), has been identified in lung cancer (COSMIC database). In the present cohort, 1 (NS168) of 17 individuals with RIT1 c.242A>G (p.Glu81Gly) developed acute lymphoblastic leukemia at the age of 5 years. The child was treated by a standard protocol and has remained in complete remission. Examining whether gain-of-function mutations in RIT1 cause tumorigenesis will require further study.
RIT1 has been isolated as a cDNA encoding highly conserved G3 and G4 domains of RAS proteins33 or identified as a gene encoding a protein related to Drosophila Ric, a calmodulin-binding RAS-related GTPase.34 RIT1 p.Gln79Leu, which corresponds to RAS p.Gln61Leu, is implicated in transforming NIH 3T3 cells, neurite outgrowth in neuronal cells, and the activation of ERK and p38 MAPK in a cell-specific manner.37,38,47 In this study, enhanced ELK1 transactivation was observed in cells expressing mutant RIT1 cDNAs. Previous studies showed that enhanced ELK transactivation was observed in NIH 3T3 cells expressing HRAS, KRAS, BRAF, and RAF1 mutations identified in individuals with Costello, CFC, and Noonan syndromes.17,18,24 Gastrulation defects observed in zebrafish embryos expressing RIT1 alterations (p.Glu81Gly, p.Gly95Ala, or p.Gln79Leu) were also reported in zebrafish embryos expressing an activating mutation in NRAS, BRAF, MAP2K1, or MAP2K2.40,48 Taken together, these results indicate that gain-of-function mutations in RIT1 cause Noonan syndrome and show a similar effect to mutations in other RASopathy-related genes in human development.
Herein, we used whole-exome sequencing to identify germline RIT1 mutations in individuals with Noonan syndrome, a disorder of the RASopathies. Mutations in PTPN11, SOS1, RAF1, KRAS, BRAF, and NRAS have been identified in 41%, 11%, 5%, 1%, 0.8%, and 0.2% of all cases, respectively,3 and thus the frequency of RIT1 mutations in Noonan syndrome might be similar to that of RAF1 mutations. Our findings will improve diagnostic accuracy of Noonan syndrome and provide a clue to understanding the disorder’s pathogenesis, including therapeutic approaches.
Acknowledgments
The authors thank the families and the doctors who participated in this study. We are grateful to Jun-ichi Miyazaki at Osaka University for supplying the pCAGGS expression vector. We thank Yoko Narumi, Tomoko Kobayashi, Shoko Komatsuzaki, Yu Abe, Yuka Saito, Rumiko Izumi, Mitsuji Moriya, and Masako Yaoita for contributing to routine diagnostic work and Yoko Tateda, Kumi Kato, and Riyo Takahashi for their technical assistance. We are grateful to Eric Haan for sending samples of Noonan syndrome and related disorders. We also acknowledge the support of the Biomedical Research Core of Tohoku University Graduate School of Medicine. This work was supported by the Funding Program for the Next Generation of World-Leading Researchers (NEXT Program) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT) to Y.A. (LS004), by Grants-in-Aids from MEXT, from the Japan Society for the Promotion of Science, and from the Ministry of Health, Labor, and Welfare to Y.M. and T.N. This work was supported in part by the National Cancer Center Research and Development Fund (23-22-11).
Supplemental Data
The heart of a zebrafish embryo beats regularly and maintains constant blood flow at 48 hpf.
The heart of a zebrafish embryo injected with WT RIT1 mRNA shows regular contraction and normal blood circulation similarly to that of the WT embryo at 48 hpf.
The heart of a zebrafish embryo injected with RIT1 c.236A>T (p.Gln79Leu) mutant mRNA shows hypoplastic chambers with an unclear atrial-ventricular junction and incomplete looping and a resultant stretched configuration of chambers. Beating is weak and arrhythmic. Blood circulation is feeble.
The heart of a zebrafish embryo injected with RIT1 c.242A>G (p.Glu81Gly) mutant mRNA shows the same morphology, heartbeat, and circulation as that of the embryo injected with c.236A>T (p.Gln79Leu) mRNA.
The heart of a zebrafish embryo injected with RIT1 c.284G>C (p.Gly95Ala) mutant mRNA shows the same morphology, heartbeat, and circulation as those of other mutants (incomplete looping, hypoplasia, and weak circulation). The atrium of this heart is abnormally dilated, and pericardial edema is formed with inflation of the pericardial sac, a sign of a circulatory defect.
The heart of a zebrafish embryo injected with antisense WT RIT1 mRNA beats regularly and maintains constant blood flow similarly to that of the WT embryo.
Web Resources
The URLs for data presented herein are as follows:
Catalogue of Somatic Mutations in Cancer (COSMIC), http://www.sanger.ac.uk/genetics/CGP/cosmic/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
The RAS/MAPK Syndromes Homepage, http://www.medgen.med.tohoku.ac.jp/RasMapk%20syndromes.html
References
- 1.Takai Y., Sasaki T., Matozaki T. Small GTP-binding proteins. Physiol. Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 2.Giehl K. Oncogenic Ras in tumour progression and metastasis. Biol. Chem. 2005;386:193–205. doi: 10.1515/BC.2005.025. [DOI] [PubMed] [Google Scholar]
- 3.Romano A.A., Allanson J.E., Dahlgren J., Gelb B.D., Hall B., Pierpont M.E., Roberts A.E., Robinson W., Takemoto C.M., Noonan J.A. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. 2010;126:746–759. doi: 10.1542/peds.2009-3207. [DOI] [PubMed] [Google Scholar]
- 4.van der Burgt I. Noonan syndrome. Orphanet J. Rare Dis. 2007;2:4. doi: 10.1186/1750-1172-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tartaglia M., Mehler E.L., Goldberg R., Zampino G., Brunner H.G., Kremer H., van der Burgt I., Crosby A.H., Ion A., Jeffery S. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001;29:465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 6.Digilio M.C., Conti E., Sarkozy A., Mingarelli R., Dottorini T., Marino B., Pizzuti A., Dallapiccola B. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am. J. Hum. Genet. 2002;71:389–394. doi: 10.1086/341528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schubbert S., Zenker M., Rowe S.L., Böll S., Klein C., Bollag G., van der Burgt I., Musante L., Kalscheuer V., Wehner L.E. Germline KRAS mutations cause Noonan syndrome. Nat. Genet. 2006;38:331–336. doi: 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- 8.Roberts A.E., Araki T., Swanson K.D., Montgomery K.T., Schiripo T.A., Joshi V.A., Li L., Yassin Y., Tamburino A.M., Neel B.G., Kucherlapati R.S. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat. Genet. 2007;39:70–74. doi: 10.1038/ng1926. [DOI] [PubMed] [Google Scholar]
- 9.Tartaglia M., Pennacchio L.A., Zhao C., Yadav K.K., Fodale V., Sarkozy A., Pandit B., Oishi K., Martinelli S., Schackwitz W. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat. Genet. 2007;39:75–79. doi: 10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- 10.Pandit B., Sarkozy A., Pennacchio L.A., Carta C., Oishi K., Martinelli S., Pogna E.A., Schackwitz W., Ustaszewska A., Landstrom A. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 2007;39:1007–1012. doi: 10.1038/ng2073. [DOI] [PubMed] [Google Scholar]
- 11.Razzaque M.A., Nishizawa T., Komoike Y., Yagi H., Furutani M., Amo R., Kamisago M., Momma K., Katayama H., Nakagawa M. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat. Genet. 2007;39:1013–1017. doi: 10.1038/ng2078. [DOI] [PubMed] [Google Scholar]
- 12.Cirstea I.C., Kutsche K., Dvorsky R., Gremer L., Carta C., Horn D., Roberts A.E., Lepri F., Merbitz-Zahradnik T., König R. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat. Genet. 2010;42:27–29. doi: 10.1038/ng.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cordeddu V., Di Schiavi E., Pennacchio L.A., Ma’ayan A., Sarkozy A., Fodale V., Cecchetti S., Cardinale A., Martin J., Schackwitz W. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat. Genet. 2009;41:1022–1026. doi: 10.1038/ng.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loh M.L., Sakai D.S., Flotho C., Kang M., Fliegauf M., Archambeault S., Mullighan C.G., Chen L., Bergstraesser E., Bueso-Ramos C.E. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 2009;114:1859–1863. doi: 10.1182/blood-2009-01-198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niemeyer C.M., Kang M.W., Shin D.H., Furlan I., Erlacher M., Bunin N.J., Bunda S., Finklestein J.Z., Sakamoto K.M., Gorr T.A. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat. Genet. 2010;42:794–800. doi: 10.1038/ng.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pérez B., Mechinaud F., Galambrun C., Ben Romdhane N., Isidor B., Philip N., Derain-Court J., Cassinat B., Lachenaud J., Kaltenbach S. Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J. Med. Genet. 2010;47:686–691. doi: 10.1136/jmg.2010.076836. [DOI] [PubMed] [Google Scholar]
- 17.Aoki Y., Niihori T., Kawame H., Kurosawa K., Ohashi H., Tanaka Y., Filocamo M., Kato K., Suzuki Y., Kure S., Matsubara Y. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat. Genet. 2005;37:1038–1040. doi: 10.1038/ng1641. [DOI] [PubMed] [Google Scholar]
- 18.Niihori T., Aoki Y., Narumi Y., Neri G., Cavé H., Verloes A., Okamoto N., Hennekam R.C., Gillessen-Kaesbach G., Wieczorek D. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat. Genet. 2006;38:294–296. doi: 10.1038/ng1749. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez-Viciana P., Tetsu O., Tidyman W.E., Estep A.L., Conger B.A., Cruz M.S., McCormick F., Rauen K.A. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–1290. doi: 10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- 20.Aoki Y., Niihori T., Narumi Y., Kure S., Matsubara Y. The RAS/MAPK syndromes: novel roles of the RAS pathway in human genetic disorders. Hum. Mutat. 2008;29:992–1006. doi: 10.1002/humu.20748. [DOI] [PubMed] [Google Scholar]
- 21.Tidyman W.E., Rauen K.A. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 2009;19:230–236. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groesser L., Herschberger E., Ruetten A., Ruivenkamp C., Lopriore E., Zutt M., Langmann T., Singer S., Klingseisen L., Schneider-Brachert W. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat. Genet. 2012;44:783–787. doi: 10.1038/ng.2316. [DOI] [PubMed] [Google Scholar]
- 23.Abe Y., Aoki Y., Kuriyama S., Kawame H., Okamoto N., Kurosawa K., Ohashi H., Mizuno S., Ogata T., Kure S., Costello and CFC syndrome study group in Japan Prevalence and clinical features of Costello syndrome and cardio-facio-cutaneous syndrome in Japan: findings from a nationwide epidemiological survey. Am. J. Med. Genet. A. 2012;158A:1083–1094. doi: 10.1002/ajmg.a.35292. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi T., Aoki Y., Niihori T., Cavé H., Verloes A., Okamoto N., Kawame H., Fujiwara I., Takada F., Ohata T. Molecular and clinical analysis of RAF1 in Noonan syndrome and related disorders: dephosphorylation of serine 259 as the essential mechanism for mutant activation. Hum. Mutat. 2010;31:284–294. doi: 10.1002/humu.21187. [DOI] [PubMed] [Google Scholar]
- 25.Komatsuzaki S., Aoki Y., Niihori T., Okamoto N., Hennekam R.C., Hopman S., Ohashi H., Mizuno S., Watanabe Y., Kamasaki H. Mutation analysis of the SHOC2 gene in Noonan-like syndrome and in hematologic malignancies. J. Hum. Genet. 2010;55:801–809. doi: 10.1038/jhg.2010.116. [DOI] [PubMed] [Google Scholar]
- 26.Narumi Y., Aoki Y., Niihori T., Neri G., Cavé H., Verloes A., Nava C., Kavamura M.I., Okamoto N., Kurosawa K. Molecular and clinical characterization of cardio-facio-cutaneous (CFC) syndrome: overlapping clinical manifestations with Costello syndrome. Am. J. Med. Genet. A. 2007;143A:799–807. doi: 10.1002/ajmg.a.31658. [DOI] [PubMed] [Google Scholar]
- 27.Narumi Y., Aoki Y., Niihori T., Sakurai M., Cavé H., Verloes A., Nishio K., Ohashi H., Kurosawa K., Okamoto N. Clinical manifestations in patients with SOS1 mutations range from Noonan syndrome to CFC syndrome. J. Hum. Genet. 2008;53:834–841. doi: 10.1007/s10038-008-0320-0. [DOI] [PubMed] [Google Scholar]
- 28.Niihori T., Aoki Y., Okamoto N., Kurosawa K., Ohashi H., Mizuno S., Kawame H., Inazawa J., Ohura T., Arai H. HRAS mutants identified in Costello syndrome patients can induce cellular senescence: possible implications for the pathogenesis of Costello syndrome. J. Hum. Genet. 2011;56:707–715. doi: 10.1038/jhg.2011.85. [DOI] [PubMed] [Google Scholar]
- 29.Saito Y., Aoki Y., Muramatsu H., Makishima H., Maciejewski J.P., Imaizumi M., Rikiishi T., Sasahara Y., Kure S., Niihori T. Casitas B-cell lymphoma mutation in childhood T-cell acute lymphoblastic leukemia. Leuk. Res. 2012;36:1009–1015. doi: 10.1016/j.leukres.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee C.H., Della N.G., Chew C.E., Zack D.J. Rin, a neuron-specific and calmodulin-binding small G-protein, and Rit define a novel subfamily of ras proteins. J. Neurosci. 1996;16:6784–6794. doi: 10.1523/JNEUROSCI.16-21-06784.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wes P.D., Yu M., Montell C. RIC, a calmodulin-binding Ras-like GTPase. EMBO J. 1996;15:5839–5848. [PMC free article] [PubMed] [Google Scholar]
- 35.van der Burgt I., Kupsky W., Stassou S., Nadroo A., Barroso C., Diem A., Kratz C.P., Dvorsky R., Ahmadian M.R., Zenker M. Myopathy caused by HRAS germline mutations: implications for disturbed myogenic differentiation in the presence of constitutive HRas activation. J. Med. Genet. 2007;44:459–462. doi: 10.1136/jmg.2007.049270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niwa H., Yamamura K., Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 37.Rusyn E.V., Reynolds E.R., Shao H., Grana T.M., Chan T.O., Andres D.A., Cox A.D. Rit, a non-lipid-modified Ras-related protein, transforms NIH3T3 cells without activating the ERK, JNK, p38 MAPK or PI3K/Akt pathways. Oncogene. 2000;19:4685–4694. doi: 10.1038/sj.onc.1203836. [DOI] [PubMed] [Google Scholar]
- 38.Shi G.X., Andres D.A. Rit contributes to nerve growth factor-induced neuronal differentiation via activation of B-Raf-extracellular signal-regulated kinase and p38 mitogen-activated protein kinase cascades. Mol. Cell. Biol. 2005;25:830–846. doi: 10.1128/MCB.25.2.830-846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai W., Rudolph J.L., Harrison S.M., Jin L., Frantz A.L., Harrison D.A., Andres D.A. An evolutionarily conserved Rit GTPase-p38 MAPK signaling pathway mediates oxidative stress resistance. Mol. Biol. Cell. 2011;22:3231–3241. doi: 10.1091/mbc.E11-05-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Runtuwene V., van Eekelen M., Overvoorde J., Rehmann H., Yntema H.G., Nillesen W.M., van Haeringen A., van der Burgt I., Burgering B., den Hertog J. Noonan syndrome gain-of-function mutations in NRAS cause zebrafish gastrulation defects. Dis Model Mech. 2011;4:393–399. doi: 10.1242/dmm.007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fürthauer M., Van Celst J., Thisse C., Thisse B. Fgf signalling controls the dorsoventral patterning of the zebrafish embryo. Development. 2004;131:2853–2864. doi: 10.1242/dev.01156. [DOI] [PubMed] [Google Scholar]
- 42.Burch M., Sharland M., Shinebourne E., Smith G., Patton M., McKenna W. Cardiologic abnormalities in Noonan syndrome: phenotypic diagnosis and echocardiographic assessment of 118 patients. J. Am. Coll. Cardiol. 1993;22:1189–1192. doi: 10.1016/0735-1097(93)90436-5. [DOI] [PubMed] [Google Scholar]
- 43.Schubbert S., Shannon K., Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 44.Kratz C.P., Rapisuwon S., Reed H., Hasle H., Rosenberg P.S. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am. J. Med. Genet. C. Semin. Med. Genet. 2011;157:83–89. doi: 10.1002/ajmg.c.30300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Denayer E., Devriendt K., de Ravel T., Van Buggenhout G., Smeets E., Francois I., Sznajer Y., Craen M., Leventopoulos G., Mutesa L. Tumor spectrum in children with Noonan syndrome and SOS1 or RAF1 mutations. Genes Chromosomes Cancer. 2010;49:242–252. doi: 10.1002/gcc.20735. [DOI] [PubMed] [Google Scholar]
- 46.Jongmans M.C.J., Hoogerbrugge P.M., Hilkens L., Flucke U., van der Burgt I., Noordam K., Ruiterkamp-Versteeg M., Yntema H.G., Nillesen W.M., Ligtenberg M.J.L. Noonan syndrome, the SOS1 gene and embryonal rhabdomyosarcoma. Genes Chromosomes Cancer. 2010;49:635–641. doi: 10.1002/gcc.20773. [DOI] [PubMed] [Google Scholar]
- 47.Hynds D.L., Spencer M.L., Andres D.A., Snow D.M. Rit promotes MEK-independent neurite branching in human neuroblastoma cells. J. Cell Sci. 2003;116:1925–1935. doi: 10.1242/jcs.00401. [DOI] [PubMed] [Google Scholar]
- 48.Anastasaki C., Estep A.L., Marais R., Rauen K.A., Patton E.E. Kinase-activating and kinase-impaired cardio-facio-cutaneous syndrome alleles have activity during zebrafish development and are sensitive to small molecule inhibitors. Hum. Mol. Genet. 2009;18:2543–2554. doi: 10.1093/hmg/ddp186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The heart of a zebrafish embryo beats regularly and maintains constant blood flow at 48 hpf.
The heart of a zebrafish embryo injected with WT RIT1 mRNA shows regular contraction and normal blood circulation similarly to that of the WT embryo at 48 hpf.
The heart of a zebrafish embryo injected with RIT1 c.236A>T (p.Gln79Leu) mutant mRNA shows hypoplastic chambers with an unclear atrial-ventricular junction and incomplete looping and a resultant stretched configuration of chambers. Beating is weak and arrhythmic. Blood circulation is feeble.
The heart of a zebrafish embryo injected with RIT1 c.242A>G (p.Glu81Gly) mutant mRNA shows the same morphology, heartbeat, and circulation as that of the embryo injected with c.236A>T (p.Gln79Leu) mRNA.
The heart of a zebrafish embryo injected with RIT1 c.284G>C (p.Gly95Ala) mutant mRNA shows the same morphology, heartbeat, and circulation as those of other mutants (incomplete looping, hypoplasia, and weak circulation). The atrium of this heart is abnormally dilated, and pericardial edema is formed with inflation of the pericardial sac, a sign of a circulatory defect.
The heart of a zebrafish embryo injected with antisense WT RIT1 mRNA beats regularly and maintains constant blood flow similarly to that of the WT embryo.